Abstract
Deficiency in the membrane cytoskeletal protein dystrophin is the underlying cause of the progressive muscle wasting disease named Duchenne muscular dystrophy. In order to detect novel disease marker candidates and confirm the complexity of the pathobiochemical signature of dystrophinopathy, mass spectrometric screening approaches represent ideal tools for comprehensive biomarker discovery studies. In this report, we describe the comparative proteomic analysis of young versus aged diaphragm muscles from wild type versus the dystrophic mdx-4cv mouse model of X-linked muscular dystrophy. The survey confirmed the drastic reduction of the dystrophin-glycoprotein complex in the mdx-4cv diaphragm muscle and concomitant age-dependent changes in key markers of muscular dystrophy, including proteins involved in cytoskeletal organization, metabolite transportation, the cellular stress response and excitation-contraction coupling. Importantly, proteomic markers of the regulation of membrane repair, tissue regeneration and reactive myofibrosis were detected by mass spectrometry and changes in key proteins were confirmed by immunoblotting. Potential disease marker candidates include various isoforms of annexin, the matricellular protein periostin and a large number of collagens. Alterations in these proteoforms can be useful to evaluate adaptive, compensatory and pathobiochemical changes in the intracellular cytoskeleton, myofiber membrane integrity and the extracellular matrix in dystrophin-deficient skeletal muscle tissues.
1. Introduction
Muscular dystrophies form a large group of progressive muscle wasting diseases in the domain of inherited neuromuscular disorders [1,2,3] and are triggered by a variety of primary abnormalities [4]. Mutations in the DMD gene cause dystrophinopathies, including severe Duchenne muscular dystrophy of early childhood and its more benign and later onset form named Becker’s muscular dystrophy [5,6]. Dystrophinopathies are characterized by the almost complete loss or abnormal size of the dystrophin protein isoform Dp427-M in skeletal muscles [7,8]. The DMD gene is extremely large and contains several promoters [9]. Dystrophin proteins of 45 kDa to 427 kDa are expressed in a tissue-specific pattern [10]. Full-length isoforms of dystrophin are mostly found in muscular and neuronal tissues [11,12,13] with major cytoskeletal functions [14,15].
In skeletal muscles, dystrophin acts as a molecular anchor of a sarcolemmal glycoprotein complex that stabilizes the cellular periphery by indirectly linking extracellular merosin (laminin-211) of the basal lamina to cortical γ-actin of the intracellular cytoskeleton [16,17,18]. The connection of the full-length dystrophin isoform to the microtubular network makes the Dp427-M protein a member of the family of giant cytolinkers in skeletal muscles [19]. The dystrophin complex also supports cellular signaling pathways and provides lateral force transmission at costamers [20]. The members of the core dystrophin-glycoprotein complex include the Dp427-M isoform of dystrophin, α/β-dystroglycans, α/β/γ/δ-sarcoglycans, sarcospan, α-dystrobrevin and α/β-syntrophins [21,22].
Importantly, the expression of the dystrophin-glycoprotein complex is greatly reduced in dystrophinopathy [23,24,25] resulting in the loss of trans-plasmalemmal linkage in dystrophic skeletal muscles [26]. This was shown to impair sarcolemmal integrity and render dystrophin-deficient fibers more susceptible to membrane rupturing, which in turn triggers abnormal ion homeostasis and impaired signaling, affecting especially calcium handling and excitation-contraction coupling in muscular dystrophy [27,28,29]. Dystrophinopathies are mainly characterized by progressive skeletal muscle weakness, cognitive impairment in a subset of Duchenne patients, articular deformities, scoliosis and the occurrence of cardio-respiratory failure, which represent the main cause of death [6,30,31]. In addition to skeletal muscle weakness, Duchenne muscular dystrophy is associated with a variety of less severe and body-wide complications affecting liver metabolism, the gastrointestinal tract, the immune system and kidney function [30,31,32]. This makes X-linked muscular dystrophy a multi-systems disease with a primary neuromuscular pathology, which is reflected by complex changes in the serum biomarker profile of Duchenne patients [33,34,35].
In order to improve the differential diagnosis, prognostic evaluation and therapeutic-monitoring of the complex pathophysiology of muscular dystrophy, a considerable number of systematic studies have been applied to screen both tissue and biofluid specimens [36,37,38]. Proteomic searches for novel biomarker candidates have included clinical samples from patients afflicted with Duchenne/Becker muscular dystrophy [39,40] and various animal models of dystrophinopathy, such as dystrophic mice, dogs and pigs [41,42,43,44,45,46]. Animal models play a key role in the initial identification of new biomarkers [47,48,49,50] that can then be utilized for the evaluation of novel therapeutic approaches to treat dystrophinopathies [51,52,53,54]. Proteome-wide changes in the skeletal musculature were established to occur in the contractile apparatus, signaling pathways, the cytoskeletal network, metabolite transportation, bioenergetic pathways, the cellular stress response and the extracellular matrix [29,55,56,57]. Building on these findings, the new study presented in this report aimed at studying the effects of aging on dystrophinopathy and establish a comprehensive biomarker signature of the advanced stages of X-linked muscular dystrophy.
