Abstract
Fengjing pigs are a Chinese native breed known for their high reproductive ability. Runs of homozygosity (ROHs) have emerged as an effective tool for evaluating inbreeding levels and identifying relevant genes in selection. However, the declining population of Fengjing pigs in recent years has raised concerns about inbreeding. Therefore, this study aimed to investigate the ROH patterns, estimate genomic inbreeding levels, and identify candidate genes associated with economic traits using whole-genome resequencing data from 105 Fengjing pigs. A total of 2448 ROHs were identified, with an average of 23.31 ROHs per individual and an average length of 9.50 Mb. The inbreeding coefficient, based on ROHs, was 0.098. Additionally, three genomic regions with a high frequency of ROHs were identified. These regions contained 64 unique genes, including 14 genes associated with important economic traits. Moreover, six overlapping quantitative trait loci (QTLs) and four candidate genes (HSPG2, CDC42, EPHB2, and GRHL3) were identified on Sus scrofa chromosome (SSC) 6. These QTLs are associated with birth traits (health and reproductive efficiency) and meat development traits (meat quality and growth). This study identified many candidate genes and QTLs that overlapped with ROHs and are associated with economically significant traits. These findings can be used in future breeding, conservation, and utilization of specific Chinese native pig breeds.
1. Introduction
Runs of homozygosity (ROHs) refer to continuous homozygous regions in the genome of diploid organisms, which are formed when offspring inherit the same haplotypes from both parents. The length and frequency of ROHs can reflect the demographic and genetic history of a livestock population []. Related studies have suggested that ROHs may originate from more closely related ancestors. The longer the ROH region, the higher the likelihood of recent inbreeding within a population. In contrast, shorter ROHs are derived from more distant ancestors, and their origins usually cannot be explained by existing genealogical records [,]. Many studies have also shown that different mating systems, selection intensities, effective population sizes, and population structures in a livestock population can lead to the formation of distinct ROH patterns in the genome [,,]. Therefore, ROHs can be used to estimate the inbreeding levels of livestock populations, infer their inbreeding history, identify selected genes and deleterious mutations, assess genetic diversity and the conservation of genetic resources, and optimize animal breeding strategies.
In recent years, with the widespread application of single nucleotide polymorphism (SNP) chips and whole-genome sequencing technology, ROH based research on livestock genomics has increased significantly []. Genes associated with growth rate, reproductive efficiency, immune function, and environmental adaptability have been identified in specific ROHs in the genomes of cattle [,], pig [,], and sheep []. Therefore, ROH detection is becoming increasingly prevalent in livestock population research and serves as an effective approach for identifying genes associated with important economic traits in livestock.
The Fengjing pig is an ancient indigenous pig breed of China, native to the Taihu Lake Basin. The Fengjing pig is characterized by early sexual maturity, good meat quality, and strong environmental adaptability. Its most prominent feature is its excellent reproductive performance, and it is also one of the breeds with the highest fecundity in the world. Owing to these outstanding breed characteristics, Fengjing pigs have been commonly used in hybrid breeding programs and scientific research projects. Over the past few decades, the population of Fengjing pigs has been declining due to the large-scale import of lean-type pigs from western countries for commercial production. Currently, the population of Fengjing pigs is estimated to be around 400–500, with the majority being concentrated in three state-supported conservation farms. A reduction in population size elevates the likelihood of inbreeding, which may increase the frequency of homozygous deleterious alleles and consequently impair individual phenotypic performance. However, there is limited research on the ROH patterns and inbreeding coefficients in the Fengjing pig population. Therefore, this study used the GeneSeek GGP Porcine 50K SNP chip to genotype 105 Fengjing pig samples, with the objectives of analyzing the genome-wide ROH patterns and inbreeding levels of Fengjing pigs, identifying high-frequency ROH regions, and detecting candidate genes associated with porcine economic traits. This study aims to contribute to the enhanced protection of the genetic diversity of Fengjing pigs and provide valuable insights for their effective utilization.
2. Materials and Methods
2.1. Population and Genotyping
The entire population of Fengjing pigs from a conservation pig farm in Jiangsu Province was used in this study, consisting of 105 individuals (99 females and 6 males). Ear tissue samples were collected from each individual, stored in 75% ethanol at −20 °C for subsequent genomic DNA extraction. Genomic DNA was isolated using a commercial DNA extraction kit (Tiangen Biotech, Beijing, China) in strict accordance with the manufacturer’s recommended protocol. The quality of the extracted DNA was assessed using a NanoDrop 2000 spectrophotometer (Thermo Fischer Scientific, Wilmington, DE, USA). DNA samples meeting the following criteria were used: a purity ratio (A260/280) between 1.7 and 2.0, and a DNA concentration higher than 50 ng/μL. Then, the qualified DNA samples were genotyped using the GeneSeek GGP Porcine 50K SNP chip (Neogen Corporation, Lansing, MI, USA).
