Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating, uniformly lethal progressive degenerative disorder of motor neurons that overlaps with frontotemporal lobar degeneration (FTLD) clinically, morphologically, and genetically. Although many distinct mutations in various genes are known to cause amyotrophic lateral sclerosis, it remains poorly understood how they selectively impact motor neuron biology and whether they converge on common pathways to cause neuronal degeneration. Many of the gene mutations are in proteins that share similar functions. They can be grouped into those associated with cell axon dynamics and those associated with cellular phagocytic machinery, namely protein aggregation and metabolism, apoptosis, and intracellular nucleic acid transport. Analysis of pathways implicated by mutant ALS genes has provided new insights into the pathogenesis of both familial forms of ALS (fALS) and sporadic forms (sALS), although, regrettably, this has not yet yielded definitive treatments. Many genes play an important role, with TARDBP, SQSTM1, VCP, FUS, TBK1, CHCHD10, and most importantly, C9orf72 being critical genetic players in these neurological disorders. In this mini-review, we will focus on the molecular mechanisms of these two diseases.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating, uniformly lethal progressive degenerative disorder of motor neurons that overlaps with frontotemporal lobar degeneration (FTLD) []. ALS is a motor neuron disorder (MND), lateral sclerosis, and spinal muscular atrophies []. ALS has a worldwide incidence of about two per 100,000 per year, and a prevalence of about five per 100,000 []. Clinical symptoms include weakness of the bulbar and limb muscles, hyperreflexia, spasticity of the arms and legs, and respiratory failure. These symptoms most commonly develop between 40 and 70 years of age, although a wider age range has been reported []. Although most cases of ALS are sporadic (sALS), ≈10% are familial (fALS), with predominantly autosomal dominant transmission []. Sporadic ALS accounts for most ALS cases, although genetic causes are also known to play a role []. fALS is linked to mutations at a specific genetic locus []. The clinical and pathological presentation of fALS and sALS are similar []. Many sALS studies (GWAS) have identified genes associated with ALS disease []. ALS is closely related to frontotemporal lobar degeneration (FTLD) []. Many studies have shown clinical, pathological, and genetic commonalities between them. Therefore, they are now considered two manifestations of one disease continuum, i.e., the ALS-FTD spectrum or the FTD-ALS spectrum with the broader name, i.e., frontotemporal lobar degeneration, being associated with motor neuron disorders (FTLD-MND) []. FTD-ALS patients have a poor prognosis with a mean survival of 2–3 years from the first onset of symptoms [,,]. The reported heritability of FTLD-ALS is high: Approximately 50% of cases are considered familial [,]. ALS shares a common molecular etiology with frontotemporal dementia (FTD) [], and approximately 15% of FTD patients develop motor neuron disease (MND), and conversely, up to 50% of MND show either direct signs of cognitive impairment [,] or minimal-mild disturbances in executive function [,]. These diseases are the extremes on a spectrum of clinically, pathologically, and genetically overlapping disorders [], which suggests an overlap of disease mechanisms [].
2. Mechanism and Molecular Pathology of ALS and FTLD
FTLD and ALS are two related neurodegenerative diseases forming the two ends of a disease spectrum. Behavioral FTLD is a form of dementia clinically characterized by progressive changes in behavior, personality, and language skills []. ALS involves the premature loss of upper and lower motor neurons, and a numeric decline in these neurons in the spinal cord, brainstem, and motor cortex [], which leads to muscle weakness and atrophy []. The large number of genes and cellular processes associated with ALS has led to the suggestion that many disease mechanisms are involved. These include disturbances in RNA metabolism, impaired protein homeostasis, nucleocytoplasmic transport defects, impaired DNA repair, excitotoxicity, mitochondrial dysfunction, oxidative stress, axonal transport disruption, neuroinflammation, oligodendrocyte dysfunction, and vesicular transport defects []. While, traditionally, FTLD and ALS were considered to be two separate disease identities, it is now thought that FTLD and ALS form one clinical continuum, in which pure forms are linked by overlapping syndromes called FTLD-MND []. Recent advances in neuropathology and molecular genetics have started to reveal the biological basis for this clinical overlap. However, recent neuropathological findings suggest that FTLD cases present with distinct TDP-43 pathologies compared with ALS and FTLD-MND, indicating divergent disease pathogenesis mechanisms that nonetheless involve the same TDP-43 protein []. Although the underlying TDP-43 pathology does not always correlate with the genetics or disease phenotype, mutations of the progranulin (GRN) gene are generally associated with type A TDP-43 pathology, whereas hexanucleotide repeat expansions in C9orf72, which is the most common genetic cause of ALS and FTLD-TDP, most frequently result in the type B pathology [,]. Although TARDBP (the gene coding protein TDP-43) mutations are rare in ALS and FTD (<1%), the pathological aggregation of TAR DNA-binding protein 43 in affected brain regions and motor neurons is characteristic of the majority of ALS and FTD patients [,].
2.1. Protein and RNA Aggregates
The presence of protein and RNA aggregates in the cytoplasm of motor neurons is the hallmark of ALS []. The histopathological characteristics of ALS and FTD include abnormal accumulation of dysfunctional protein aggregations in the affected parts of the nervous systems []. Inclusions in ALS and FTD suggest that they share common pathogenic mechanisms leading to neurodegeneration and aggregation of specific inclusion proteins []. Similar ubiquitin-positive inclusions were observed in degenerating motor neurons of ALS patients [], and aggregations of FUS protein have been reported in rare cases of ALS [,]. The most common protein inclusion in ALS is TDP-43 (encoded by the TARDBP gene) []; however, TDP-43 deposits have been observed in other neurodegenerative diseases such as Alzheimer’s disease (A.D.) [,], Lewy bodies dementia (DLB) [,], and corticobasal degeneration (CBD) [,]. The relevance of the concomitant TDP-43 pathology remains unclear, and attempts to correlate a concurrent TDP-43 pathology with clinical phenotypes have provided mixed results [,]. C9orf72 (chromosome 9 open reading frame 72) has an essential role in stress granule formation, microglial function, and autophagy [,,]. The accumulation of (GGGGCC) might lead to sequestration of RNA binding proteins and disruption of the translation of diverse mRNA or increased nucleolar stress []. These abnormal protein aggregates are thought to be the mechanism by which C9orf72 expanded repeats contribute to ALS. One of the most frequently found proteins in neuropathological lesions is ubiquitin-binding protein p62 (sequestosome 1). Many experimental and clinical studies have shown that p62 plays a significant role in autophagy, an evolutionarily conserved pathway for the degradation of long-lived proteins and organelles. Dysfunction of the autophagy pathway may contribute to the pathology of various neurodegenerative disorders characterized by abnormal protein accumulation [,]. Recently, p62 was also shown to deliver ubiquitinated proteins, such as tau, and other crucial proteins involved in neurodegeneration, to proteasomes for degradation []. The build-up of p62-positive inclusions suggests defects in protein clearance pathways. Finally, a new role for p62 in maintaining mitochondrial integrity has recently been described []. A portion of p62 directly localizes within the mitochondria and supports stable electron transport by forming heterogeneous protein complexes []. Mutations in Superoxide Dismutase 1 (SOD1) produce an unstable protein deposited in the cytoplasm; oligomerization of unstable SOD1 leads to aggregate formation []. Fused in sarcoma (FUS) is another RNA binding protein in which mutations can result in cytoplasmic aggregates. It is also a component of stress granules and may form p62 and TDP-43 positive aggregates []. It has been postulated that impaired autophagy could contribute to the accumulation of cytoplasmic aggregates []. Mutations in several autophagy genes have been associated with ALS, including Sequestosome 1 (SQSTM1), SOD1, optineurin (OPTN), valosin-containing protein (VCP), ubiquitin-2 (UBQLN2), and TANK-binding kinase 1 (TBK1) []. The removal of misfolded or damaged protein is critical for optimal cell functioning. In both the cytosol and the nucleus, major proteolytic pathways exist to recycle misfolded or damaged proteins, i.e., the UPS (ubiquitin proteasome system) and endosomal-lysosomal system (ELS) []. An impaired UPS is thought to be associated with the formation of proteinaceous inclusions in many neurodegenerative disorders [].
2.2. Mitochondrial Dysfunction
Mitochondria dysfunction is at least partly responsible for the broad clinical spectrum of ALS and FTD. Mitochondrial dysfunction has been implicated in ALS motor neuron death []. Fragmentation of mitochondria and changes in mitochondrial morphology and expression of fusion/fission proteins are well described in ALS and have pronounced effects on normal mitochondrial function []. Mitochondria from ALS patients have impaired Ca2+ homeostasis and increased production of reactive oxygen species (ROS). ROS are associated with oxidative-related damage, including changes in protein carbonylates and tyrosine nitration []. Mitochondria are essential for cellular respiration, calcium buffering, and apoptosis. Neurons are particularly sensitive to mitochondrial dysfunction given their high metabolic rate []; as such, the presence of abnormal or dysfunctional mitochondria in neurons is thought to be a contributing factor in ALS. Mitochondria are of particular importance in neurons. Neurons have high metabolic requirements, with the brain consuming 20% of the body’s resting ATP production despite being only 2% of the body’s mass [,]. Moreover, mitochondria are essential calcium buffering organelles in neurons and function to modulate local calcium dynamics, for example, modulating neurotransmitter release []. Many proteins that have been linked to familial and sporadic ALS, including SOD1, TDP-43, FUS, and C9orf72, show interactions with mitochondria [,,]. Direct evidence that disruption of mitochondrial structure (and as a result disruption of mitochondrial function; see below) may contribute to the etiology of ALS comes from the discovery of causative mutations in the mitochondrial protein Coiled-coil-helix-coiled-coil-helix domain-containing protein 10 (CHCHD10); this protein is localized to contact sites between the inner and outer mitochondrial membrane []. ALS-associated mutations in CHCHD10 can disrupt mitochondrial cristae and profoundly affect the mitochondrial structure []. Mitochondrial DNA instability disorders are responsible for frontotemporal dementia []. In recent years, a growing list of FTD genes responsible for mitochondrial DNA instability has been reported []. The c.176C>T mutation in the CHCHD10 gene was described in an FTD-ALS patient whose family was originally from Catalonia (Spain), with affected individuals carrying a missense mutation in the CHCHD10 gene. Functional characterization of the CHCHD10 mutant identified in the family showed fragmentation of the mitochondrial network and the loss of cristae junctions []. CHCHD10 is a novel gene responsible for the clinical spectrum of ALS-FTD, which raises the intriguing prospect of an underlying mitochondrial basis for this group of disorders.