The mass spectrometry-based proteomic screening of young versus aged mdx-4cv diaphragm muscle described in this report has revealed complex proteome-wide changes in relation to dystrophin deficiency. Especially interesting was the identification of drastic and age-related increases in crucial markers of membrane repair regulation, tissue regeneration and reactive myofibrosis, i.e., annexins [58,59,60], dysferlin [61,62,63], caveolins [64,65,66], integrins [67,68,69], cadherin [70,71,72], CD34 [73,74,75], periostin [76,77,78] and collagens [79,80,81]. Changes in annexin 2, periostin and collagen VI were independently confirmed by comparative immunoblot analysis.
This is of considerable biomedical interest since annexins are intrinsically involved in the regulation of membrane repair [82,83,84], and the extracellular collagen network and its associated components play a crucial role in the stabilization, force transmission and functioning of skeletal muscles [85,86,87]. Considerable crosstalk occurs between the extracellular matrix and contractile fibers in various myopathies [88,89,90]. Thus, the elevated expression levels of these crucial markers of regeneration, membrane repair and reactive myofibrosis at advanced stages of X-linked muscular dystrophy could be useful for the establishment of robust and informative indicators of changes in the sarcolemma and extracellular matrix region during the progression of dystrophinopathy [91,92,93].
2. Materials and Methods
2.1. Materials
General materials and analytical reagents for the comparative proteomic survey of normal versus dystrophic diaphragm muscles were obtained from Bio-Rad Laboratories (Hemel-Hempstead, Hertfordshire, UK), GE Healthcare (Little Chalfont, Buckinghamshire, UK) and Sigma Chemical Company (Dorset, UK). Protease inhibitor cocktail tablets were from Roche (Mannheim, Germany). For digestion of protein samples, MS-grade trypsin protease was purchased from ThermoFisher Scientific (Dublin, Ireland). Filter-aided sample preparations were carried out with Vivacon 500 spin filters (VN0H22; 30,000 MWCO) from Sartorius (Göttingen, Germany). Precast Invitrogen Bolt 4–12% Bis-Tris gels and Whatman nitrocellulose transfer membranes were used for one-dimensional gel electrophoresis and immunoblotting, respectively, and obtained from Bio-Science Ltd. (Dun Laoghaire, Ireland). For the visualization of gel-separated protein bands, InstantBlue Coomassie Protein Stain was purchased from Expedeon (Heidelberg, Germany). Primary antibodies for immunoblotting were from Invitrogen, Waltham, MA, USA (mAb SD83-03 against collagen VI), Abcam, Cambridge, UK (pAb ab41803 to annexin ANXA2) and Novus Biologicals, Cambridge, UK (mAb NBP1-30042 against periostin). Secondary peroxidase-conjugated anti-IgG were purchased from Sigma Chemical Company (Dorset, UK). Chemiluminescence kits were from Roche (Mannheim, Germany). The Pierce 660 nm Protein Assay Reagent was purchased from ThermoFisher Scientific (Dublin, Ireland).
2.2. Wild Type and Dystrophic Diaphragm Muscle Specimens
The dissection of freshly prepared postmortem diaphragm specimens from 3-month and 15-month old wild type C57BL6 mice and aged-matched samples from the mdx-4cv mouse model of Duchenne muscular dystrophy was performed according to institutional regulations. Normal controls and dystrophic mice were handled in strict adherence to local governmental and institutional animal care regulations and were approved by the Institutional Animal Care and Use Committee (Amt für Umwelt, Verbraucherschutz und Lokale Agenda der Stadt Bonn, North Rhine-Westphalia, Germany). Frozen specimens were transported on dry ice to Maynooth University in accordance with the regulations of the Department of Agriculture (animal by-product register number 2016/16 to the Department of Biology, National University of Ireland, Maynooth). Comparative proteomic analyses were carried out with muscle samples from 6 wild type and 6 dystrophic mice. Verification analyses using immunoblotting were carried out with specimens derived from a minimum of 4 wild type and 4 dystrophic mice.
2.3. Preparation of Muscle Tissue Extracts
The preparation of tissue specimens and subsequent analysis of extracted proteins by bottom-up proteomics was performed by a standardized procedure, which has recently been described in a detailed methods paper [94]. Wild type C57BL6 and dystrophic mdx-4cv mice of differing age were sacrificed in the Bioresource Unit of the University of Bonn and diaphragm muscle specimens then quick-frozen in liquid nitrogen [45]. Frozen samples were transported on dry ice to Maynooth University and stored at −80 °C prior to comparative proteomic analysis. Both, young and aged diaphragm samples were homogenized in a comparative way using lysis buffer containing 4% (w/v) sodium dodecyl sulfate, 0.1 M dithiothreitol and 100 mM Tris-Cl, pH 7.6, as well as a protease inhibitor cocktail [95]. Homogenization was carried out with a handheld homogenizer (Kimble Chase, Rockwood, TN, USA) [96]. Following homogenization, samples were briefly treated in a sonicating water bath, then heated for 3 min at 95 °C and centrifuged at 16,000× g for 5 min. The protein-containing supernatant was extracted and used for subsequent proteomic studies [95]. The Pierce 660 nm Protein Assay system was used to determine protein concentration [97]. Extracted diaphragm proteins were further processed for mass spectrometric analysis. Samples were mixed with 200 μL of 8 M urea, 0.1 M Tris pH 8.9 in Vivacon 500 spin filter units and centrifuged at 14,000× g for 15 min. The filter-aided sample preparation (FASP) method, developed by Wiśniewski and co-workers [98], was used for sample processing, buffer switching and protein trypsination prior to mass spectrometric peptide analysis [94].