For genotypic data processing, individuals with a SNP missing rate higher than 5% were removed, and only autosomal SNP markers were used for further analysis. The quality of the dataset was evaluated using the PLINK software (version 1.90) [], with the following criteria: (1) individual call rate higher than 90%; (2) minor allele frequency (MAF) higher than 0.01; (3) Hardy–Weinberg equilibrium (HWE) p-value higher than 1.00 × 10−6. The observed heterozygosity (Ho) and expected heterozygosity (He) were also calculated using the PLINK software (version 1.90) [].
2.2. ROHs Identification and Inbreeding Coefficient Estimation
ROHs were identified on the autosomes of each individual using the R package detectRUNS (version 0.9.6) (https://cran.r-project.org/web/packages/detectRUNS/vignettes/detectRUNS.vignette.html (accessed on 10 July 2024)), with the following specific detection criteria: (1) Minimum ROH length higher than 1 Mb; (2) Minimum number of consecutive SNPs per ROH higher than 50; (3) Maximum gap between consecutive SNPs in a ROH less than 1 Mb; (4) Maximum allowed number of missing genotypes per ROH is 5, and maximum allowed number of heterozygous genotypes per ROH is 1; (5) Sliding window threshold set to 0.05; (6) Minimum SNP density set to 1 SNP/50 kb.
Based on the length of ROH regions, all identified ROHs were categorized into three groups: 1–5 Mb, 5–10 Mb, and >10 Mb. For the entire population, the number of ROHs in each length category was counted, and the distribution of ROHs across individual chromosomes was determined. Additionally, for each individual, the total number of ROHs and the total length of all ROH regions were calculated and summarized. The genomic inbreeding coefficient based on ROHs (FROH) for each individual was calculated using the formula described in published literature []: FROH = ΣLroh/Lauto. Here, ΣLroh represents the total length of all ROH regions in an individual, and Lauto denotes the total length of the autosomal genome covered by SNPs, which was determined to be 2.45 Gb in this study.
2.3. Detection of Genomic Regions with a High Frequency of ROHs
To identify genomic regions with a high frequency of ROHs, the proportion of SNPs located within ROHs was calculated. This was achieved by counting the number of times each SNP was found in ROH regions across all individuals. Then, the top 0.1% of SNPs with the highest frequency of occurrence in ROHs was selected as the threshold for defining genomic regions with a high frequency of ROHs.
Based on the pig reference genome (Sscrofa11.1), the genes located in these high-frequency ROH regions were annotated using ANNOVAR software (version 20200608) []. Subsequently, these annotated genes were subjected to Gene Ontology (GO) functional enrichment analysis and Kyoto Encyclopaedia of Genes and Genomes (KEGG) enrichment pathway analysis. The results were visualized using the clusterProfiler R package (version 4.4.4), with a p-value < 0.05 considered to indicate significant enrichment. Furthermore, the specific genes with significant functional enrichments were compared against the pig quantitative trait loci (QTL) database (Pig QTLdb) to identify candidate functional genes.
3. Results
3.1. Genetic Diversity and Inbreeding Coefficient
After the analysis of the sequence datasets in this study, a total of 50,697 SNPs were identified. Following quality control, 21,442 informative SNPs were obtained. The distribution of these SNPs on chromosomes is illustrated in Figure 1. The number of SNPs varied across chromosomes, with the highest number (2047 SNPs) located on Sus scrofa chromosome (SSC) 1 and the lowest number (547 SNPs) located on SSC10.
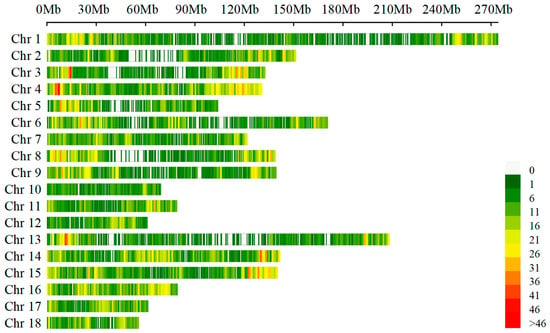
Figure 1.
