
Familial Partial Lipodystrophy—Literature Review and Report of a Novel Variant in PPARG Expanding the Spectrum of Disease-Causing Alterations in FPLD3

 , and
, and
Abstract
:1. Introduction
2. Familial Partial Lipodystrophy (FPLD)
2.1. Familial Partial Lipodystrophy Type 2 (Dunnigan Type)
2.2. Familial Partial Lipodystrophy Type 3
3. Metabolic Abnormalities in Lipodystrophy
3.1. Insulin Resistance (IR) and Diabetes Mellitus
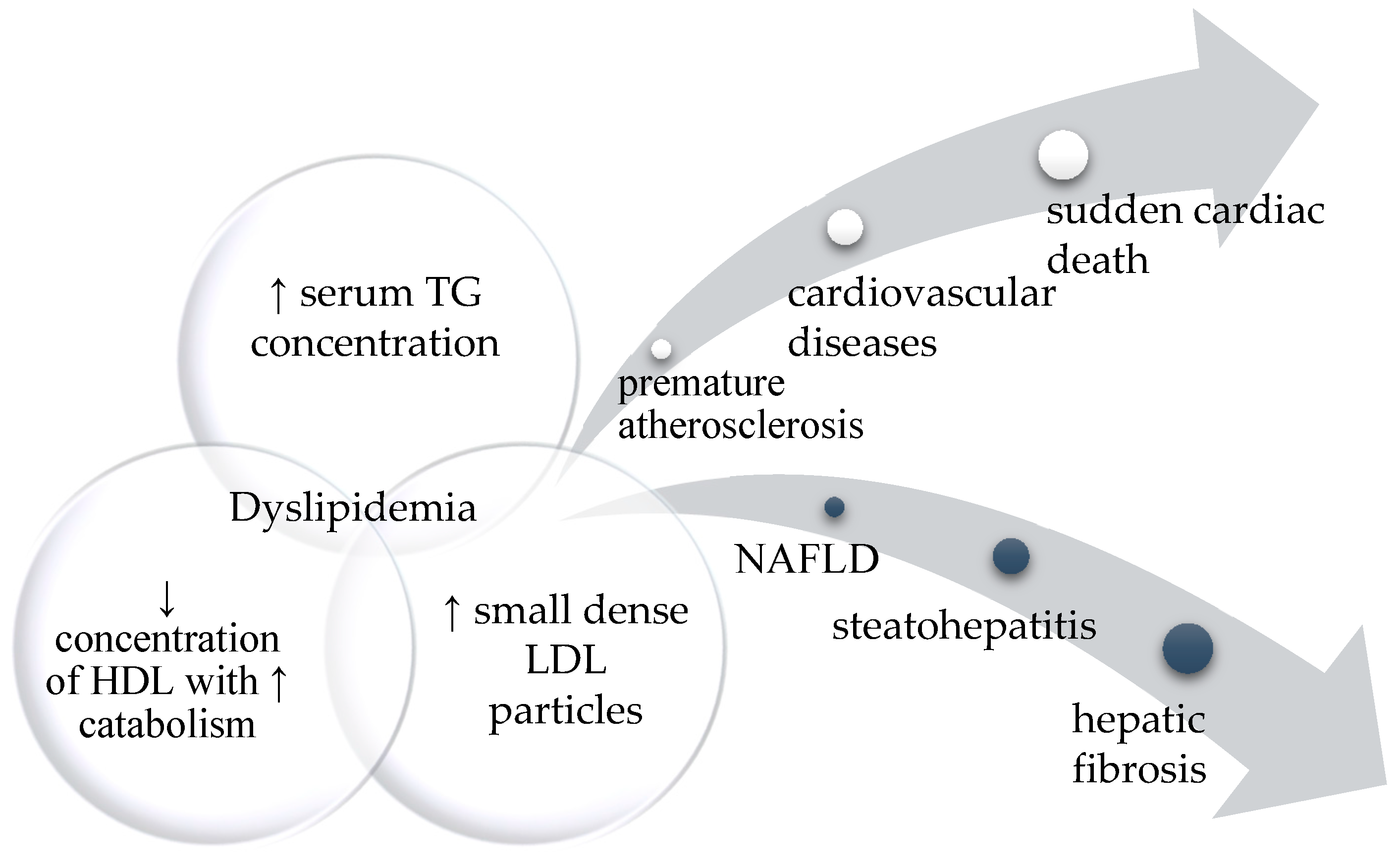
3.2. Hyperlipidemia with Hypertriglyceridemia
4. Results
4.1. Clinical Characteristics of Index Patient
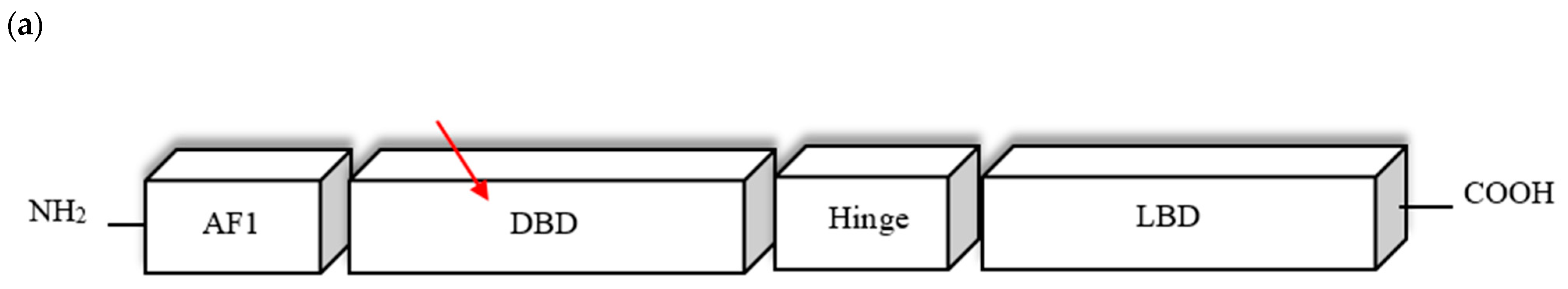
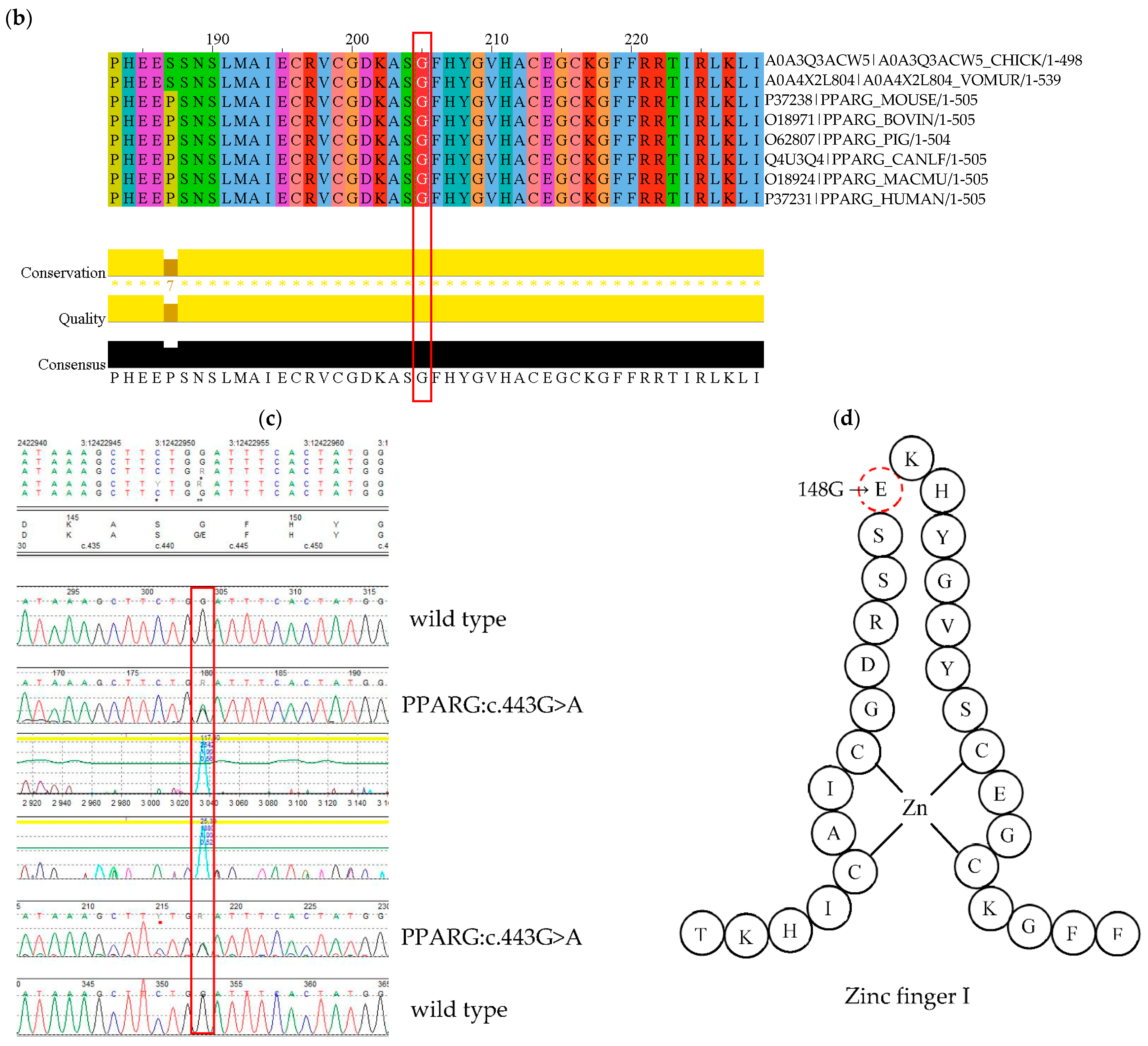
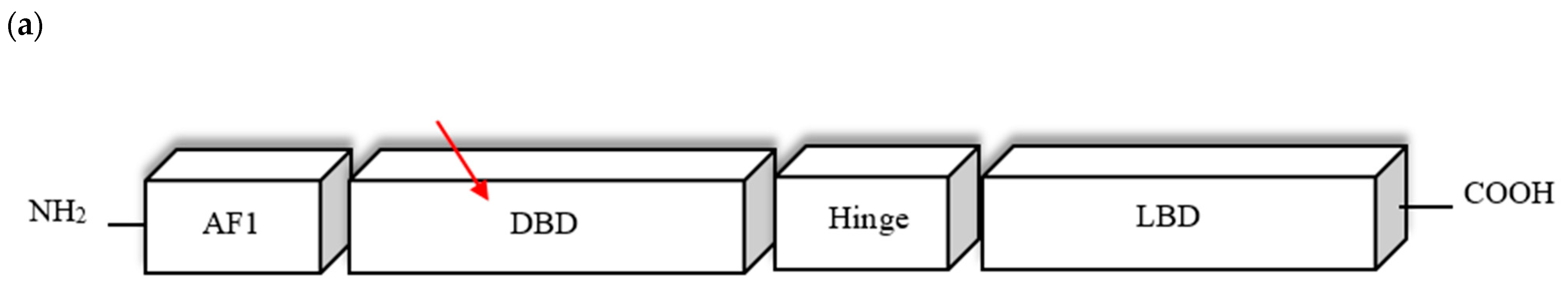
4.2. PPARG Mutation
5. Discussion
6. Materials and Methods
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bagias, C.; Xiarchou, A.; Bargiota, A.; Tigas, S. Familial Partial Lipodystrophy (FPLD): Recent Insights. Diabetes Metab. Syndr. Obesity Targets Ther. 2020, 13, 1531–1544. [Google Scholar] [CrossRef]
- Mann, J.P.; Savage, D.B. What lipodystrophies teach us about the metabolic syndrome. J. Clin. Investig. 2019, 129, 4009–4021. [Google Scholar] [CrossRef] [Green Version]
- Gonzaga-Jauregui, C.; Ge, W.; Staples, J.; Van Hout, C.; Yadav, A.; Colonie, R.; Leader, J.B.; Kirchner, H.L.; Murray, M.F.; Reid, J.G.; et al. Clinical and Molecular Prevalence of Lipodystrophy in an Unascertained Large Clinical Care Cohort. Diabetes 2019, 69, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Lightbourne, M.; Brown, R.J. Genetics of Lipodystrophy. Endocrinol. Metab. Clin. N. Am. 2017, 46, 539–554. [Google Scholar] [CrossRef]
- Broekema, M.; Savage, D.; Monajemi, H.; Kalkhoven, E. Gene-gene and gene-environment interactions in lipodystrophy: Lessons learned from natural PPARγ mutants. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2019, 1864, 715–732. [Google Scholar] [CrossRef]
- Knebel, B.; Müller-Wieland, D.; Kotzka, J. Lipodystrophies—Disorders of the Fatty Tissue. Int. J. Mol. Sci. 2020, 21, 8778. [Google Scholar] [CrossRef]