2.4. Mass Spectrometry and Proteomic Data Analysis
The comparative label-free liquid chromatography mass spectrometric analysis of young versus aged diaphragm muscles from wild type versus the dystrophic mdx-4cv mouse model of dystrophinopathy was performed with a Thermo Orbitrap Fusion Tribrid mass spectrometer from Thermo Fisher Scientific (Waltham, MA, USA). Details of the proteomic workflow describing all preparative steps and analytical procedures using data-dependent acquisition, as well as bioinformatic data handling, were recently outlined in detail [94]. Reverse-phased capillary high-pressure liquid chromatography was carried out with a Thermo UltiMate 3000 nano system and directly coupled in-line with the Thermo Orbitrap Fusion Tribrid mass spectrometer. The UniProtKB-SwissProt Mus musculus database with Proteome Discoverer 2.2 using Sequest HT (Thermo Fisher Scientific) and Percolator were employed for the qualitative data analysis of mass spectrometric files. For the identification of diaphragm proteins, the following crucial search parameters were employed: (i) a value of 0.02 Da for MS/MS mass tolerance, (ii) a value of 10 ppm for peptide mass tolerance, (iii) variable modification settings for methionine oxidation, (iv) fixed modification settings in relation to carbamido-methylation and (v) tolerance for the occurrence of up to two missed cleavages. Peptide probability was set to high confidence. A minimum XCorr score of 1.5 for 1, 2.0 for 2, 2.25 for 3 and 2.5 for 4 charge state was employed for the filtering of peptides. The software analysis programme Progenesis QI for Proteomics (version 2.0; Nonlinear Dynamics, a Waters company, Newcastle upon Tyne, UK) was used to carry out quantitative label-free data analysis. Proteome Discoverer 2.2 using Sequest HT (Thermo Fisher Scientific) and a percolator were employed for the identification of peptides and proteins. Datasets were imported into Progenesis QI software for further analysis. Following the review of protein identifications, only those data that agreed with a crucial set of criteria were deemed as differentially expressed protein species between experimental groups based on statistical significance and high confidence. The criteria included an ANOVA p-value of ≤0.01 between experimental groups, and proteins with ≥2 unique peptides contributing to the identification. The Progenesis QI programme calculated the mean abundance for individual protein species in each experimental condition to determine the maximum fold change for particular proteins. Condition-vs-condition matrixes with mean values were then used to determine the maximum fold change between any two condition’s mean protein abundances [94]. The multi-consensus MS files and listings of altered proteins in wild type versus dystrophic diaphragm muscle that were generated by the comparative proteomic study shown in this report have been deposited (15 September 2022) under the title ‘Proteomic analysis of aged mdx-4cv diaphragm’ with the unique identifier ‘qmtre’ to the Open Science Foundation (https://osf.io/qmtre/, accessed on 15 September 2022). The standard bioinformatic analysis tool PANTHER [99] was utilized for the identification of protein classes.
2.5. Comparative Immunoblot Analysis
Primary antibodies to the matricellular protein periostin (POSTN), the extracellular matrix component collagen (COL-VI) and the Ca2+-dependent membrane repair protein annexin (ANXA2) were used for the independent verification of key findings from the mass spectrometry-based proteomic screening of young versus aged mdx-4cv diaphragm muscle preparations. One-dimensional gel electrophoresis and immunoblot analysis were carried out by standardized methodology [95]. Diaphragm samples were incubated in Laemmli-type sample buffer and heated for 30 min at 37 °C. Per lane, 20 μg of protein were ran on Invitrogen Bolt 4–12% Bis-Tris gels. Coomassie staining of protein gels was performed with InstantBlue Coomassie Protein Stain. Gel electrophoretically separated proteins were transferred to nitrocellulose membranes for immunoblot analysis. Blocking was performed with a fat-free milk solution and membrane sheets were incubated overnight with 1:1000 (v/v) diluted primary antibodies. Detection of labelled protein bands was carried out by incubation with 1:1000 (v/v) diluted peroxidase-conjugated secondary antibodies and enhanced chemiluminescence. Statistical analysis of immunoblots (n = 4) was performed with ImageJ software (NIH, Bethesda, MD, USA), along with Microsoft Excel, in which statistical significance was based on a p-value ≤ 0.05.