The SNPs density distribution on chromosomes. The x-axis represents the genomic position (Mb), and the y-axis represents the chromosomes. The density of SNPs within each genomic region is indicated by color.
The average observed heterozygosity (Ho) and expected heterozygosity (He) of the Fengjing pig population were calculated by genome-wide SNPs to be 0.330 ± 0.161 and 0.313 ± 0.148, respectively. To determine the inbreeding coefficient, FROH was used to assess the inbreeding status of the Fengjing pig population (Table 1). At the individual level, the average FROH_all value was 0.098, ranging from 0.008 to 0.200. Among the three inbreeding coefficients based on different ROH lengths, the FROH 1–5Mb displayed the lowest values. Meanwhile, FROH 1–5Mb had low correlations with FROH 5–10Mb and FROH>10Mb (Table S1).

Table 1.
The inbreeding coefficient of the Fengjing pig population.
3.2. ROHs Distribution Across the Genome
Among the 105 individuals, a total of 2448 ROHs were identified (Table S2). The average number of ROHs per individual was 23.31, with an average length of 9.50 Mb. The longest ROH, located on SSC1, spanned 68.95 Mb and contained 772 SNPs, while the shortest ROH, located on SSC13, measured 1.34 Mb and contained 54 SNPs. The ROHs were divided into three groups based on their length, as shown in Table 2. Of the total ROHs, 802 ROHs were shorter than 5 Mb, 869 ROHs ranged from 5 to 10 Mb, and 777 ROHs were longer than 10 Mb. The largest group was the ROHs with a length of 5 to 10 Mb, accounting for 35.50% of total ROHs.
Table 2.
Statistics of ROH number and length based on ROH length.
The distribution of ROHs on each chromosome is shown in Figure 2. SSC10 had the lowest number of ROHs (23 ROHs), while SSC6 had the highest number of ROHs (233 ROHs) (Figure 2a). Furthermore, the coverage of ROHs for each chromosome was estimated. SSC10 displayed the lowest coverage of ROHs, while SSC16 displayed the highest coverage of ROHs (Figure 2b).
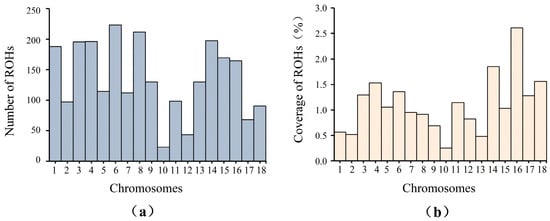
Figure 2.
Distribution of ROHs on each chromosome. (a) Number of ROHs on each chromosome; (b) Coverage of ROHs on each chromosome.
3.3. Genomic Regions with a High Frequency of ROHs
To identify genomic regions with a high frequency of ROHs, the frequency of SNPs in ROHs was calculated by tallying the number of times a SNP appeared in a specific ROH across all individuals. Based on this, the top 0.1% of SNPs with the highest frequency of occurrence in ROHs was selected (Table S3). The genomic positions of these SNPs on their respective chromosomes are presented in Figure 3. Three genomic regions with a high frequency of ROHs were identified (Table 3). These regions, located on SSC3, SSC6, and SSC16, contain 60, 32, and 33 SNPs, respectively, with lengths of 7.89 Mb, 3.46 Mb, and 5.32 Mb.
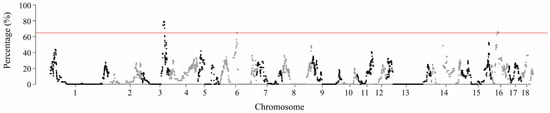
Figure 3.
Manhattan plot of SNP frequency within ROHs across all individuals. The x-axis represents the genomic position on each chromosome; the y-axis represents the percentage of SNPs located within ROHs. The horizontal line indicates the threshold for the top 0.1% of SNPs with the highest frequency of occurrence in ROHs.
Table 3.
Statistics of the genomic regions with a high frequency of ROHs. A high-frequency ROH region was defined as a genomic segment containing SNPs ranked within the top 0.1% for frequency of occurrence in ROHs.
3.4. Candidate Gene Annotation and Enrichment Analysis
To assess the selection signatures of genomic regions with a high frequency of ROHs, candidate genes located in these regions were annotated (Table S3). A total of 64 genes were identified, including 3 protein-coding genes and 5 long non-coding RNAs (lncRNAs). Functional enrichment analyses were then performed on these annotated genes (Figure 4). GO functional enrichment analysis revealed that these genes were involved in galactose catabolic process, galactose metabolic process, monosaccharide catabolic process, and others. Additionally, KEGG enrichment pathway analysis showed that these genes were significantly enriched in Galactose metabolism, Chemokine signaling pathway, Glycerophospholipid metabolism, T cell receptor signaling pathway, and others.