2.3. Impaired DNA Repair
Impair DNA repair is another suggested mechanism that may contribute to ALS pathogenesis. Two of the best-studied ALS-linked proteins, TDP-43 and FUS, function to prevent or repair transcription-associated DNA damage []. FUS, in particular, seems to play an essential role in this regard and is involved in the repair of double-stranded DNA breaks via both homologous recombination and non-homologous end-joining repair mechanisms [,]. Variations in the genes of other ALS-linked RNA-binding proteins, including TATA-box binding protein associated factor 15 (TAF15), senataxin (SETX), and RNA-binding protein EWS (EWSR1), have also been linked to impaired DNA damage repair, further implicating the breakdown of this process in ALS pathogenesis [,,].
2.4. Axonal Transport Defects
Axonal transport defects are a common observation in various neurodegenerative diseases, and mutations in components of the axonal transport machinery have unequivocally shown that impaired axonal transport can cause neurodegeneration []. The underlying cause of axonal transport defects in ALS is not fully understood []. Several mechanisms by which axonal transport may be perturbed in sporadic ALS and familial ALS by mutations in non-axonal transport genes have been proposed based mainly on studies of mutant SOD1-related ALS []. These include reductions in [] microtubule stability, [] mitochondrial damage, [] pathogenic signaling (which alters phosphorylation of molecular motors and thereby regulate their function or through phosphorylation of cargo, such as neurofilaments, to disrupt their association with motors), and [] protein aggregation [,]. There is evidence that TDP-43 may also be involved in the axonal transport defects seen in ALS []; axonal transport defects are commonly seen in neurodegenerative diseases []. TDP-43 has an important role in regulating axonal growth and impairment in posttranscriptional regulation of mRNAs in the cytoplasm of motor neurons []. Mutations in the genes coding for axonal transport first came to light when LaMonte et al. showed that disruption of the dynein/dynactin interaction by postnatal overexpression of p50/dynamitin, a 50-kDa subunit of dynactin encoded by DCTN2, caused reduced axonal transport in motor neurons and consequently led to a late-onset progressive motor neuron disease phenotype in the transgenic mice []. This study was followed by several studies showing that loss-of-function mutations in DCTN1 cause a slowly progressive autosomal dominant distal hereditary motor neuropathy with vocal paresis (HMN7B) and ALS [,,]. One key role for dynein in the neuron may be the removal of misfolded or degraded proteins from the cell periphery and the transport of these proteins back to the cell body for degradation. Dynein is also involved in the accumulation of misfolded proteins into aggresomes []. The next major family of microtubule-based molecular motors is kinesin. Kinesin moves mostly toward the plus end of microtubules, and was the first axonal transport motor to be identified. Now known as kinesin-1, it is a unidirectional motor, driving plus end-directed motility along microtubules in vitro []. Microtubules (e.g., α-tubulin) play a pivotal role in developing and maintaining neuronal cell structure and function, and they serve as essential tracks for both fast and slow long-distance axonal transport []. Several variants of the α-tubulin gene (i.e., TUBA4A), which destabilize the microtubule network and diminish its re-polymerization capability, have been identified as possible causes of ALS []. Whether these mutations affect axonal transport has not yet been determined, but since axonal transport prefers stable microtubules, they will likely have a detrimental effect [].
2.5. Altered RNA Metabolism
As key regulators of RNA metabolism, RNA-binding proteins (RBP) play a critical role in maintaining the normal function of neuronal systems. RNA-binding proteins are involved in several aspects of RNA metabolism, including splicing, transcription, transport, translation, and storage in stress granules []. The aggregation of RBP is a pathological hallmark of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Interestingly, many of the ALS-linked RNA-binding proteins contain prion-like domains that are involved in stress granule formation or dynamics, including TDP-43, FUS, TAF15, ESWR1, hnRNPA1, and hnRNPA2B1 []. Mutations in ALS genes contribute to the etiology of FTD and vice versa []. Many ALS-causing mutations impact proteins involved in RNA metabolism, including RNA-binding proteins such as TDP-43, FUS, and heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) []. These and related RNA-binding proteins are components of organelles without membranes found in the nucleus (e.g., nuclear speckles and nucleoli) and cytoplasm (e.g., processing bodies and stress granules) in neurons and other cell types [,,].
2.6. Mechanisms Leading to Dysregulation of RBP in ALS
Mutations in genes encoding many RBP are highly associated with ALS. In addition, dysregulation of RBP as a result of compromised nucleocytoplasmic trafficking, posttranslational modification (PTM), aggregation, and sequestration by abnormal RNAs also contribute significantly to disease pathogenesis. This section will briefly discuss the underlying mechanisms resulting in RBP dysregulation in ALS [].
In response to a variety of stressors such as heat shock and oxidative insult, TDP-43 and FUS translocate from the nucleus and associate with cytoplasmic stress granules (SG), which are dense aggregations of protein-RNA complexes [,]. RBPs recruited to stress granules under conditions of chronic stress are capable of forming insoluble protein aggregates, even when other components of the stress granules have dissociated from the complex []. These granules facilitate cell survival by the translational arrest of non-essential transcripts and pro-apoptotic proteins when under stress []. Prion-like domains are thought to be vital for the reversible assembly of stress granules due to their capacity to form multiple transient weak interactions []. RBP also contains low complexity sequence domains (LCD), i.e., a glycine-rich domain that promotes protein aggregation [] and contains RNA-recognition motifs (RRM) necessary for the nucleic acid binding functions of the protein []. Each protein also contains a nuclear localization sequence (NLS) that directs the subcellular localization of the protein to the nucleus under normal conditions []. Mutations in genes encoding NLS and LCD (Figure 1) lead to cytoplasmic retention and inclusion formation in cultured cells []. More than 250 proteins with aggregation-prone properties that are likely to contribute to neurodegeneration have been identified [].
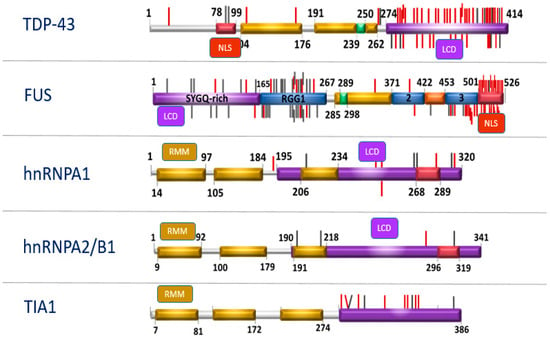
Figure 1.
Structure of RNA-binding protein (RBP) genes.
Low Complexity sequence Domains (LCD), Nuclear Localization Sequence (NLS). Mutations that occur in these domains (LCD and NLS) can trigger the same pathological cascade, which leads to a deterioration in the dynamics of stress granules (updated following original citation Baradaran-Heravi et al., 2019).
These studies suggest that such RBP could or should be considered as potential functional candidate genes in genetic studies. RBM45, an RNA-binding protein, is most likely a causal gene for ALS-FTD. In addition, a novel and evolutionary conserved structural element homo-oligomer assembly (HOA) domain has been identified. It is located within the linker region between RNA-recognition motifs (RRM2 and RRM3), which are essential for the self-association and oligomerization of RBM45 (Figure 2). Since RBM45 contains three RRM domains, it may associate with TDP-43 and FUS through RNA-protein interactions [].
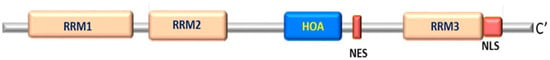
Figure 2.
Structure of the RBM45 gene.
RBM45 lacks the typical low complexity domain (LCD), which is actually common in RBPs; it has a suspicious homo oligomerization domain that, similar to LCD, mediates self-assembly through homo oligomerization and interaction with other proteins (updated following original citation Li et al. (2015).
Another RBP is TIA-1 that promotes the assembly of stress granules discrete cytoplasmic inclusions into which stalled translation initiation complexes are dynamically recruited in cells subjected to environmental stress []. TIA-1 is a modular protein composed of three RNA recognition motifs and a carboxy-terminal glutamine-rich motif that is structurally related to prion protein (PRD). Overexpressed TIA-1 induces SG formation and represses reporter gene expression, whereas the isolated prion-related domain (PRD) of TIA-1 forms cytoplasmic microaggregates []. These data suggest that the PRD is capable of self-oligomerization, just like RBM45.