3. Results
The comparative proteomic profiling of the dystrophin-deficient mdx-4cv diaphragm was carried out with a Thermo Orbitrap Fusion Tribrid mass spectrometer and multi-consensus analysis identified 2421 and 2790 protein species in wild type versus mdx-4cv preparations, respectively. Throughout the manuscript, names of proteins, protein subunits or protein isoforms that were identified by proteomics are abbreviated in capital letters. Differential protein expression patterns were analyzed by bioinformatics and are displayed in this report according to association with the (i) dystrophin-glycoprotein complex, (ii) the established marker signature of dystrophinopathy, (iii) excitation-contraction coupling, (iv) the annexin family, (v) the collagen family, and (vi) the extracellular matrix. The increased abundance of collagen COL-VI as marker of reactive myofibrosis, the multi-functional periostin (POSTN) as a prototype of a matricellular component and the membrane repair protein annexin ANXA2 were verified by immunoblotting.
3.1. Mass Spectrometric Analysis of Young versus Aged mdx-4cv Diaphragm Muscle
Overall changes in protein classes were determined by bioinformatic PANTHER analysis [99]. The findings are displayed in Figure 1 and Figure 2, which show drastic alterations in a variety of protein types, including RNA metabolism proteins, cytoskeletal proteins, metabolite interconversion enzymes, protein modifiers and translational proteins. An interesting change between 3-month and 15-month old mdx-4cv diaphragm muscle is the drastic increase in the changed density of metabolite interconversion enzymes during aging (Figure 2).
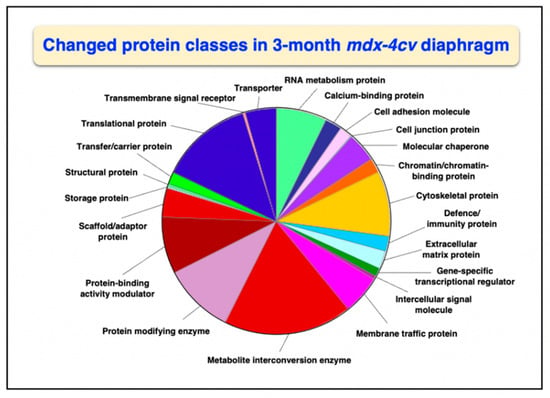
Figure 1.
Bioinformatic analysis of changed protein classes in 3−month old mdx-4cv diaphragm muscle. The analysis was carried out with the PANTHER program [99].
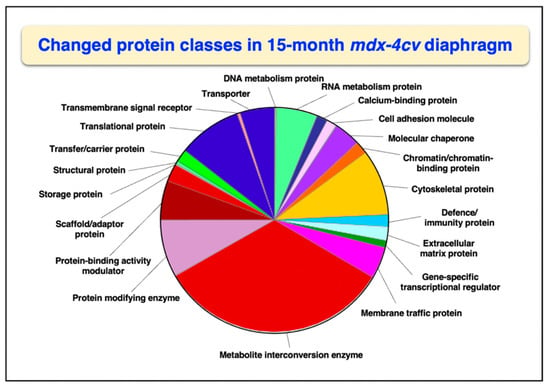
Figure 2.
Bioinformatic analysis of changed protein classes in 15−month old mdx-4cv diaphragm muscle. The analysis was carried out with the PANTHER program [99].
3.2. Reduced Dystrophin Complex in mdx-4cv Diaphragm Muscle
Following the proteomic analysis of young versus aged mdx-4cv diaphragm muscle, the mutant status of the analyzed dystrophic muscle specimens was confirmed by comparison between wild type and mdx-4cv samples. As diagrammatically shown in Figure 3, the dystrophin complex consists of the cytoskeletal Dp427-M isoform of dystrophin, integral β-dystroglycan, extracellular α-dystroglycan, α/β/γ/δ-sarcoglycans, sarcospan, α-dystrobrevin and α/β-syntrophins [20]. Linkage to this sarcolemma-associated complex occurs to extracellular collagen isoforms via laminin-211, and intracellularly dystrophin binds to γ-actin filaments and tubulins of the microtubular network.
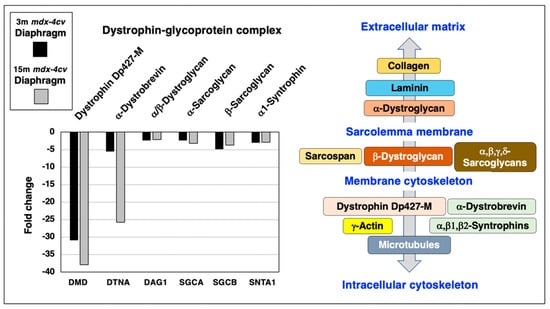
Figure 3.
Proteomic analysis of the expression of the dystrophin-glycoprotein complex in 3−month (3 m, black bars) versus 15−month (15 m; grey bars) old mdx-4cv diaphragm muscle. On the left is shown a bar diagram of the fold change in abundance of representative members of the dystrophin-glycoprotein complex, as determined by comparative proteomics. Identified proteins included Dystrophin (DMD), α/β-Dystroglycan (DAG1), α-Sarcoglycan (SGCA), β-Sarcoglycan (SGCB), α-Dystrobrevin (DTNA) and α1-Syntrophin (SNTA1). On the right side is shown a diagrammatic presentation of the dystrophin-glycoprotein complex [20].