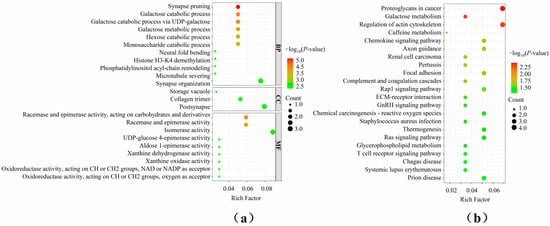
Figure 4.
Functional enrichment analysis of candidate genes annotation in high-frequency ROH regions. (a) GO functional enrichment analysis of candidate genes; (b) KEGG enrichment pathway analysis of candidate genes.
Furthermore, based on significant functional enrichments, genes associated with specific traits relevant to livestock breeding were screened. A total of 14 annotated genes were found to be associated with important economic traits (Table 3). Additionally, based on the pig QTLdb, eight QTLs associated with porcine economic traits were found to be overlapped with high-frequency regions (Table 4). Interestingly, six of these overlapping QTLs were located on SSC6, including QTL:21528 and QTL:21527 (both in HSPG2 gene sequences), QTL:178770 (in CDC42 gene sequences), QTL:179298 and QTL:179297 (both in EPHB2 gene sequences), and QTL:284953 (in GRHL3 gene sequences).
Table 4.
Statistics of overlapping QTLs associated with porcine economic traits based on the pig QTLdb.
4. Discussion
The occurrence and distribution of ROHs in a population are influenced by selection, recombination, and population history [,]. ROHs are commonly used to assess the level of inbreeding across the entire genome in animal populations and to identify special genomic regions under selection [,]. In this study, we used the GeneSeek GGP Porcine 50K chip to investigate the occurrence and distribution of ROHs in the Fengjing pig population. Our goal was to analyze ROH patterns in Fengjing pigs and to detect candidate genes associated with economic traits.
4.1. Analysis of Genetic Diversity and Inbreeding Coefficient
Previous studies have suggested that the value of Ho and He are indicative of population genetic diversity []. In this study, we observed that Ho was slightly higher than He in the Fengjing pig population, and both values were close to 0.3. This may indicate higher genetic diversity in the Fengjing pig population. Moreover, genome-wide SNPs are widely used for assessing population inbreeding [], and FROH mainly determined by the ratio of the total length of homozygous regions to the autosomal genome length []. In our study, the inbreeding coefficients were estimated based on ROHs. The results showed that the FROH value for the all ROH length category in the Fengjing pig population was relatively high, at 0.098. This result may be attributed to the small effective population size, as a reduced population size is known to promote increased levels of inbreeding []. However, the FROH value for the 0.5–1 Mb ROH length category was lower than those of the other length categories. This suggesting that a reduced effective population size in recent generations was the primary reason for the increase in inbreeding coefficient. When compared to commercial pig breeds, the FROH_all value of the Fengjing pigs was lower than that of the Large White pigs (0.140) [] and Pietrain pigs (0.205) []. This phenomenon may indicate that the Fengjing pig population has not undergone intensive artificial selection, thereby retaining a certain level of genetic diversity.
4.2. Analysis of General Characteristics of ROHs
Many studies have demonstrated that ROHs may originate from more closely related ancestors, and the genomic distribution patterns of ROHs serve as an indicator of differences in breed management and formation [,]. The length of ROHs reflects the historical origins of breed populations: short ROHs are indicative of ancient inbreeding, while long ROHs suggest recent inbreeding [,]. In our study, the majority (approximately 68%) of ROHs detected in the Fengjing pig population were classified as short or medium length (<10 Mb). This finding is consistent with previous studies on pigs [,,]. However, long ROHs (>10 Mb) still accounted for a relatively large proportion (31.74%) of the total ROHs, exceeding the corresponding proportions reported for Jinhua pigs (7.56%), Erhualian pigs (10.58%), and Meishan pigs (10.08%) [,]. This result suggests that the effective population size of the Fengjing pig population has been small in recent generations, resulting in a higher frequency of long ROHs. In fact, conservation efforts for Jinhua pigs, Meishan pigs, and Erhualian pigs have been more successful in recent generations, which is accompanied by their larger effective population sizes. Additionally, it is worth noting that population bottlenecks and genetic drift can also have an impact on the formation of ROHs [].