- Savage, D.B. Mouse models of inherited lipodystrophy. Dis. Model Mech. 2009, 2, 554–562. [Google Scholar] [CrossRef] [Green Version]
- Wojtanik, K.M.; Edgemon, K.; Viswanadha, S.; Lindsey, B.; Haluzik, M.; Chen, W.; Poy, G.; Reitman, M.; Londos, C. The role of LMNA in adipose: A novel mouse model of lipodystrophy based on the Dunnigan-type familial partial lipodystrophy mutation. J. Lipid Res. 2009, 50, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Nolis, T. Exploring the pathophysiology behind the more common genetic and acquired lipodystrophies. J. Hum. Genet. 2013, 59, 16–23. [Google Scholar] [CrossRef]
- Cao, H. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2000, 9, 109–112. [Google Scholar] [CrossRef] [Green Version]
- Varlet, A.-A.; Helfer, E.; Badens, C. Molecular and Mechanobiological Pathways Related to the Physiopathology of FPLD2. Cells 2020, 9, 1947. [Google Scholar] [CrossRef]
- Garg, A.; Misra, A. Lipodystrophies: Rare disorders causing metabolic syndrome. Endocrinol. Metab. Clin. N. Am. 2004, 33, 305–331. [Google Scholar] [CrossRef]
- Foss-Freitas, M.C.; Akinci, B.; Luo, Y.; Stratton, A.; Oral, E.A. Diagnostic strategies and clinical management of lipodystrophy. Expert Rev. Endocrinol. Metab. 2020, 15, 95–114. [Google Scholar] [CrossRef]
- Gerbino, A.; Procino, G.; Svelto, M.; Carmosino, M. Role of Lamin A/C Gene Mutations in the Signaling Defects Leading to Cardiomyopathies. Front. Physiol. 2018, 9, 1356. [Google Scholar] [CrossRef]
- Jeninga, E.H.; Gurnell, M.; Kalkhoven, E. Functional implications of genetic variation in human PPARγ. Trends Endocrinol. Metab. 2009, 20, 380–387. [Google Scholar] [CrossRef]
- Pap, A.; Cuaranta-Monroy, I.; Peloquin, M.; Nagy, L. Is the Mouse a Good Model of Human PPARγ-Related Metabolic Diseases? Int. J. Mol. Sci. 2016, 17, 1236. [Google Scholar] [CrossRef]