Representative members of the core dystrophin-glycoprotein complex were identified by mass spectrometry, i.e., dystrophin (DMD), α/β-dystroglycan (DAG1), α-sarcoglycan (SGCA), β-sarcoglycan (SGCB), α-dystrobrevin (DTNA) and α1-syntrophin (SNTA1), and shown to be greatly reduced in both young and aged mdx-4cv diaphragm (Figure 3). The most drastic change between young and old muscle specimens were observed for α-dystrobrevin in 15-month old mdx-4cv diaphragm muscle.
3.3. Established Changes of Dystrophic Biomarkers in mdx-4cv Diaphragm Muscle
In order to put this new study into perspective, a variety of established proteomic markers of X-linked muscular dystrophy were analyzed. As shown in Figure 4, increases in the intermediate filament component vimentin (VIM), the microtubular protein isoform tubulin-alpha-1c (TUBA1C), the intracellular iron regulatory protein ferritin light chain (FTL1), heat shock protein A5 (HSPA5), small heat shock protein B7 (HSPB7), large heat shock protein 90AA1 (HSP90AA1) and nuclear lamin isoform B2 (LMNB2) were confirmed. Characteristic decreases were established for the cytosolic calcium-signaling protein parvalbumin (PVALB) and the metalloenzyme carbonic anhydrase isoform 3 (CA3).
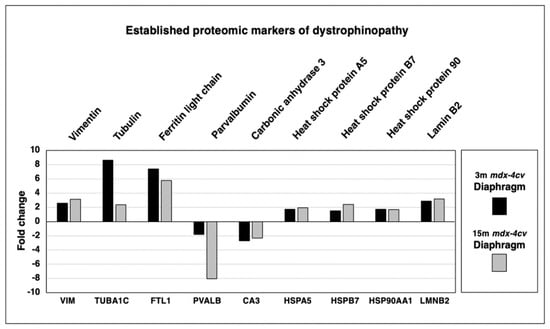
Figure 4.
Proteomic analysis of the expression of established proteomic markers of X-linked muscular dystrophy in 3−month (3 m, black bars) versus 15−month (15 m; grey bars) old mdx-4cv diaphragm muscle. Identified proteins included Vimentin (VIM), Tubulin-alpha-1c (TUBA1C), Ferritin light chain (FTL1), Heat shock protein A5 (HSPA5), Heat shock protein B7 (HSPB7), Heat shock protein 90AA1 (HSP90AA1), Lamin isoform B2 (LMNB2), Parvalbumin (PVALB) and Carbonic anhydrase isoform 3 (CA3).
3.4. Proteomics of Excitation-Contraction Coupling and Calcium Handling in mdx-4cv Diaphragm
Since micro-rupturing of the dystrophin-deficient sarcolemma membrane causes disturbed calcium homeostasis and abnormal cellular signaling in X-linked muscular dystrophy, it was of interest to evaluate the status of proteins involved in excitation-contraction coupling and intracellular calcium handling. This included the mass spectrometric survey of the ryanodine receptor calcium-release channel (RYR1) of the sarcoplasmic reticulum, the transverse tubular L-type calcium channel (Caᵥ1.1) with its voltage-sensing α1S-subunit (CACNA1S) and the auxiliary subunits β1 (CACNB1) and α2/δ (CACNA2D1), the calcium-binding protein calsequestrin (CASQ1) of the terminal cisternae region, the fast SERCA1 isoform of the sarco(endo)plasmic reticulum calcium-ATPase (ATP2A1), and the junctophilins 1 (JPH1) and 2 (JPH2) of the triad junction (Figure 5).
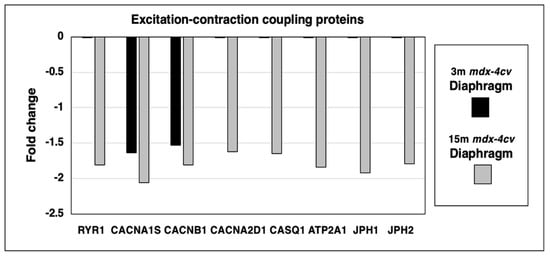
Figure 5.
Proteomic identification of changes in the expression levels of key proteins involved in excitation-contraction coupling, calcium homeostasis and triad integrity in 3−month (3 m, black bars) versus 15−month (15 m; grey bars) old mdx-4cv diaphragm muscle. This included the Ryanodine receptor calcium-release channel (RYR1), the voltage-sensing L-type calcium channel with its α1S-subunit (CACNA1S), β1-subunit (CACNB1) and α2/δ-subunits (CACNA2D1), fast Calsequestrin (CASQ1), the fast SERCA1 Calcium-ATPase (ATP2A1), Junctophilin 1 (JPH1) and Junctophilin 2 (JPH2).