4.3. Analysis of Genomic Regions with a High Frequency of ROHs and Candidate Genes
Previous studies have established consensuses regarding the role of ROHs in animal populations. Specifically, the distinct distribution patterns of ROHs occurred by artificial selection or domestication, and genomic regions with a high frequency of ROHs may indicate potential selection signatures for specific traits [,]. In this study, we detected three genomic regions with a high frequency of ROHs (The top 0.1% of SNPs with the frequency of occurrence in ROHs), and 64 candidate genes were identified in these regions. Functional enrichment analyses showed that these genes were enriched in the GO term “galactose metabolic process” and in KEGG pathways, including “Galactose metabolism, “Chemokine signaling pathway”, “Glycerophospholipid metabolism”, and “T cell receptor signaling pathway”. These results indicate that the functional genes detected in this study are closely related to the meat quality, growth, reproduction and health of pigs.
Based on significant functional enrichments, we identified several candidate genes in Fengjing pigs that may be associated with the traits of interest. Three candidate genes related to growth traits: LTBP1, BIRC6, and ITGA2. Among them, LTBP1 is an important signaling molecule in the TGF-β pathway, involved in regulating the growth and physiological processes of skeletal muscle in mammalian bodies [,]. The BIRC6 gene plays a role in cell apoptosis and cytoplasmic division and has been shown to affect animal fertility, average daily gain, and feed efficiency [,]. Four candidate genes related to reproduction traits: BIRC6, KIF17, CDC42, and EPHB2. Among them, BIRC6 has been confirmed to be involved in the development of animal follicles and early embryonic development [,]. KIF17 belongs to the kinesin-2 family and plays an indispensable role in mammalian spermatogenesis []. Previous studies have demonstrated that KIF17 colocalizes with its cargo proteins in the manchette and the principal piece of the sperm tail and was implicated in spermatogenesis []. Four candidate genes related to meat traits: LCLAT1, FGF10, HSPG2, and NNT. Among them, LCLAT1, a microsomal enzyme involved in phospholipid remodeling, has been identified as a candidate gene associated with pork quality (Intramuscular fat content) []. FGF10, a member of the FGF family involved in various developmental processes, has been identified as a candidate gene associated with meat color traits in pigs []. Four candidate genes related to health traits: MUL1, CCL28, C5orf34, and GRHL3. Among them, MUL1 was an immune-related protein gene that participates in regulating the body’s innate immune response to against viral infections by inhibiting the RIG-I-dependent antiviral response []. CCL28 functions as an immune mucosal protein that binds to CCR3 receptors, driving lymphocyte migration, initiating immune response, and clearing pathogens []. These results indicate that genomic regions with a high frequency of ROHs in Fengjing pigs are associated with key selected production traits (growth, reproduction, meat quality, and health traits), these traits are directly shaped by long-term environmental conditions and artificial selection.
Finally, by searching the pig QTLdb, we have identified eight QTLs overlapping with high-frequency ROH regions that are associated with economically important traits in pigs. Interestingly, six of these QTLs were located on SSC6, residing within the HSPG2, CDC42, EPHB2, and GRHL3 gene sequences, respectively. The HSPG2 gene encodes a large proteoglycan that is a component of the extracellular matrix and has been identified as a candidate gene for fat deposition and growth traits, QTL (QTL:21528) in HSPG2 gene sequence associated with growth (Days to 113 kg) were identified []. Moreover, Duroc pigs with the GG genotype (QTL:21527 in HSPG2 gene sequence) grew more slowly and had higher marbling scores []. CDC42 is expressed in the trophoblast cells of the placenta and participates in the migration of trophoblast cells [], QTL (QTL:178770) in CDC42 gene sequence associated with litter size have been reported [,]. EPHB2 belongs to the Eph family of receptors, which plays an important role in pig embryo implantation and may affect reproductive efficiency [], and two QTLs (QTL:179298; QTL:179297) in EPHB2 gene sequence associated with litter weight were identified []. GRHL3 belongs to the Grainyhead-like (GRHL) transcription factor family and plays a regulatory role in cell proliferation and apoptosis [], QTL(QTL:284953) in GRHL3 gene sequence showed a significant association with susceptibility to umbilical hernia in pigs []. These results reveal that the overlapping QTLs detected on SSC6 in the present study are strongly associated with birth traits (health and reproductive efficiency) and meat development traits (meat quality and growth). This finding also reflects the unique characteristics and historical selection effects of Chinese native pigs. Meanwhile, these QTLs are all located in the high-frequency ROH regions on SSC6. This indicates that the homozygous genomic region on SSC6 is a core region under selection in the Fengjing pig population and a key target for future pig breeding.