- Sokołowska, M.; Kowalski, M.L.; Pawliczak, R. Peroxisome proliferator-activated receptors-g (PPAR-γ) and their role in immunoregulation and inflammation control. Postępy Hig. Med. Dosw. 2005, 3, 472–484. [Google Scholar]
- Kroker, A.J.; Bruning, J.B. Review of the Structural and Dynamic Mechanisms of PPARγ Partial Agonism. PPAR Res. 2015, 2015, 816856. [Google Scholar] [CrossRef] [Green Version]
- Vasandani, C.; Li, X.; Sekizkardes, H.; Brown, R.; Garg, A. SUN-LB111 Comparison of Phenotype and Metabolic Abnormalities Among Familial Partial Lipodystrophy Due to LMNA or PPARG Variants. J. Endocr. Soc. 2020, 4. [Google Scholar] [CrossRef]
- Brown, R.J.; Araujo-Vilar, D.; Cheung, P.T.; Dunger, P.D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.A.; Patni, N.; Rother, K.I.; et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 4500–4511. [Google Scholar] [CrossRef]
- Akinci, B.; Sahinoz, M.; Oral, E. Lipodystrophy Syndromes: Presentation and Treatment. In Endotext; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Stears, A.; Hames, C. Diagnosis and management of lipodystrophy: A practical update. Clin. Lipidol. 2014, 9, 235–259. [Google Scholar] [CrossRef]
- Phiske, M.M. An approach to acanthosis nigricans. Indian Dermatol. Online J. 2014, 5, 239–249. [Google Scholar] [CrossRef]
- Simha, V.; Garg, A. Inherited lipodystrophies and hypertriglyceridemia. Curr. Opin. Lipidol. 2009, 20, 300–308. [Google Scholar] [CrossRef]
- Joseph, J.; Shamburek, R.D.; Cochran, E.K.; Gorden, P.; Brown, R.J. Lipid Regulation in Lipodystrophy versus the Obesity-Associated Metabolic Syndrome: The Dissociation of HDL-C and Triglycerides. J. Clin. Endocrinol. Metab. 2014, 99, E1676–E1680. [Google Scholar] [CrossRef]
- Semple, R.K.; Savage, D.B.; Cochran, E.K.; Gorden, P.; O’Rahilly, S. Genetic Syndromes of Severe Insulin Resistance. Endocr. Rev. 2011, 32, 498–514. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Perakakis, N.; Mantzoros, C.S. Fatty liver in lipodystrophy: A review with a focus on therapeutic perspectives of adiponectin and/or leptin replacement. Metabolism 2019, 96, 66–82. [Google Scholar] [CrossRef]