2.7. Neuroinflammation
The inflammatory environment associated with ALS changes with disease progression and involves both neurotoxic and neuroprotective aspects. Neuroinflammation associated with neuronal loss is characterized by microglia and astrocyte activation, overproduction of inflammatory cytokines, and infiltration of T lymphocytes []. The secretion of inflammatory proteins by activated microglia leads to the potentially neurotoxic activation of astrocytes, which may contribute to the death of neurons and oligodendrocytes []. Genes that influence these functions are highly expressed in microglia and include C9orf72, TBK1, OPTN, SQSTM1, and PGRN [,,,]. Recent preclinical studies suggest that dysfunction of the gastrointestinal tract may also play a role in ALS pathogenesis by modifying the gut microbiota-brain axis []. Microglia and astrocytes in the central nervous system were shown to be regulated by metabolites derived from symbiotic gut microbes; the pathway inhibited neuroinflammation and neurodegeneration in an experimental autoimmune encephalomyelitis model [,]. Generation of low molecular weight metabolites by the gut microbiome is one postulated mechanism; these compounds are capable of passing through the blood-brain barrier and influencing neuronal function [,,]. A correlation between ALS and altered composition of the gut microbiota was previously tested in animal models [,]. Several preliminary studies have analyzed the fecal microbiota in ALS patients, but with no conclusive results [,]. Whether the gut microbiome influences ALS is still controversial and remains a matter of debate.
3. Overview of ALS and FTD Genes
The advent of next-generation sequencing technologies, such as whole-genome sequencing (WGS) and whole-exome sequencing (WES), has led to a wave of novel genes associated with ALS [,]. A recent study found that multiple genetic variants can interact simultaneously to increase ALS susceptibility; these oligogenic cases of ALS may not appear familial in a conventional Mendelian sense; nonetheless, they may underlie the sporadic form of the disease [,,]. More than forty-six ALS-related genes overlap with ALS genes linked to hereditary spastic paraplegia (HSP), FTD, mitochondrial disease, and lower motor neuropathies (LMN) [] (Figure 3). Most of the heritability of FTD is accounted for by autosomal dominant mutations in three genes: Progranulin (GRN), microtubule-associated protein tau (MAPT), and C9orf72 []. In recent years, an increasing number of mutations in other genes have been associated with autosomal dominant FTD, e.g., VCP (2004), CHMP2B (2005), TARDBP (2008), FUS (2009), SQSTM1 (2012), CHCHD10 (2014), TBK1 (2015), OPTN (2015), CCNF (2016), and TIA1 (2017). Recent studies have identified TBK1 as probably the fourth most common genetic cause overall of FTD, accounting for between 1% and 2% of all cases (although the pathogenic nature of many of the reported missense variants remains unclear) []. The first ALS gene, cytosolic superoxide dismutase (SOD1), was reported in 1993 [] as well as other genes such as TAR DNA binding protein (TARDBP) [,,,], angiogenin (ANG) [], fused in sarcoma (FUS) [,], optineurin (OPTN) [], and the recently described chromosome 9 open reading frame 72 (C9orf72) [,]. An overview of recently proposed ALS genes that were identified based on rare genetic variants (TBK1, CHCHD10, TUBA4A, CCNF, MATR3, NEK1, C21orf2, ANXA11, TIA1) and their potential relevance to the genetic etiology of frontotemporal dementia have also been described [].
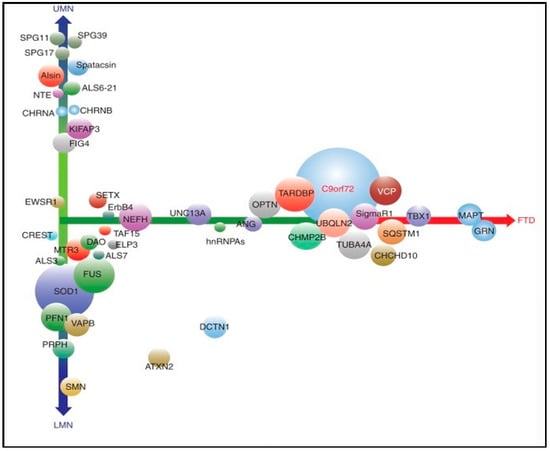
Figure 3.
ALS-related genes. Symbols: LMN: Lower motor neuropathies, FTD: Frontotemporal dementia, UMN: Upper motor neurons. X-axis: Overlap with frontotemporal dementia, Y-axis: The extent to which corticospinal versus lower motor neurons are involved (updated following Ghasemi M. 2018).
3.1. SOD1
Mutations in SOD1 account for the second most common cause of fALS after C9orf72 []. SOD1 mutations account for 15–20% of fALS pedigrees [,] and, until the discovery of C9orf72, was the most commonly identified gene in ALS. In most families harboring SOD1 gene mutations, disease penetrance is >90% by age 70 yrs [], and more than 170 mutations have now been detected in the fALS SOD1 gene []. This likely remains true in many non-white populations, where C9orf72 is much less common. Although multiple hypotheses have been proposed to explain mutant SOD1-mediated toxicity [], the exact mechanism(s) responsible for motor neuron degeneration remains unresolved. Mitochondrial dysfunction is thought to contribute to the pathogenesis [], and a proportion of the predominantly cytosolic SOD1 has been reported to localize to mitochondria under certain conditions [,,]. Pickles et al. reported that wild-type SOD1 proteins are only partially located in the mitochondria, while mutant proteins show an increased propensity to be located in mitochondria, suggesting mitochondria involvement in the ALS etiology []. Another key player that directly interacts with SOD1 is voltage-dependent anion channel 1 (VDAC1), a mitochondrial channel protein. Translocation of ions and proteins between mitochondria and the cytoplasm may be affected by mitochondria-associated misfolded mutant SOD1 [], leading to increased mitochondria dynamic abnormalities and fragmentation []. Pure upper motor neuron (UMN) and lower motor neuron (LMN) forms have also been described, representing opposite clinical ends of the MND spectrum []. Mutations in the SOD1 gene could be associated with significant LMN involvement with or without signs of UMN. The A4V missense mutation occurs in around 40% of patients in North America, but is rare in the European population. LMN signs predominate, with features of UMN being mild or absent. Disease progression is particularly rapid, with a median survival of 1.2 years from disease onset []. The A4T mutation is also associated with a similarly rapid disease course for LMN predominant syndrome []. In contrast, the G93C mutation has been associated with a pure clinical phenotype of LMN, i.e., without bulbar involvement and a more favorable prognosis (i.e., a median survival of 153 months) [].
3.2. TARDBP and FUS
One consistent pathologic finding in ALS and FTD is the presence of heavily ubiquitinated neuronal cytoplasmic inclusions, which in 2006 were found to contain TDP-4 []. TDP-43, an RNA-binding protein, is implicated in multiple aspects of RNA processing, including regulation of transcription, splicing, transport, and mRNA stabilization []. Significant modifications of TDP-43 have been identified as being hyperphosphorylated and proteolytically cleaved by caspases. Activation and cleavage of TDP-43 is a key molecular step linking cellular redistribution and toxicity to the neurodegeneration observed in TDP-43 proteinopathies []. TDP-43 has a promiscuous protein interaction pattern with more than 200 targets reported, suggesting an involvement in a vast array of intracellular events []. Abnormal molecular weight TDP-43 fragments have been observed in neurons and astrocytes in patients with a spectrum of neurodegenerative diseases, including 95% of familial and sporadic ALS [], making it an interesting candidate for all forms of the disease. TARDBP mutations were initially identified [] as a direct consequence of the identification of the TDP-43-derived protein species as the principal constituent of the aggregates found in the upper and lower motor neurons of ALS patients without SOD1 mutations and in FTLD-UPS [,]. Whereas 5% of familial ALS patients have the TARDBP mutation, mutations are rarely found in FTLD and FTD-MND [,].
3.3. C9orf72
The protein encoded by C9orf72 is mainly related to autophagy, endosomal transport, and immune function. According to statistics, about 40–50% of fALS and 10% of sALS patients carry the C9orf72 expanded alleles. In one study, the C9orf72 expansion accounted for 11.7% of familial FTD []. The pathogenic alleles of C9orf72 may have hundreds or even thousands of GGGGCC hexanucleotide repeats. A large number of clinical studies have shown that about 700–1600 GGGGCC hexanucleotide repeats are inserted into the intron located between the two untranslated optional exons 1a and 1b of the C9orf72 gene [,,,]. Disease penetrance of C9orf72-related ALS is thought to be nearly 100% by the age of 80 yrs. No predictions for individual phenotypes, i.e., ALS, FTD, or ALS/FTD, the exact age at onset, the disease course, and disease duration is currently possible [].
4. Novel ALS Genes
4.1. KIF5A
Kinesins are microtubule-based motor proteins involved in the intracellular transport of organelles within eukaryotic cells. KIF5 genes are expressed in neurons, and transcription products function to transport cargo by binding to distinct adaptor proteins []. The central role of kinesins in axonal transport leads us to speculate that mutations in KIF5A cause disease by disrupting axonal transport. KIF5 is responsible for the axonal transport of neurofilaments [], and KIF5A knockout mice display abnormal neurofilament transport []. Abnormal accumulation of neurofilaments is a pathological hallmark of ALS, and rare mutations in the neurofilament heavy polypeptide (NEFH) are associated with ALS []. KIF5 also contributes to the transport of mitochondria [], and the impaired mitochondrial transport and function is another common hallmark of ALS patients [,,,].
4.2. TBK1 and OPTN
The TBK1 and OPTN genes encode functionally related proteins that recently gained increased attention from the ALS research community. TBK1 (tumor necrosis factor (TNF) receptor-associated factor NF-κB activator (TANK)-binding kinase 1), also known as NAK or T2K, recently attracted the attention of human geneticists, immunologists, and neurologists alike for its critical role in pathologies of the central nervous system (CNS). TBK1 is involved in the activation of various cellular pathways leading to IFN and pro-inflammatory cytokine production following infection [], autophagic degradation of protein aggregates or pathogens [,], and homeostatic cellular functions such as cell growth and proliferation []. The majority of TBK1 mutations are loss-of-function (LOF), leading to loss of the mutant transcript through nonsense-mediated mRNA decay (NMD) []. TBK1 LOF mutations account for 3–4% of ALS-FTD patients []. Nonsense and frameshift mutations cause major disruptions to TBK1 and may decrease its expression at both the mRNA and protein level, implying that TBK1 haploinsufficiency contributes to the development of ALS [,].