5. Conclusions
In this study, we investigated the ROH patterns and genomic inbreeding level of the Fengjing pig population using whole-genome resequencing data. A total of 2448 ROHs were identified. Through the analysis of these ROHs, we identified three genomic regions with a high frequency of ROHs. These regions contained 64 unique genes, including 14 genes associated with important economic traits. By searching the pig QTLdb, we identified eight QTLs that overlapped with the high-frequency ROH regions; six of these QTLs were located on SSC6, residing within the HSPG2, CDC42, EPHB2, and GRHL3 gene sequences. These QTLs are associated with various traits such as growth, reproduction, meat quality, and health. Our findings enhance the understanding of genetic diversity, genome structure, and the genetic mechanisms underlying economically important traits in the Fengjing pig population.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/life15121823/s1, Table S1: Pearson correlation coefficients between inbreeding coefficients; Table S2: Detection of ROHs in each genome; Table S3: The list of the top 0.1% of SNPs with the highest frequency of occurrence in ROHs.
Author Contributions
Conceptualization, L.N. and P.X.; methodology, L.N. and H.G.; software, J.Z.; validation, L.N., H.G. and Z.G.; formal analysis, L.N., S.L. and P.X.; investigation, C.Z.; resources, X.W. and C.Z.; data curation, Z.G., J.Z. and S.L.; writing—original draft preparation, L.N. and H.G.; writing—review and editing, P.X.; visualization, L.N.; supervision, X.W.; project administration, P.X. and X.W.; funding acquisition, P.X. and X.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Technology Project of Jiangsu Agri-animal Husbandry Vocational College (NSF2024JB02), the Project of Open Bidding for Selecting the Best Candidates (JBGS2021028), the Project of Taizhou 311 High-level Talent Training (2024SE136) and the 2024 University-level Undergraduate Science and Technology Innovation Fund Project of Yangzhou University (XCX20240780).
Institutional Review Board Statement
This study was performed according to the Regulations for the Administration of Affairs Concerning Experimental Animals and approved by the ethics committee of Jiangsu Agri-animal Husbandry Vocational College on 12 January 2024 (SYXK(Su) IACUC2024-JB02).
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article and Supplementary Materials. Further inquiries can be directed to the corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.L.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Rebelato, A.B.; Caetano, A.R. Runs of homozygosity for autozygosity estimation and genomic analysis in production animals. Pesqui. Agropecuária Bras. 2018, 53, 975–984. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Q.; Xiao, Q.; Sun, H.; Gao, H.; Yang, Y.; Chen, J.; Li, Z.; Xue, M.; Ma, P.; et al. Distribution of runs of homozygosity in Chinese and Western pig breeds evaluated by reduced-representation sequencing data. Anim. Genet. 2018, 49, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Wang, Y.; Zhou, P.; Shi, H.; Ma, X.; Yin, Z.; Liu, Y. Genomic Scan for Runs of Homozygosity and Selective Signature Analysis to Identify Candidate Genes in Large White Pigs. Int. J. Mol. Sci. 2023, 24, 12914. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tong, S.F.; Zhu, M.; Xie, R.; Dong-Feng, L.I.; Zhang, L.F.; Liu, Y. Genome-wide detection for runs of homozygosity analysis in three pig breeds from Chinese Taihu Basin and Landrace pigs by SLAF-seq data. J. Integr. Agric. 2022, 21, 3293–3301. [Google Scholar] [CrossRef]