- Ozen, S.; Akinci, B.; Oral, E.A. Current Diagnosis, Treatment and Clinical Challenges in the Management of Lipodystrophy Syndromes in Children and Young People. J. Clin. Res. Pediatr. Endocrinol. 2020, 12, 17–28. [Google Scholar] [CrossRef]
- Arshad, M.; Bhatti, A.; John, P. Identification and in silico analysis of functional SNPs of human TAGAP protein: A comprehensive study. PLoS ONE 2018, 13, e0188143. [Google Scholar] [CrossRef] [Green Version]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Tang, Q.; Xiao, F.; Fu, N. Regulation of Lipid Metabolism by Lamin in Mutation-Related Diseases. Front. Pharmacol. 2022, 13, 820857. [Google Scholar] [CrossRef]
- Lazarte, J.; Wang, J.; McIntyre, A.D.; Hegele, R.A. Prevalence of severe hypertriglyceridemia and pancreatitis in familial partial lipodystrophy type 2. J. Clin. Lipidol. 2021, 15, 653–657. [Google Scholar] [CrossRef]
- Hegele, R.A.; Joy, T.R.; Al-Attar, S.A.; Rutt, B.K. Thematic review series: Adipocyte Biology. Lipodystrophies: Windows on adipose biology and metabolism. J. Lipid Res. 2007, 48, 1433–1444. [Google Scholar] [CrossRef] [Green Version]
- Araújo-Vilar, D.; Santini, F. Diagnosis and treatment of lipodystrophy: A step-by-step approach. J. Endocrinol. Investig. 2019, 42, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Cortes, V.A.; Fernández-Galilea, M. Lipodystrophies: Adipose tissue disorders with severe metabolic implications. J. Physiol. Biochem. 2015, 71, 471–478. [Google Scholar] [CrossRef]
- Agostini, M.; Schoenmakers, E.; Mitchell, C.; Szatmari, I.; Savage, D.; Smith, A.; Rajanayagam, O.; Semple, R.; Luan, J.; Bath, L.; et al. Non-DNA binding, dominant-negative, human PPARγ mutations cause lipodystrophic insulin resistance. Cell Metab. 2006, 4, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Visser, M.E.; Kropman, E.; Kranendonk, M.E.; Koppen, A.; Hamers, N.; Stroes, E.S.; Kalkhoven, E.; Monajemi, H. Characterisation of non-obese diabetic patients with marked insulin resistance identifies a novel familial partial lipodystrophy-associated PPARγ mutation (Y151C). Diabetologia 2011, 54, 1639–1644. [Google Scholar] [CrossRef] [Green Version]