4.3. CHCHD10
Many additional Coiled-coil-helix-coiled-coil-helix domain-containing protein 10 (CHCHD10) variants are now known to cause ALS, FTD, and other related degenerative diseases [,]. However, the degree of their pathogenicity and penetrance is undetermined. Variants causing ALS have been described; however, experimental evidence does not support the assumption that all disease-causing variants have the same mode of action []. CHCHD10 G58R and CHCHD10 G66V were identified in mitochondrial myopathy and late-onset spinal muscular atrophy [,,] such as VCP and Matrin 3 (MATR3), which also exhibit clinical pleiotropy, including myopathy [,]. CHCHD10 expression in patient tissues is unaffected, and CHCHD10 S59L overexpression causes mitochondrial defects similar to those in affected patients []. This suggests that CHCHD10 S59L is a dominant gain-of-function mutation []. To date, no published study supports routine diagnostic or predictive testing for CHCHD10 variants in pure ALS [].
4.4. MATR3
Matrin 3 is a highly conserved, inner nuclear matrix protein with two zinc finger domains and two RNA recognition motifs (RRM). It has been proposed that it stabilizes certain messenger RNA species []. Another study suggests that MATR3 regulates alternative splicing events by binding to introns flanking repressed exons [,]. MATR3 mutations are observed in 0.5–2% of ALS patients [,,], but no studies so far have identified any MATR3 mutations in FTLD [].
4.5. HNRNPA1 and HNRNPA2B1
Over the last decade, dysfunction of hnRNPs has become closely linked to neurodegenerative diseases, most prominently amyotrophic lateral sclerosis and frontotemporal dementia, two diseases with significant genetic and pathological overlap []. hnRNP A1 and hnRNP A2B1 share similar domain architectures and are primarily localized in the nucleus [,,]. hnRNP A2B1 are components of RNA transport granules found in neurons []. In addition, hnRNP A1 and hnRNP A2B1 translocate to the cytoplasm in response to stress and are recruited to stress granules [,]. hnRNP A1 and hnRNP A2B1 are associated with <1% of familial and sporadic forms of ALS; instead, they are more frequently associated with the broader spectrum multisystem proteinopathy (MSP) disorder [,].
4.6. TIA1
As a gene that has only recently been associated with ALS, TIA1 harbors several ALS- and ALS/FTD-associated mutations in the low-complexity sequence domain (LCD) []. Notably, the LCD of TIA1 is also the site of a mutation that causes Welander distal myopathy, a myopathy characterized by TDP-43-positive inclusions [] and p62 [,]. TIA1 assembles in organelles without membranes, e.g., stress granules [].
4.7. NEK1 and C21orf2
NEK1 encodes a member of the highly conserved NIMA (never in mitosis gene A) kinase family. It is a serine/threonine kinase involved in cell-cycle regulation, ciliogenesis, mitochondrial membrane permeability, and DNA damage repair [,,]. Interestingly, in DNA damage repair, NEK1 was shown to interact with C21orf2, which was recently associated with an increased ALS risk [,,] (Table 1). Mutations in both NEK1 and C21orf2 are linked to skeletal disorders and axial spondylometaphyseal dysplasia []. Additionally, C21orf2 may participate in the DNA repair process via interaction with NEK1 []. Loss of function (LOF) variants account for about 1% of patients, and an interpretation of the pathogenicity and penetrance is complicated by the observation of occasional LOF variants in asymptomatic carriers []. In neurons, NEK proteins take part in maintaining the cytoskeleton network [,,], which was previously linked to an ALS etiology via TUBA4A and PFN1 [,,]. With only 5–10% of sporadic ALS cases harboring disease-associated mutations in known ALS genes [], the remainder of sALS cases are presumed to represent a complex disease process influenced by both genetic and environmental exposures. Efforts to identify genetic risk factors have largely focused on genome-wide, and candidate gene association studies have met with only occasional success. Variants of the UNC13A gene, for example, have been associated with susceptibility to ALS and shorter survival times []. Intermediate length trinucleotide repeat expansions of both the ATXN1 and ATXN2 genes also increase the risk of disease, particularly for C9orf72 expansion carriers in the case of ATXN1 [,]. A copy number variation in the EPHA3 gene, in contrast, has been flagged as a potential protective factor for ALS [].

Table 1.
Overview of recent ALS genes in different form ALS and TDP.
5. Oligogenic/Polygenic Sporadic ALS
Variants in these genes are more likely to have a significant effect, and having the genotype greatly increases the probability of ALS; in other words, these variants show moderate to high penetrance; however, gene variants with low penetrance are also of interest, even though they only modestly increase the risk for any individual []. In at least some cases, ALS can be oligogenic, with affected individuals carrying more than one rare variant implicated in ALS [,]. An oligogenic basis of amyotrophic lateral sclerosis is debatable, since the presence of some variants have an uncertain significance []. However, a combination of a known pathogenic variant with one of uncertain significance is considered oligogenic inheritance by some []. What we do know is that oligogenic/polygenic sporadic ALS cases showed an earlier age of onset [].
6. Future Questions
Our understanding of the biological and genetic basis of ALS, as well as our ability to care for ALS patients, has improved substantially over the last few years. Genetic causes of ALS have been identified in both sporadic and familial patients, and the number of disease-associated genes is still increasing []. One might assume that most of the monogenic forms have already been identified, but the issue of “heritability” is still unresolved, which might be explained by rare variants with large effect sizes []. The identification of genetic causes of ALS will help develop new therapeutic approaches, either through the identification of shared disease pathways such as the TDP-43 pathology or by targeted therapies for known mutations []. Currently, antisense oligonucleotide trials in SOD1- and C9orf72-related ALS are being conducted []. Besides riluzole, a second medication, edaravone, was approved by the FDA last year []. Whether it is beneficial to all ALS patients or just to subgroups needs to be evaluated over the next few years []. In the future, biomarkers will hopefully help monitor disease progression and genomics, and transcriptomics will help to further personalize treatment based on each patient’s individual disease subtype. For now, we need to define the molecular mechanisms that link specific disease-causing mutations to stress granule dysfunction and the accumulation of pathological inclusions.
Funding
This study was supported by the Ministry of Health, Czech Republic (Conceptual development of research organization VFN64165, General University Hospital in Prague and Thomayer Hospital in Prague, TN64190), by the Grants Agency of the Ministry of Health (NV19-04-00090), by Charles University (Project Progress Q27/LF1, Q35/LF3, and Erasmus Charles University, Third Faculty of Medicine), and by the AVASTipendium (Alzheimer Foundation Czech Republic).