The principal ion channel-containing α1S-subunit of the transverse tubular L-type calcium channel and its auxiliary β1-subunit were shown to be reduced in both young and aged dystrophic diaphragm muscle. In contrast, the ryanodine receptor, the α2/δ-subunit of the transverse tubular calcium channel, fast calsequestrin, the fast sarcoplasmic reticulum calcium ATPase SERCA1 and junctophilins exhibited a reduced expression level only in 15-month old mdx-4cv diaphragm.
3.5. Proteomics of Membrane Repair and Calcium Sensing in mdx-4cv Diaphragm
A protein of central importance in the calcium-dependent membrane repair process of damaged skeletal muscles is dysferlin (DYSF). The mass spectrometric survey of wild type versus mdx-4cv diaphragm preparations revealed an increase of this sarcolemmal repair protein. Since the dysferlin/myoferlin system closely interacts with caveolins and annexins, the expression levels of these crucial components of the caveolae structures and calcium regulation, respectively, were also examined. The concentration of caveolin 1 (CAV1) and caveolin 2 (CAV2) increased drastically in aged and dystrophic diaphragm, while the muscle-specific caveolin-3 (CAV3) isoform showed elevated levels in 3-month old mdx-4cv diaphragm but only marginal changes in 15-month old dystrophic muscles. Integrins are useful markers of proliferation and the main type present in the skeletal muscle periphery, α7β1-integrin (ITGA7/ITGB1), was identified to be increased in both young and aged mdx-4cv diaphragm. Interestingly, the expression of the myogenic markers cadherin-13 (CDH13) and CD34 were both elevated in muscular dystrophy.
The calcium-dependent annexin isoforms A1 to A7 (ANXA1, ANXA2, ANXA3, ANXA4, ANXA5, ANXA7) of which some are involved in calcium-related and dysferlin-associated repair mechanisms of the muscle surface membrane system, were analyzed in 3-month versus 15-month old mdx-4cv diaphragm muscle preparations. The degree of increased expression levels in young versus aged mdx-4cv diaphragm was found to be relatively comparable (Figure 6).
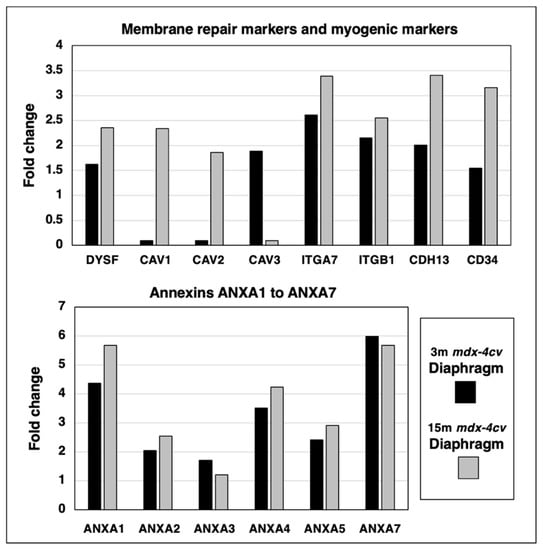
Figure 6.
Proteomic identification of changes in the expression levels of key proteins involved in membrane repair and calcium sensing in 3−month (3 m, black bars) versus 15−month (15 m; grey bars) old mdx-4cv diaphragm muscle. This included the sarcolemmal membrane repair protein Dysferlin (DYSF), Caveolin 1 (CAV1), Caveolin 2 (CAV2), muscle-specific Caveolin 3 (CAV3), Integrin alpha-7 (ITGA7), Integrin beta-1 (ITGB1), Cadherin-13/T-cadherin (CDH13) and the myogenic marker CD34, as well as Annexin isoforms A1 (ANXA1), A2 (ANXA2), A3 (ANXA3), A4 (ANXA4), A5 (ANXA5) and A7 (ANXA7).
3.6. Proteomics of Collagens and the Extracellular Matrix in mdx-4cv Diaphragm Muscle
Skeletal muscles contain a considerable number of collagen isoforms and its extracellular matrix is formed by a highly complex mesh of diverse proteins. The comparative proteomic analysis of 3-month versus 15-month old mdx-4cv diaphragm muscle revealed increases in collagen I (COL1A1 and COL1A2 chains), collagen IV (COL4A1, COL4A2 and COL4A3 chains), collagen V (COL5A1 and COL5A2 chains), collagen VI (COL6A1, COL6A2, COL6A5 and COL6A6 chains), collagen XII (COL12A1 chain), collagen XIV (COL14A chain), collagen XV (COL15A1 chain) and collagen XVIII (COL18A1 chain), as shown in Figure 7. Increased levels of crucial extracellular matrix proteins were identified in the case of the matricellular protein periostin (POSTN), the small leucine-rich proteoglycans asporin (ASPN), biglycan (BGN), lumican (LUM) and mimecan/osteoglycin (OGN), the adhesive glycoprotein fibronectin (FN1), the proteoglycan decorin (DCN), the tyrosine-rich acidic matrix protein dermatopontin (DPT), the hemopexin-type glycoprotein vitronectin (VTN) and the basal lamina component nidogen-2/osteonidogen (NID2). The most drastic differences in the degree of increased expression levels between young versus aged mdx-4cv diaphragm were found for the collagen chains COL5A1, COL5A2, COL6A5 and COL-XIV, as well as the matricellular protein periostin (Figure 7).