- Lüdtke, A.; Buettner, J.; Wu, W.; Muchir, A.; Schroeter, A.; Zinn-Justin, S.; Spuler, S.; Schmidt, H.H.-J.; Worman, H.J. Peroxisome Proliferator-Activated Receptor-y C190S Mutation Causes Partial Lipodystrophy. J. Clin. Endocrinol. Metab. 2007, 92, 2248–2255. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Major Genetic Background | Manner of Inheritance | OMIM Number | Observed Phenotype |
|---|---|---|---|---|
| FPLD type 1, Kobberling | unknown/polygenetic origin | - | %608600 | Loss of subcutaneous fat from the limbs with truncal obesity Reduction of gluteal AT Normal or increased facial and neck AT |
| FPLD type 2, Dunnigan | LMNA | dominant | #151660 | Loss of subcutaneous fat from the limbs and trunk Reduction of gluteal AT Excess fat accumulation in the face and neck Increased muscularity |
| FPLD type 3 | PPARG | dominant | #604367 | Loss of subcutaneous fat from the lower limbs Normal or increased abdominal, facial and neck AT Increased muscularity |
| FPLD type 4 | PLIN1 | dominant | #613877 | Loss of subcutaneous fat primarily in gluteal and lower limb regions Muscular appearance |
| FPLD type 5 | CIDEC | recessive | #615238 | Lack of AT on limbs and gluteal region Presence of visceral, neck and axillary fat pads Increased muscularity |
| FPLD type 6 | LIPE | recessive | #615980 | Reduced lower limbs subcutaneous fat In some patients abnormal fat accumulation in the back and axillae |
| FPLD type 7 | CAV1 | dominant | #606721 | Absence of AT over entire body except buttocks, hips and thighs |
| Structural Domains | Functional Domains | Role | Degree of Conservation |
|---|---|---|---|
| N-terminus A/B | AF-1 Ligand-independent transactivation function 1 | Regulates the ligand-independent transcriptional PPARG activity | Poorly conserved |