Acknowledgments
The authors wish to thank Thomas Secrest for the revision of the English version of this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kang, S.H.; Li, Y.; Fukaya, M.; Lorenzini, I.; Cleveland, D.W.; Ostrow, L.W.; Rothstein, J.D.; Bergles, D.E. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 2013, 5, 571–579. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Andersen, P.M.; Chandran, S.; Chio, A.; Corcia, P.; Couratier, P.; Danielsson, O.; De Carvaiho, M.; Desnuelle, C.; Grehl, T.; et al. July 2017 ENCALS statement on edaravone. Amyotroph Lateral Scler Front. Degener. 2017, 18, 471–474. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 22, 1688–1700. [Google Scholar] [CrossRef]
- Chiò, A.; Moglia, C.; Canosa, A.; Manera, U.; D’Ovidio, F.; Vasta, R.; Grassano, M.; Brunetti, M.; Barberis, M.; Corrado, L.; et al. ALS phenotype is influenced by age, sex, and genetics: A population-based study. Neurology 2020, 8, e802–e810. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M.; Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011, 11, 603–615. [Google Scholar] [CrossRef]
- Yrne, S.; Walsh, C.; Lynch, C.; Bede, P.; Elamin, M.; Kenna, K.; McLaughlin, R.; Hardiman, O. Rate of familial amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 6, 623–627. [Google Scholar]
- Deivasigamani, S.; Verma, H.K.; Ueda, R.; Ratnaparkhi, A.; Ratnaparkhi, G.S. A genetic screen identifies Tor as an interactor of VAPB in a Drosophila model of amyotrophic lateral sclerosis. Biol. Open 2014, 11, 1127–1138. [Google Scholar] [CrossRef]
- Hardiman, O.; Van den Berg, L.H.; Kiernan, M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 11, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Van Broeckhoven, C.; Van der Zee, J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018, 34, 404–423. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Hodges, J.R.; Davies, R.; Xuereb, J.; Kril, J.; Halliday, G. Survival in frontotemporal dementia. Neurology 2003, 61, 349–354. [Google Scholar] [CrossRef]
- Josephs, K.A.; Hodges, J.R.; Snowden, J.S.; Mackenzie, I.R.; Neumann, M.; Mann, D.M.; Dickson, D.W. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011, 122, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Rusina, R.; Ridzon, P.; Kulist’ák, P.; Keller, O.; Bartos, A.; Buncová, M.; Fialová, L.; Koukolík, F.; Matej, R. Relationship between ALS and the degree of cognitive impairment, markers of neurodegeneration and predictors for poor outcome. A prospective study. Eur. J. Neurol. 2010, 17, 23–30. [Google Scholar] [CrossRef]
- Rohrer, J.D.; Guerreiro, R.; Vandrovcova, J.; Uphill, J.; Reiman, D.; Beck, J.; Isaacs, A.M.; Authier, A.; Ferrari, R.; Fox, N.C.; et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology 2009, 73, 1451–1456. [Google Scholar] [CrossRef]
- Seelaar, H.; Schelhaas, H.J.; Azmani, A.; Küsters, B.; Rosso, S.; Majoor-Krakauer, D.; de Rijk, M.C.; Rizzu, P.; Brummelhuis, M.; van Doorn, P.A.; et al. TDP-43 pathology in familial frontotemporal dementia and motor neuron disease without Progranulin mutations. Brain 2007, 130, 1375–1385. [Google Scholar] [CrossRef]
- Van Langenhove, T.; Van der Zee, J.; Van Broeckhoven, C. The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann. Med. 2012, 44, 817–828. [Google Scholar] [CrossRef]
- Cruts, M.; Gijselinck, I.; Van Langenhove, T.; Van der Zee, J.; Van Broeckhoven, C. Current insights into the C9 or f72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013, 36, 450–459. [Google Scholar] [CrossRef]
- Perrone, F.; Nguyen, H.P.; Van Mossevelde, S.; Moisse, M.; Sieben, A.; Santens, P.; De Bleecker, J.; Vandenbulcke, M.; Engelborghs, S.; Baets, J.; et al. Investigating the role of ALS genes CHCHD10 and TUBA4A in Belgian FTD-ALS spectrum patients. Neurobiol. Aging 2017, 51, e9–e177. [Google Scholar] [CrossRef]
- Lomen-Hoerth, C.; Murphy, J.; Langmore, S.; Kramer, J.H.; Olney, R.K.; Miller, B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology 2003, 8, 1094–1097. [Google Scholar] [CrossRef]
- Ringholz, G.M.; Appel, S.H.; Bradshaw, M.; Cooke, N.A.; Mosnik, D.M.; Schulz, P.E. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 2005, 65, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Lillo, P.; Hodges, J.R. Frontotemporal dementia and motor neurone disease: Overlapping clinic-pathological disorders. J. Clin. Neurosci. 2009, 9, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 1998, 51, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, F.; Petit, H. Frontotemporal dementia: Its rediscovery. Eur. Neurol. 1997, 8, 1–6. [Google Scholar] [CrossRef]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A.; et al. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- De Jesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Tan, R.H.; Yang, Y.; Kim, W.S.; Dobson-Stone, C.; Kwok, J.B.; Kiernan, M.C.; Halliday, G.M. Distinct TDP-43 inclusion morphologies in frontotemporal lobar degeneration with and without amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2017, 5, 76. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Neumann, M.; Baborie, A.; Sampathu, D.M.; Du Plessis, D.; Jaros, E.; Perry, R.H.; Trojanowski, J.Q.; Mann, D.M.; Lee, V.M. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011, 122, 111–113. [Google Scholar] [CrossRef]
- Boeve, B.F.; Boylan, K.B.; Graff-Radford, N.R.; De Jesus-Hernandez, M.; Knopman, D.S.; Pedraza, O.; Vemuri, P.; Jones, D.; Lowe, V.; Murray, M.E.; et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012, 3, 765–783. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95, 808–816. [Google Scholar] [CrossRef]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Albert, M.S.; Grossman, M.; Miller, B.; Dickson, D.; Trojanowski, J.Q.; Work Group on Frontotemporal Dementia and Pick’s Disease. Clinical and pathological diagnosis of frontotemporal dementia: Report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Clinical and pathological diagnosis of frontotemporal dementia: Report of the Work Group on Frontotemporal Dementia and Pick‘s Disease. Arch. Neurol. 2001, 58, 1803–1809. [Google Scholar] [PubMed]
- Leigh, P.N.; Anderton, B.H.; Dodson, A.; Gallo, J.M.; Swash, M.; Power, D.M. Ubiquitin deposits in anterior horn cells in motor neurone disease. Neurosci. Lett. 1988, 93, 197–203. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Amador-Ortiz, C.; Lin, W.L.; Ahmed, Z.; Personett, D.; Davies, P.; Duara, R.; Graff-Radford, N.R.; Hutton, M.L.; Dickson, D.W. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer‘s disease. Ann. Neurol. 2007, 61, 435–445. [Google Scholar] [CrossRef]
- Uryu, K.; Nakashima-Yasuda, H.; Forman, M.S.; Kwong, L.K.; Clark, C.M.; Grossman, M.; Miller, B.L.; Kretzschmar, H.A.; Lee, V.M.; Trojanowski, J.Q.; et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 2008, 67, 555–664. [Google Scholar] [CrossRef]
- Higashi, S.; Iseki, E.; Yamamoto, R.; Minegishi, M.; Hino, H.; Fujisawa, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer‘s disease and dementia with Lewy bodies. Brain Res. 2007, 1184, 284–294. [Google Scholar] [CrossRef]
- Yokota, O.; Davidson, Y.; Arai, T.; Hasegawa, M.; Akiyama, H.; Ishizu, H.; Terada, S.; Sikkink, S.; Pickering-Brown, S.; Mann, D.M.; et al. Effect of topographical distribution of α-synuclein pathology on TDP-43 accumulation in Lewy body disease. Acta Neuropathol. 2010, 120, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Geser, F.; Martinez-Lage, M.; Kwong, L.K.; Lee, V.M.; Trojanowski, J.Q. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: The TDP-43 diseases. J. Neurol. 2009, 256, 1205–1214. [Google Scholar] [CrossRef]
- Josephs, K.A.; Whitwell, J.L.; Knopman, D.S.; Hu, W.T.; Stroh, D.A.; Baker, M.; Rademakers, R.; Boeve, B.F.; Parisi, J.E.; Smith, G.E.; et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 2008, 70, 1850–1857. [Google Scholar] [CrossRef]
- Maharjan, N.; Künzli, C.; Buthey, K.; Saxena, S. C9ORF72 Regulates Stress Granule Formation and Its Deficiency Impairs Stress Granule Assembly, Hypersensitizing Cells to Stress. Mol. Neurobiol. 2017, 54, 3062–3077. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351, 324–329. [Google Scholar] [CrossRef]
- Sullivan, P.M.; Zhou, X.; Robins, A.M.; Paushter, D.H.; Kim, D.; Smolka, M.B.; Hu, F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 2016, 4, 51. [Google Scholar] [CrossRef]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Matěj, R.; Rohan, Z.; Holada, K.; Olejár, T. The contribution of proteinase-activated receptors to intracellular signaling, transcellular transport and autophagy in Alzheimer‘s disease. Curr. Alzheimer Res. 2015, 12, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Seibenhener, M.L.; Du, Y.; Diaz-Meco, M.T.; Moscat, J.; Wooten, M.C.; Wooten, M.W. A role for sequestosome 1/p62 in mitochondrial dynamics, import and genome integrity. Biochim. Biophys. Acta 2013, 1833, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Rubino, E.; Rainero, I.; Chiò, A.; Rogaeva, E.; Galimberti, D.; Fenoglio, P.; Grinberg, Y.; Isaia, G.; Calvo, A.; Gentile, S.; et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 2012, 79, 1556–1562. [Google Scholar] [CrossRef]
- Lindberg, M.J.; Byström, R.; Boknäs, N.; Andersen, P.M.; Oliveberg, M. Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc. Natl. Acad. Sci. USA 2005, 102, 9754–9759. [Google Scholar] [CrossRef] [PubMed]