- Peripolli, E.; Munari, D.P.; Silva, M.V.G.B.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Sun, H.; Lu, S.; Gou, X.; Yan, D.; Xu, Z.; Zhang, Z.; Qadri, Q.R.; Zhang, Z.; Wang, Z.; et al. Genetic Diversity and Selection Signatures Within Diannan Small-Ear Pigs Revealed by Next-Generation Sequencing. Front. Genet. 2020, 11, 733. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wirth, A.; Duda, J.; Emmerling, R.; Götz, K.U.; Birkenmaier, F.; Distl, O. Analyzing Runs of Homozygosity Reveals Patterns of Selection in German Brown Cattle. Genes 2024, 15, 1051. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Falchi, L.; Cesarani, A.; Criscione, A.; Hidalgo, J.; Garcia, A.; Mastrangelo, S.; Macciotta, N.P.P. Effect of genotyping density on the detection of runs of homozygosity and heterozygosity in cattle. J. Anim. Sci. 2024, 102, 147. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, S.; Yang, J.; Li, G.; Ding, R.; Zhuang, Z.; Ruan, D.; Wu, J.; Yang, H.; Zheng, E.; Cai, G.; et al. Identification of Homozygous Regions with Adverse Effects on the Five Economic Traits of Duroc Pigs. Front. Vet. Sci. 2022, 9, 855933. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, Z.; Sun, H.; Zhang, Z.; Zhao, Q.; Olasege, B.S.; Li, Q.; Yue, Y.; Ma, P.; Zhang, X.; Wang, Q.; et al. Assessment of Autozygosity Derived from Runs of Homozygosity in Jinhua Pigs Disclosed by Sequencing Data. Front. Genet. 2019, 10, 274. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, Q.; Huang, C.; Chen, Q.; Su, Y.; Zhang, Y.; Wang, R.; Su, R.; Xu, H.; Liu, S.; Ma, Y.; et al. Genomic Inbreeding and Runs of Homozygosity Analysis of Cashmere Goat. Animals 2024, 14, 1246. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mcquillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xie, R.; Shi, L.; Liu, J.; Deng, T.; Wang, L.; Liu, Y.; Zhao, F. Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes in Three Pig Breeds. Animals 2019, 9, 518. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Keller, M.C.; Visscher, P.M.; Goddard, M.E. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 2011, 189, 237–249. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kaviriri, D.K.; Zhang, Q.; Zhang, X.; Jiang, L.; Zhang, J.; Wang, J.; Khasa, D.P.; You, X.; Zhao, X. Phenotypic Variability and Genetic Diversity in a Pinus koraiensis Clonal Trial in Northeastern China. Genes 2020, 11, 673. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Toro, M.A.; Fernández, J.; Caballero, A. Molecular characterization of breeds and its use in conservation. Livest. Sci. 2009, 120, 174–195. [Google Scholar] [CrossRef]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Curik, I.; Ferencakovic, M.; Solkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Shi, L.; Wang, L.; Liu, J.; Deng, T.; Yan, H.; Zhang, L.; Liu, X.; Gao, H.; Hou, X.; Wang, L.; et al. Estimation of inbreeding and identification of regions under heavy selection based on runs of homozygosity in a Large White pig population. J. Anim. Sci. Biotechnol. 2020, 11, 46. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, S.; Zhang, K.; Peng, X.; Xie, S.; Li, X.; Zhao, S.; Ma, Y. Genome-Wide Patterns of Homozygosity and Relevant Characterizations on the Population Structure in Pietrain Pigs. Genes 2020, 11, 577. [Google Scholar] [CrossRef]
- Liu, G.E.; Hou, Y.; Zhu, B.; Cardone, M.F.; Jiang, L.; Cellamare, A.; Mitra, A.; Alexander, L.J.; Coutinho, L.L.; Dell’Aquila, M.E.; et al. Analysis of copy number variations among diverse cattle breeds. Genome Res. 2010, 20, 693–703. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kollias, H.D.; McDermott, J.C. Transforming growth factor-beta and myostatin signaling in skeletal muscle. J. Appl. Physiol. 2008, 104, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Tatti, O.; Vehviläinen, P.; Lehti, K.; Keski-Oja, J. MT1-MMP releases latent TGF-beta1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp. Cell Res. 2008, 314, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Serao, N.V.; González-Peña, D.; Beever, J.E.; Bollero, G.A.; Southey, B.R.; Faulkner, D.B. Bivariate genome-wide association analysis of the growth and intake components of feed efficiency. PLoS ONE 2013, 8, e78530. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guan, D.; Luo, N.; Tan, X.; Zhao, Z.; Huang, Y.; Na, R.; Zhang, J.; Zhao, Y. Scanning of selection signature provides a glimpse into important economic traits in goats (Capra hircus). Sci. Rep. 2016, 6, 36372. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Salilew-Wondim, D.; Holker, M.; Rings, F.; Phatsara, C.; Mohammadi-Sangcheshmeh, A.; Tholen, E.; Schellander, K.; Tesfaye, D. Depletion of BIRC6 leads to retarded bovine early embryonic development and blastocyst formation in vitro. Reprod. Fertil. Dev. 2010, 22, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Lin, Z.; Song, X.; Hu, C.; Qiu, M.; Yang, L.; Zhang, Z.; Pen, H.; Chen, J.; Xiong, X.; et al. Whole transcriptome analysis reveals the key genes and noncoding RNAs related to follicular atresia in broilers. Anim. Biotechnol. 2023, 34, 3144–3153. [Google Scholar] [CrossRef] [PubMed]