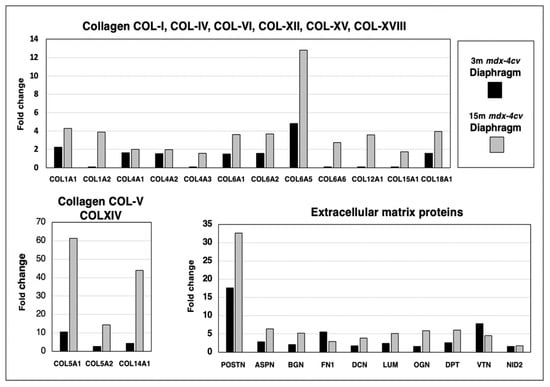
Figure 7.
Proteomic identification of changes in the expression levels of collagens and related extracellular matrix proteins in 3−month (3 m, black bars) versus 15−month (15 m; grey bars) old mdx-4cv diaphragm muscle. This included Collagen I (COL1A1, COL1A2), Collagen IV (COL4A1, COL4A2, COL4A3), Collagen V (COL5A1, COL5A2), Collagen VI (COL6A1, COL6A2, COL6A5, COL6A6), Collagen XII (COL12A1), Collagen XIV (COL14A), Collagen XV (COL15A1), Collagen XVIII (COL18A1), Periostin (POSTN), Asporin (ASPN), Biglycan (BGN), Lumican (LUM), Mimecan/osteoglycin (OGN), Fibronectin (FN1), Decorin (DCN), Dermatopontin (DPT), Vitronectin (VTN) and Nidogen-2/osteonidogen (NID2).
3.7. Immunoblotting of Collagen VI, Periostin and Annexin 2 in mdx-4cv Diaphragm
Trends in changed protein expression pattern as determined by the above-described comparative mass spectrometric investigation of protein extracts from 3-month versus 15-month old mdx-4cv diaphragm muscle were independently verified by immunoblotting, as shown in Figure 8.
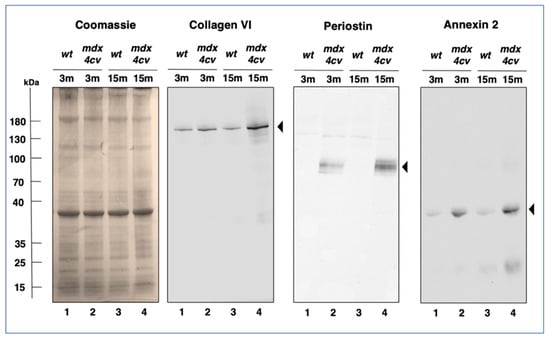
Figure 8.
Immunoblot analysis of Collagen VI, Periostin and Annexin 2 in mdx-4cv diaphragm. Shown is a Coomassie-stained gel plus identical immunoblots labelled with antibodes to Collagen isoform COL-VI, the matricellular protein Periostin and Annexin isoform ANXA2. Lanes 1 to 4 contain protein extracts from 3−month (3 m) versus 15−month (15 m) old wild type versus mdx-4cv diaphragm, respectively. Molecular weight standards are marked on the left.
Coomassie Blue staining of one-dimensionally separated diaphragm proteins revealed no major differences in the band pattern of 3-month versus 15-month old wild type versus mdx-4cv diaphragm preparations. However, immunoblotting with antibodies to collagen COL-VI, periostin and annexin isoform ANXA2 clearly showed a significant increase of these proteins in dystrophic diaphragm muscle. Both, collagen and periostin exhibited high expression levels in aged and dystrophin-deficient muscles (Figure 8).
4. Discussion
The comparative proteomic analysis described in this report was carried out with young versus aged diaphragm muscles from wild type versus the dystrophic mdx-4cv mouse model of Duchenne muscular dystrophy. The mdx-type models of Duchenne muscular dystrophy are based on the spontaneous mdx-23 mouse [100] which is characterized by a point mutation in exon 23 of the DMD gene [101]. This results in the almost complete deficiency of dystrophin isoform Dp427-M [47] and causes an X-linked myopathy in association with muscular hypotrophy, hypertrophy and hyperplasia in mdx-23 fibres [102]. Especially the diaphragm muscle is severely affected in mdx-type mice and exhibits progressive fiber degeneration and reactive myofibrosis [103,104,105] making the dystrophin-deficient diaphragm a suitable tissue for studying the molecular and cellular pathogenesis of Duchenne muscular dystrophy [47,49]. An alternative model of dystrophinopathy is the mdx-4cv mouse [106] that has been generated by chemical mutagenesis with N-ethylnitrosourea which induced a C-to-T transition at position 7916 in exon 53 [107]. This nonsense point mutation results in the premature termination of translation and the production of a truncated and non-functional dystrophin protein [108]. Importantly, skeletal muscles from the mdx-4cv mouse display approximately 10-fold fewer revertant and dystrophin-positive contractile fibers as compared to the mdx-23 mouse [109]. The drastically reduced presence of revertant fibers makes the mdx-4cv mouse an attractive model and has therefore been widely used for (i) the detailed analysis of dystrophic changes in the skeletal musculature [95,96,110,111,112], (ii) studying dystrophinopathy-related cardiomyopathy [113], (iii) evaluating the protective phenotype of extraocular muscle [114], (iv) surveying of bodily fluids for biomarker candidates including serum, urine and saliva [115,116,117], (v) the screening of body-wide changes in the multi-systems pathology of X-linked muscular dystrophy including proteome-wide alterations in liver, spleen, kidney, stomach and brain [118,119,120,121,122] and (vi) the testing of new therapeutic procedures to treat muscular dystrophy [123,124,125].