| C | DBD DNA-binding domain | Binding PPARγ to the promoter region of the targeted genes | Highly conserved |
| D | HINGE flexible hinge region | Involved in interaction with coactivators and corepressors | Poorly conserved |
| C-terminus E/F | LBD Ligand binding domain AF-2 Ligand-dependent transactivation function 2 | Regulates the ligand-dependent transcriptional PPARG activity; Responsible for dimerization with RXR | Highly conserved |
| TC [mg/dL] | LDL [mg/dL] | HDL [mg/dL] | TG [mg/dL] | |
|---|---|---|---|---|
| Index case | 299 | 111 | 25 | 870 |
| Mother | 232 (* 573) | 102 | 54 | 382 (* 1989) |
| Son | 141 | 88 | 28 | 360 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rutkowska, L.; Salachna, D.; Lewandowski, K.; Lewiński, A.; Gach, A. Familial Partial Lipodystrophy—Literature Review and Report of a Novel Variant in PPARG Expanding the Spectrum of Disease-Causing Alterations in FPLD3. Diagnostics 2022, 12, 1122. https://doi.org/10.3390/diagnostics12051122
Rutkowska L, Salachna D, Lewandowski K, Lewiński A, Gach A. Familial Partial Lipodystrophy—Literature Review and Report of a Novel Variant in PPARG Expanding the Spectrum of Disease-Causing Alterations in FPLD3. Diagnostics. 2022; 12(5):1122. https://doi.org/10.3390/diagnostics12051122
Chicago/Turabian StyleRutkowska, Lena, Dominik Salachna, Krzysztof Lewandowski, Andrzej Lewiński, and Agnieszka Gach. 2022. "Familial Partial Lipodystrophy—Literature Review and Report of a Novel Variant in PPARG Expanding the Spectrum of Disease-Causing Alterations in FPLD3" Diagnostics 12, no. 5: 1122. https://doi.org/10.3390/diagnostics12051122
APA StyleRutkowska, L., Salachna, D., Lewandowski, K., Lewiński, A., & Gach, A. (2022). Familial Partial Lipodystrophy—Literature Review and Report of a Novel Variant in PPARG Expanding the Spectrum of Disease-Causing Alterations in FPLD3. Diagnostics, 12(5), 1122. https://doi.org/10.3390/diagnostics12051122