- Hogeveen, K.N.; Sassone-Corsi, P. Regulation of gene expression in post-meiotic male germ cells: CREM-signalling pathways and male fertility. Hum. Fertil. 2006, 9, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Saade, M.; Irla, M.; Govin, J.; Victorero, G.; Samson, M.; Nguyen, C. Dynamic distribution of Spatial during mouse spermatogenesis and its interaction with the kinesin KIF17b. Exp. Cell Res. 2007, 313, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Nonneman, D.J.; Shackelford, S.D.; King, D.A.; Wheeler, T.L.; Wiedmann, R.T.; Snelling, W.M.; Rohrer, G.A. Genome-wide association of meat quality traits and tenderness in swine. J. Anim. Sci. 2013, 91, 4043–4050. [Google Scholar] [CrossRef] [PubMed]
- Zha, C.; Liu, K.; Wu, J.; Li, P.; Hou, L.; Liu, H.; Huang, R.; Wu, W. Combining genome-wide association study based on low-coverage whole genome sequencing and transcriptome analysis to reveal the key candidate genes affecting meat color in pigs. Anim. Genet. 2023, 54, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, K.; Khoo, J.J.; Sadler, A.; Piganis, R.; Wang, D.; Borg, N.A.; Hjerrild, K.; Gould, J.; Thomas, B.J.; Nagley, P.; et al. Mitochondrially localised MUL1 is a novel modulator of antiviral signaling. Immunol. Cell Biol. 2013, 91, 321–330. [Google Scholar] [CrossRef] [PubMed]
- John, A.E.; Thomas, M.S.; Berlin, A.A.; Lukacs, N.W. Temporal production of CCL28 corresponds to eosinophil accumulation and airway hyperreactivity in allergic airway inflammation. Am. J. Pathol. 2005, 166, 345–353. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Choi, I.; Bates, R.O.; Raney, N.E.; Steibel, J.P.; Ernst, C.W. Evaluation of QTL for carcass merit and meat quality traits in a US commercial Duroc population. Meat Sci. 2012, 92, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.L.; Liu, Y.W.; Dang, Y.L.; Jiang, X.X.; Xu, H.; Huang, X.; Wang, Y.L.; Wang, H.; Zhu, C.; Xue, L.Q.; et al. PLAC8, a new marker for human interstitial extravillous trophoblast cells, promotes their invasion and migration. Development 2018, 145, 148932. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hernandez, S.C.; Finlayson, H.A.; Ashworth, C.J.; Haley, C.S.; Archibald, A.L. A genome-wide linkage analysis for reproductive traits in F2 Large White × Meishan cross gilts. Anim. Genet. 2014, 45, 191–197. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, R.; Deng, D.; Liu, X.; Xiao, Y.; Huang, J.; Wang, F.; Li, X.; Yu, M. A miR-18a binding-site polymorphism in CDC42 3’UTR affects CDC42 mRNA expression in placentas and is associated with litter size in pigs. Mamm. Genome 2019, 30, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Firat-Karalar, E.N.; Sante, J.; Elliott, S.; Stearns, T. Proteomic analysis of mammalian sperm cells identifies new components of the centrosome. J. Cell Sci. 2014, 127, 4128–4133. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, Z.; Chen, Z.; Ye, S.; He, Y.; Huang, S.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J. Genome-Wide Association Study for Reproductive Traits in a Duroc Pig Population. Animals 2019, 9, 732. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dworkin, S.; Auden, A.; Partridge, D.D.; Daglas, M.; Medcalf, R.L.; Mantamadiotis, T.; Georgy, S.R.; Darido, C.; Jane, S.M.; Ting, S.B. Grainyhead-like 3 (Grhl3) deficiency in brain leads to altered locomotor activity and decreased anxiety-like behaviors in aged mice. Dev. Neurobiol. 2017, 77, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, L.; Yin, L.; Tang, Z.; Wang, Z.; Liu, X.; Xiang, T.; Yu, M.; Liu, X.; Li, C. Searching for new signals for susceptibility to umbilical hernia through genome-wide association analysis in three pig breeds. Anim. Genet. 2023, 54, 798–802. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).