- Peters, O.M.; Ghasemi, M.; Brown, R.H., Jr. Emerging mechanisms of molecular pathology in ALS. J. Clin. Invest. 2015, 25, 1767–1779. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. The Function of Autophagy in Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 16, 26797–26812. [Google Scholar] [CrossRef]
- Forsberg, K.; Graffmo, K.; Pakkenberg, B.; Weber, M.; Nielsen, M.; Marklund, S.; Brännström, T.; Andersen, P.M. Misfolded SOD1 inclusions in patients with mutations in C9orf72 and other ALS/FTD-associated genes. J. Neurol. Neurosurg. Psychiatry 2019, 90, 861–869. [Google Scholar] [CrossRef]
- Matej, R.; Botond, G.; László, L.; Kopitar-Jerala, N.; Rusina, R.; Budka, H.; Kovacs, G.G. Increased neuronal Rab5 immunoreactive endosomes do not colocalize with TDP-43 in motor neuron disease. Exp. Neurol. 2010, 225, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; O’Connor, T. Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 2010, 9, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Sasaki, S.; Horie, Y.; Iwata, M. Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2007, 114, 633–639. [Google Scholar] [CrossRef]
- Beal, M.F.; Ferrante, R.J.; Browne, S.E.; Matthews, R.T.; Kowall, N.W.; Brown, R.H., Jr. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann. Neurol. 1997, 42, 644–654. [Google Scholar] [CrossRef]
- Kann, O.; Kovács, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef] [PubMed]
- Engl, E.; Attwell, D. Non-signalling energy use in the brain. J. Physiol. 2015, 593, 3417–3429. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet. 2015, 11, 1005357. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly (GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef]
- Genin, E.C.; Plutino, M.; Bannwarth, S.; Villa, E.; Cisneros-Barroso, E.; Roy, M.; Ortega-Vila, B.; Fragaki, K.; Lespinasse, F.; Pinero-Martos, E.; et al. CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med. 2016, 8, 58–72. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- Hill, S.J.; Mordes, D.A.; Cameron, L.A.; Neuberg, D.S.; Landini, S.; Eggan, K.; Livingston, D.M. Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. Proc. Natl. Acad. Sci. USA 2016, 113, E7701–E7709. [Google Scholar] [CrossRef]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef]
- Paronetto, M.P.; Miñana, B.; Valcárcel, J. The Ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol. Cell 2011, 43, 353–568. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Izhar, L.; Adamson, B.; Ciccia, A.; Lewis, J.; Pontano-Vaites, L.; Leng, Y.; Liang, A.C.; Westbrook, T.F.; Harper, J.W.; Elledge, S.J. A Systematic Analysis of Factors Localized to Damaged Chromatin Reveals PARP-Dependent Recruitment of Transcription Factors. Cell Rep. 2011, 11, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.A.; Rizzo, V.; Puthanveettil, S.V. Pathologies of axonal transport in neurodegenerative diseases. Transl. Neurosci. 2012, 3, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Millecamps, S.; Julien, J.P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal transport and neurological disease. Nat. Rev. Neurol. 2019, 15, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.N.; White, J.A.; Gunawardena, S. Axonal transport and neurodegenerative disease: Vesicle-motor complex formation and their regulation. Degener. Neurol. Neuromuscul. Dis. 2014, 4, 29–47. [Google Scholar] [PubMed]
- La Monte, B.H.; Wallace, K.E.; Holloway, B.A.; Shelly, S.S.; Ascaño, J.; Tokito, M.; Van Winkle, T.; Howland, D.S.; Holzbaur, E.L. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron 2002, 347, 15–27. [Google Scholar]
- Puls, I.; Oh, S.J.; Sumner, C.J.; Wallace, K.E.; Floeter, M.K.; Mann, E.A.; Kennedy, W.R.; Wendelschafer-Crabb, G.; Vortmeyer, A.; Powers, R.; et al. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann. Neurol. 2005, 57, 687–694. [Google Scholar] [CrossRef]
- Münch, C.; Sedlmeier, R.; Meyer, T.; Homberg, V.; Sperfeld, A.D.; Kurt, A.; Prudlo, J.; Peraus, G.; Hanemann, C.O.; Stumm, G.; et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004, 637, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, C.J.; Dawe, R.K.; Christie, K.R.; Cleveland, D.W.; Dawson, S.C.; Endow, S.A.; Goldstein, L.S.; Goodson, H.V.; Hirokawa, N.; Howard, J.; et al. A standardized kinesin nomenclature. J. Cell Biol. 2004, 167, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.A.; Yeaman, E.J.; Blizzard, C.A.; Chuckowree, J.A.; Dickson, T.C. A Case for Microtubule Vulnerability in Amyotrophic Lateral Sclerosis: Altered Dynamics During Disease. Front. Cell Neurosci. 2016, 13, 204. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Ticozzi, N.; Fallini, C.; Gkazi, A.S.; Topp, S.; Kenna, K.P.; Scotter, E.L.; Kost, J.; Keagle, P.; Miller, J.W.; et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 2014, 84, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; McEwen, D.P.; Martens, J.R.; Meyhofer, E.; Verhey, K.J. Single molecule imaging reveals differences in microtubule track selection between Kinesin motors. PLoS Biol. 2009, 7, 1000216. [Google Scholar] [CrossRef]
- Khalil, B.; Morderer, D.; Price, P.L.; Liu, F.; Rossoll, W. mRNP assembly, axonal transport, and local translation in neurodegenerative diseases. Brain Res. 2018, 1693, 75–91. [Google Scholar] [CrossRef]
- Harrison, A.F.; Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4334–4339. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Spector, D.L. U1 and U2 small nuclear RNAs are present in nuclear speckles. Proc. Natl. Acad. Sci. USA 1992, 89, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Sheth, U.; Parker, R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 2003, 300, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.C.; Ng, C.S.; Xiang, P.; Liu, H.; Zhang, K.; Mohamud, Y.; Luo, H. Dysregulation of RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2020, 29, 78. [Google Scholar] [CrossRef] [PubMed]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, 13250. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.J.; Meyerowitz, J.; James, J.L.; Liddell, J.R.; Crouch, P.J.; Kanninen, K.M.; White, A.R. Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochem. Int. 2012, 60, 415–424. [Google Scholar] [CrossRef]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 5, 46–64. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Cleveland, D.W. Rethinking ALS: The FUS about TDP-43. Cell 2009, 136, 1001–1004. [Google Scholar] [CrossRef]
- Dormann, D.; Haass, C. TDP-43 and FUS: A nuclear affair. Trends Neurosci. 2011, 34, 339–348. [Google Scholar] [CrossRef]
- Baradaran-Heravi, Y.; Van Broeckhoven, C.; Van der Zee, J. Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol. Dis. 2020, 134, 104639. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.; Riascos, D.; Kovalik, T.; An, J.; Krupa, K.; Hood, B.L.; Conrads, T.P.; Renton, A.E.; Traynor, B.J.; Bowser, R. The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 2012, 124, 717–732. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Visibly stressed: The role of eIF2, TIA-1, and stress granules in protein translation. Cell Stress Chaperones 2002, 7, 213–221. [Google Scholar] [CrossRef]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 1999, 1471, 431–442. [Google Scholar]
- Komine, O.; Yamanaka, K. Neuroinflammation in motor neuron disease. Nagoya J. Med. Sci. 2015, 77, 537–549. [Google Scholar] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Irwin, D.; Lippa, C.F.; Rosso, A. Progranulin (PGRN) expression in ALS: An immunohistochemical study. J. Neurol. Sci. 2009, 276, 9–13. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Lall, D.; Baloh, R.H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Invest. 2017, 127, 3250–3258. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; Von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef]
- Sharon, G.; Cruz, N.J.; Kang, D.W.; Gandal, M.J.; Wang, B.; Kim, Y.M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J.; et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019, 177, 1600–1618. [Google Scholar] [CrossRef] [PubMed]
- Blacher, E.; Levy, M.; Tatirovsky, E.; Elinav, E. Microbiome-Modulated Metabolites at the Interface of Host Immunity. J. Immunol. 2017, 198, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kasper, L.H. The role of microbiome in central nervous system disorders. Brain Behav. Immun. 2014, 38, 1–12. [Google Scholar] [CrossRef]
- Longstreth, W.T., Jr.; Meschke, J.S.; Davidson, S.K.; Smoot, L.M.; Smoot, J.C.; Koepsell, T.D. Hypothesis: A motor neuron toxin produced by a clostridial species residing in gut causes ALS. Med. Hypotheses 2005, 64, 1153–1156. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Hiergeist, A.; Adis, C.; Mayer, B.; Gessner, A.; Ludolph, A.C.; Weishaupt, J.H. The fecal microbiome of ALS patients. Neurobiol. Aging 2018, 61, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Rowin, J.; Xia, Y.; Jung, B.; Sun, J. Gut inflammation and dysbiosis in human motor neuron disease. Physiol. Rep. 2017, 5, 13443. [Google Scholar] [CrossRef]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Sieben, A.; Van Langenhove, T.; Engelborghs, S.; Martin, J.J.; Boon, P.; Cras, P.; De Deyn, P.P.; Santens, P.; Van Broeckhoven, C.; Cruts, M. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012, 124, 353–372. [Google Scholar] [CrossRef]
- Luigetti, M.; Lattante, S.; Zollino, M.; Conte, A.; Marangi, G.; Del Grande, A.; Sabatelli, M. SOD1 G93D sporadic amyotrophic lateral sclerosis (SALS) patient with rapid progression and concomitant novel ANG variant. Neurobiol. Aging 2011, 32, e15–e18. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Restagno, G.; Brunetti, M.; Ossola, I.; Calvo, A.; Canosa, A.; Moglia, C.; Floris, G.; Tacconi, P.; Marrosu, F.; et al. ALS/FTD phenotype in two Sardinian families carrying both C9ORF72 and TARDBP mutations. J. Neurol. Neurosurg. Psychiatry 2012, 83, 730–733. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Blitterswijk, M.; Van Es, M.A.; Hennekam, E.A.; Dooijes, D.; Van Rheenen, W.; Medic, J.; Bourque, P.R.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 3776–3784. [Google Scholar] [CrossRef]
- Ghasemi, M.; Brown, R.H., Jr. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, 024125. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, C.J.; Beck, J.; Rohrer, J.D.; Lashley, T.; Mok, K.; Shakespeare, T.Y.; Yeatman, T.; Warrington, E.K.; Schott, J.M.; Fox, N.C.; et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: Clinical, neuroanatomical and neuropathological features. Brain 2012, 135, 736–750. [Google Scholar] [CrossRef] [PubMed]
- De Majo, M.; Topp, S.D.; Smith, B.N.; Nishimura, A.L.; Chen, H.J.; Gkazi, A.S.; Miller, J.; Wong, C.H.; Vance, C.; Baas, F.; et al. ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol. Aging 2018, 71, 266. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Borroni, B.; Bonvicini, C.; Alberici, A.; Buratti, E.; Agosti, C.; Archetti, S.; Papetti, A.; Stuani, C.; Di Luca, M.; Gennarelli, M.; et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 2009, 30, E974–E983. [Google Scholar] [CrossRef]
- Gelpi, E.; Van der Zee, J.; Turon Estrada, A.; Van Broeckhoven, C.; Sanchez-Valle, R. TARDBP mutation p.Ile383Val associated with semantic dementia and complex proteinopathy. Neuropathol. Appl. Neurobiol. 2014, 402, 225–230. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; Mc Conkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Greenway, M.J.; Andersen, P.M.; Russ, C.; Ennis, S.; Cashman, S.; Donaghy, C.; Patterson, V.; Swingler, R.; Kieran, D.; Prehn, J.; et al. ANG mutations segregate with familial and ’sporadic’ amyotrophic lateral sclerosis. Nat. Genet. 2006, 38, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Gijselinck, I.; Van Langenhove, T.; Van der Zee, J.; Sleegers, K.; Philtjens, S.; Kleinberger, G.; Janssens, J.; Bettens, K.; Van Cauwenberghe, C.; Pereson, S.; et al. A C9 or f72 promoter repeat expansion in a Flanders-Belgian cohortwith disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 2012, 11, 54–65. [Google Scholar] [CrossRef]
- Chiò, A.; Traynor, B.J.; Lombardo, F.; Fimognari, M.; Calvo, A.; Ghiglione, P.; Mutani, R.; Restagno, G. Prevalence of SOD1 mutations in the Italian ALS population. Neurology 2008, 70, 533–537. [Google Scholar] [CrossRef]
- Battistini, S.; Giannini, F.; Greco, G.; Bibbò, G.; Ferrera, L.; Marini, V.; Causarano, R.; Casula, M.; Lando, G.; Patrosso, M.C.; et al. SOD1 mutations in amyotrophic lateral sclerosis. Results from a multicenter Italian study. J. Neurol. 2005, 252, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Cudkowicz, M.E.; Mc Kenna-Yasek, D.; Sapp, P.E.; Chin, W.; Geller, B.; Hayden, D.L.; Schoenfeld, D.A.; Hosler, B.A.; Horvitz, H.R.; Brown, R.H. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann. Neurol. 1997, 41, 210–221. [Google Scholar] [CrossRef]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef]
- Hirano, A.; Donnenfeld, H.; Sasaki, S.; Nakano, I. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 1984, 43, 461–470. [Google Scholar] [CrossRef]
- Bergemalm, D.; Jonsson, P.A.; Graffmo, K.S.; Andersen, P.M.; Brännström, T.; Rehnmark, A.; Marklund, S.L. Overloading of stable and exclusion of unstable human superoxide dismutase-1 variants in mitochondria of murine amyotrophic lateral sclerosis models. J. Neurosci. 2006, 26, 4147–4154. [Google Scholar] [CrossRef]
- Deng, H.X.; Shi, Y.; Furukawa, Y.; Zhai, H.; Fu, R.; Liu, E.; Gorrie, G.H.; Khan, M.S.; Hung, W.-Y.; Huang, W.-Y.; et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7142–7147. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Semmler, S.; Broom, H.R.; Destroismaisons, L.; Legroux, L.; Arbour, N.; Meiering, E.; Cashman, N.R.; Vande Velde, C. ALS-linked misfolded SOD1 species have divergent impacts on mitochondria. Acta Neuropathol. Commun. 2016, 4, 43. [Google Scholar] [CrossRef]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef]
- Bali, T.; Self, W.; Liu, J.; Siddique, T.; Wang, L.H.; Bird, T.D.; Ratti, E.; Atassi, N.; Boylan, K.B.; Glass, J.D.; et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J. Neurol. Neurosurg. Psychiatry 2017, 88, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, H.; Dean, G.; Elian, M.; Deng, H.X.; Deng, G.; Juneja, T.; Storey, E.; McKinlay Gardner, R.J.; Jacob, R.L.; Laing, N.G.; et al. A4T mutation in the SOD1 gene causing familial amyotrophic lateral sclerosis. Neuroepidemiology 2003, 22, 235–238. [Google Scholar] [CrossRef]
- Régal, L.; Vanopdenbosch, L.; Tilkin, P.; Van den Bosch, L.; Thijs, V.; Sciot, R.; Robberecht, W. The G93C mutation in superoxide dismutase 1: Clinicopathologic phenotype and prognosis. Arch. Neurol. 2006, 63, 262–267. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Pesiridis, G.S.; Lee, V.M.; Trojanowski, J.Q. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum. Mol. Genet. 2009, 18, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Rohn, T.T. Cytoplasmic inclusions of TDP-43 in neurodegenerative diseases: A potential role for caspases. Histol. Histopathol. 2009, 24, 1081–1086. [Google Scholar]
- Freibaum, B.D.; Chitta, R.K.; High, A.A.; Taylor, J.P. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 2010, 9, 1104–1120. [Google Scholar] [CrossRef] [PubMed]
- Liscic, R.M.; Grinberg, L.T.; Zidar, J.; Gitcho, M.A.; Cairns, N.J. ALS and FTLD: Two faces of TDP-43 proteinopathy. Eur. J. Neurol. 2008, 15, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, N.; Ince, P.G.; Smith, M.O.; Highley, R.; Skibinski, G.; Andersen, P.M.; Morrison, K.E.; Pall, H.S.; Hardiman, O.; Collinge, J.; et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 2006, 67, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Benajiba, L.; Le Ber, I.; Camuzat, A.; Lacoste, M.; Thomas-Anterion, C.; Couratier, P.; Legallic, S.; Salachas, F.; Hannequin, D.; Decousus, M.; et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann. Neurol. 2009, 65, 470–473. [Google Scholar] [CrossRef]
- Recabarren-Leiva, D.; Alarcón, M. New insights into the gene expression associated to amyotrophic lateral sclerosis. Life Sci. 2018, 193, 110–123. [Google Scholar] [CrossRef]
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med. Genet. 2018, 30, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P.; et al. Causative Genes in Amyotrophic Lateral Sclerosis and Protein Degradation Pathways: A Link to Neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef]
- Kanai, Y.; Okada, Y.; Tanaka, Y.; Harada, A.; Terada, S.; Hirokawa, N. KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci. 2000, 20, 6374–6384. [Google Scholar] [CrossRef]
- Wang, L.; Brown, A. A hereditary spastic paraplegia mutation in kinesin-1A/KIF5A disrupts neurofilament transport. Mol. Neurodegener. 2010, 15, 52. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.H.; Roberts, E.A.; Her, L.S.; Liu, X.; Williams, D.S.; Cleveland, D.W.; Goldstein, L.S. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J. Cell Biol. 2003, 161, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Andersen, P.M.; Nilsson, P.; Chioza, B.; Andersson, J.L.; Russ, C.; Shaw, C.E.; Powell, J.F.; Leigh, P.N. Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum. Mol. Genet. 1999, 8, 157–164. [Google Scholar] [CrossRef]
- Chevalier-Larsen, E.; Holzbaur, E.L. Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta 2006, 762, 1094–1108. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef]
- Alomo, G.M.; Manfredi, G. Exploring new pathways of neurodegeneration in ALS: The role of mitochondria quality control. Brain Res. 2015, 1607, 36–46. [Google Scholar]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Mc Whirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Thurston, T.L.; Ryzhakov, G.; Bloor, S.; Von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef]
- Clément, J.F.; Meloche, S.; Servant, M.J. The IKK-related kinases: From innate immunity to oncogenesis. Cell Res. 2008, 18, 889–899. [Google Scholar] [CrossRef]
- Pottier, C.; Bieniek, K.F.; Finch, N.; Van de Vorst, M.; Baker, M.; Perkersen, R.; Brown, P.; Ravenscroft, T.; van Blitterswijk, M.; Nicholson, A.M.; et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015, 130, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Saw, W.T.; Tan, E.K. Mitochondrial CHCHD-Containing Proteins: Physiologic Functions and Link with Neurodegenerative Diseases. Mol. Neurobiol. 2017, 54, 5534–5546. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Glynn, S.M.; Gibbs, J.R.; Nalls, M.A.; Sabatelli, M.; Restagno, G.; Drory, V.E.; Chiò, A.; Rogaeva, E.; Traynor, B.J. Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain 2014, 137, e311. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.; et al. Reply: Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain 2014, 137, e310. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Müller, K.; Andersen, P.M.; Hübers, A.; Marroquin, N.; Volk, A.E.; Danzer, K.M.; Meitinger, T.; Ludolph, A.C.; Strom, T.M.; Weishaupt, J.H. Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain 2014, 137, e309. [Google Scholar] [CrossRef] [PubMed]
- Ajroud-Driss, S.; Fecto, F.; Ajroud, K.; Lalani, I.; Calvo, S.E.; Mootha, V.K.; Deng, H.X.; Siddique, N.; Tahmoush, A.J.; Heiman-Patterson, T.D.; et al. Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics 2015, 16, 1–9. [Google Scholar] [CrossRef]
- Penttilä, S.; Jokela, M.; Bouquin, H.; Saukkonen, A.M.; Toivanen, J.; Udd, B. Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann. Neurol. 2015, 77, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.D.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Senderek, J.; Garvey, S.M.; Krieger, M.; Guergueltcheva, V.; Urtizberea, A.; Roos, A.; Elbracht, M.; Stendel, C.; Tournev, I.; Mihailova, V.; et al. Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am. J. Hum. Genet. 2009, 84, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Liu, T.; Trotter, C.; Fang, C.C.; De Narvaez, E.; LePochat, P.; Maslar, D.; Bukhari, A.; Zhao, X.; Deonarine, A.; et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 2017, 8, 15558. [Google Scholar] [CrossRef]
- Salton, M.; Elkon, R.; Borodina, T.; Davydov, A.; Yaspo, M.-L.; Halperin, E.; Shiloh, Y. Matrin 3 binds and stabilizes mRNA. PLoS ONE 2011, 6, e23882. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.B.; Attig, J.; Bellora, N.; König, J.; Hallegger, M.; Kayikci, M.; Eyras, E.; Ule, J.; Smith, C.W. Nuclear matrix protein Matrin3 regulates alternative splicing and forms overlapping regulatory networks with PTB. EMBO J. 2015, 34, 653–668. [Google Scholar] [CrossRef]
- Borghero, G.; Pugliatti, M.; Marrosu, F.; Marrosu, M.G.; Murru, M.R.; Floris, G.; Cannas, A.; Parish, L.D.; Occhineri, P.; Cau, T.B.; et al. Genetic architecture of ALS in Sardinia. Neurobiol. Aging 2014, 35, 2882. [Google Scholar] [CrossRef]
- Origone, P.; Verdiani, S.; Bandettini Di Poggio, M.; Zuccarino, R.; Vignolo, M.; Caponnetto, C.; Mandich, P. A novel Arg147Trp MATR3 missense mutation in a slowly progressive ALS Italian patient. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 530–531. [Google Scholar] [CrossRef]
- Lin, K.P.; Tsai, P.C.; Liao, Y.C.; Chen, W.T.; Tsai, C.P.; Soong, B.W.; Lee, Y.C. Mutational analysis of MATR3 in Taiwanese patients with amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, e1–e4. [Google Scholar] [CrossRef]
- Jean-Philippe, J.; Paz, S.; Caputi, M. hnRNP A1: The Swiss army knife of gene expression. Int. J. Mol. Sci. 2013, 14, 18999–19024. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Wall, M.L.; Lewis, S.M. Methylarginines within the RGG-Motif Region of hnRNP A1 Affect Its IRES Trans-Acting Factor Activity and Are Required for hnRNP A1 Stress Granule Localization and Formation. J. Mol. Biol. 2017, 429, 295–307. [Google Scholar] [CrossRef]
- Elvira, G.; Wasiak, S.; Blandford, V.; Tong, X.K.; Serrano, A.; Fan, X.; del Rayo Sánchez-Carbente, M.; Servant, F.; Bell, A.W.; Boismenu, D.; et al. Characterization of an RNA granule from developing brain. Mol. Cell Proteom. 2006, 5, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Long, J.C.; Cáceres, J.F. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell Biol. 2006, 26, 5744–5758. [Google Scholar] [CrossRef] [PubMed]
- Mc Donald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Vande Velde, C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P. Multisystem proteinopathy: Intersecting genetics in muscle, bone, and brain degeneration. Neurology 2015, 85, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Klar, J.; Sobol, M.; Melberg, A.; Mäbert, K.; Ameur, A.; Johansson, A.C.; Feuk, L.; Entesarian, M.; Orlén, H.; Casar-Borota, O.; et al. Welander distal myopathy caused by an ancient founder mutation in TIA1 associated with perturbed splicing. Hum. Mutat. 2013, 34, 572–577. [Google Scholar] [PubMed]
- Brand, P.; Dyck, P.J.; Liu, J.; Berini, S.; Selcen, D.; Milone, M. Distal myopathy with coexisting heterozygous TIA1 and MYH7 Variants. Neuromuscul. Disord. 2016, 26, 511–515. [Google Scholar] [CrossRef]
- Hackman, P.; Sarparanta, J.; Lehtinen, S.; Vihola, A.; Evilä, A.; Jonson, P.H.; Luque, H.; Kere, J.; Screen, M.; Chinnery, P.F.; et al. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann. Neurol. 2013, 73, 500–509. [Google Scholar] [CrossRef]
- Shalom, O.; Shalva, N.; Altschuler, Y.; Motro, B. The mammalian Nek1 kinase is involved in primary cilium formation. FEBS Lett. 2008, 582, 1465–1470. [Google Scholar] [CrossRef]
- Chen, Y.; Craigen, W.J.; Riley, D.J. Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell Cycle 2009, 8, 257–267. [Google Scholar] [CrossRef]
- Pelegrini, A.L.; Moura, D.J.; Brenner, B.L.; Ledur, P.F.; Maques, G.P.; Henriques, J.A.; Saffi, J.; Lenz, G. Nek1 silencing slows down DNA repair and blocks DNA damage-induced cell cycle arrest. Mutagenesis 2010, 25, 447–454. [Google Scholar] [CrossRef]
- Fang, X.; Lin, H.; Wang, X.; Zuo, Q.; Qin, J.; Zhang, P. The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim. Biophys. Sin. 2015, 47, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.; Võsa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Van Mossevelde, S.; Dillen, L.; De Bleecker, J.L.; Moisse, M.; Van Damme, P.; Van Broeckhoven, C.; van der Zee, J.; BELNEU Consortium. NEK1 genetic variability in a Belgian cohort of ALS and ALS-FTD patients. Neurobiol. Aging 2018, 61, e1–e255. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Baloh, R.H.; Milbrandt, J. The NIMA-family kinase Nek3 regulates microtubule acetylation in neurons. J. Cell Sci. 2009, 122, 2274–2282. [Google Scholar] [CrossRef] [PubMed]
- Helferich, A.M.; Brockmann, S.J.; Reinders, J.; Deshpande, D.; Holzmann, K.; Brenner, D.; Andersen, P.M.; Petri, S.; Thal, D.R.; Michaelis, J.; et al. Dysregulation of a novel miR-1825/TBCB/TUBA4A pathway in sporadic and familial ALS. Cell Mol. Life Sci. 2018, 75, 4301–4319. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Montiel, M.J.; Chaineau, M.; Durcan, T.M. The Neglected Genes of ALS: Cytoskeletal Dynamics Impact Synaptic Degeneration in ALS. Front. Cell. Neurosci. 2020, 14, 594975. [Google Scholar] [CrossRef]
- Wu, C.H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012, 488, 499–503. [Google Scholar] [CrossRef]
- Wicks, P.; Abrahams, S.; Papps, B.; Al-Chalabi, A.; Shaw, C.E.; Leigh, P.N.; Goldstein, L.H. SOD1 and cognitive dysfunction in familial amyotrophic lateral sclerosis. J. Neurol. 2009, 256, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Diekstra, F.P.; Van Vught, P.W.; Van Rheenen, W.; Koppers, M.; Pasterkamp, R.J.; Van Es, M.A.; Schelhaas, H.J.; de Visser, M.; Robberecht, W.; Van Damme, P.; et al. UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 630. [Google Scholar] [CrossRef] [PubMed]
- Sproviero, W.; Shatunov, A.; Stahl, D.; Shoai, M.; Van Rheenen, W.; Jones, A.R.; Al-Sarraj, S.; Andersen, P.M.; Bonini, N.M.; Conforti, F.L.; et al. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol. Aging 2017, 51, e1–e178. [Google Scholar] [CrossRef] [PubMed]
- Lattante, S.; Pomponi, M.G.; Conte, A.; Marangi, G.; Bisogni, G.; Patanella, A.K.; Meleo, E.; Lunetta, C.; Riva, N.; Mosca, L.; et al. ATXN1 intermediate-length polyglutamine expansions are associated with amyotrophic lateral sclerosis. Neurobiol. Aging 2018, 64, e1–e157. [Google Scholar] [CrossRef] [PubMed]
- Uyan, Ö.; Ömür, Ö.; Ağım, Z.S.; Özoğuz, A.; Li, H.; Parman, Y.; Deymeer, F.; Oflazer, P.; Koç, F.; Tan, E.; et al. Genome-wide copy number variation in sporadic amyotrophic lateral sclerosis in the Turkish population: Deletion of EPHA3 is a possible protective factor. PLoS ONE 2013, 8, 72381. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morgan, S.; Shatunov, A.; Sproviero, W.; Jones, A.R.; Shoai, M.; Hughes, D.; Al Khleifat, A.; Malaspina, A.; Morrison, K.E.; Shaw, P.J.; et al. A comprehensive analysis of rare genetic variation in amyotrophic lateral sclerosis in the UK. Brain 2017, 140, 1611–1618. [Google Scholar] [CrossRef]
- Cady, J.; Allred, P.; Bali, T.; Pestronk, A.; Goate, A.; Miller, T.M.; Mitra, R.D.; Ravits, J.; Harms, M.B.; Baloh, R.H.; et al. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol. 2015, 77, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Nakamura, R.; Sone, J.; Atsuta, N.; Tohnai, G.; Watanabe, H.; Yokoi, D.; Nakatochi, M.; Watanabe, H.; Ito, M.; Senda, J.; et al. Next-generation sequencing of 28 ALS-related genes in a Japanese ALS cohort. Neurobiol. Aging 2016, 39, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Mc Cann, E.P.; Henden, L.; Fifita, J.A.; Zhang, K.Y.; Grima, N.; Bauer, D.C.; Chan Moi Fat, S.; Twine, N.A.; Pamphlett, R.; Kiernan, M.C.; et al. Evidence for polygenic and oligogenic basis of Australian sporadic amyotrophic lateral sclerosis. J. Med. Genet. 2020, 14, 06866. [Google Scholar]
- Vasilopoulou, C.; Morris, A.P.; Giannakopoulos, G.; Duguez, S.; Duddy, W. What Can Machine Learning Approaches in Genomics Tell Us about the Molecular Basis of Amyotrophic Lateral Sclerosis? J. Pers. Med. 2020, 10, 247. [Google Scholar] [CrossRef]
- Tsuji, S. Genetics of neurodegenerative diseases: Insights from high-throughput resequencing. Hum. Mol. Genet. 2010, 19, R65–R70. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Riboldi, G.; Zanetta, C.; Ranieri, M.; Nizzardo, M.; Simone, C.; Magri, F.; Bresolin, N.; Comi, G.P.; Corti, S. Antisense oligonucleotide therapy for the treatment of C9ORF72 ALS/FTD diseases. Mol. Neurobiol. 2014, 50, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Cappella, M.; Ciotti, C.; Cohen-Tannoudji, M.; Biferi, M.G. Gene Therapy for ALS-A Perspective. Int. J. Mol. Sci. 2019, 20, 4388. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).