Building on these studies, we have used the aged mdx-4cv diaphragm muscle as a model system to study the advanced stages of X-linked muscular dystrophy. Crucial findings from the mass spectrometric survey include the detection of increased levels of key marker proteins that are involved in the regulation of membrane repair, tissue regeneration and reactive myofibrosis. Annexin isoform ANXA2 [58,59,60], the matricellular protein periostin [76,77,78] and collagen isoform COL-VI [79,80,81], as well as the caveolins CAV1 and CAV2 [64,65,66], α7β1-integrin [67,68,69], cadherin-13/T-cadherin [70,71,72] and CD34 [73,74,75] were shown to be drastically increased in aged and dystrophin-lacking muscle preparations. Interestingly, the muscle-specific caveolin isoform CAV3 was only elevated in young and dystrophic diaphragm. Immunoblotting clearly verified the increased concentration of specific isoforms of annexin, periostin and collagen in muscular dystrophy. This suggests that these markers of membrane repair, tissue regeneration and myofibrosis are suitable to characterize muscle biopsy specimens from genetic animal models of dystrophinopathy. Since the spontaneous mdx-23 mouse is the most frequently used animal model in muscular dystrophy research [47], it was of interest to compare the proteomic changes in the chemically induced mdx-4cv mouse [106,107,108] to the naturally occurring mdx-23 mutant [100,101,102]. The proteome-wide changes identified in this report on established markers of X-linked muscular dystrophy, i.e., decreases in all members of the core dystrophin complex and concomitant increases in vimentin, tubulin, ferritin, various molecular chaperones and lamin, and decreases in parvalbumin and carbonic anhydrase isoform CA3, agree with previous studies of the dystrophic mdx-23 mouse model [41,42,43,44,45,46,126,127]. An increased abundance of collagen COL-VI was also observed in mdx-23 muscle preparations using two-dimensional gel electrophoresis combined with staining by the fluorescent dye ruthenium II tris bathophenanthroline disulfonate [128].
New biomarker candidates can now be used to improve diagnostic procedures, the accuracy of prognosis, therapeutic-monitoring and the evaluation of potential side effects due to novel pharmacological treatments or new gene therapeutic approaches, such as gene substitution, exon-skipping or gene editing [51,52,53,54]. Proteomic biomarkers can be utilized in a variety of crucial biomedical and clinical applications and be useful to improve the evaluation of susceptibility to muscular dystrophy, differential diagnosis, prognostics, prediction of patient sensitivity, pharmacodynamics, monitoring of therapeutic success and drug safety [35]. Figure 9 summarizes some of the key findings of this proteomic survey of aged and dystrophin-deficient mdx-4cv diaphragm muscle.
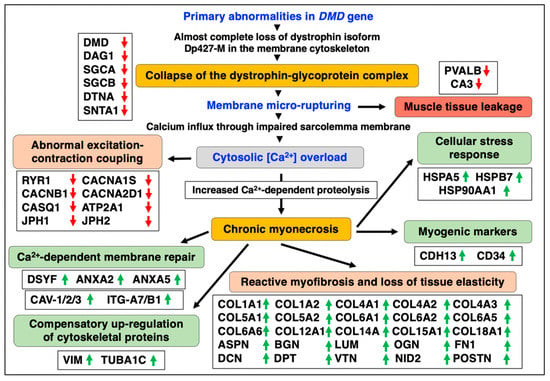
Figure 9.
Overview of pathobiochemical and adaptive changes in the aged and dystrophin-deficient mdx-4cv diaphragm muscle as revealed by mass spectrometry-based proteomics. Listed are protein markers of crucial aspects of the molecular and cellular pathogenesis of dystrophinopathy, including the collapse of the dystrophin-glycoprotein complex, membrane rupturing, abnormal excitation-contraction coupling, reactive myofibrosis, calcium-dependent membrane repair, myogenic activation, the apparent compensatory up-regulation of cytoskeletal proteins, the cellular stress response and muscle tissue regeneration. Detected decreases in muscle-associated proteins are symbolized by red downward arrows and increases in proteins are marked by green upward arrows.
In relation to bioenergetic and metabolic enzymes, the bioinformatic PANTHER analysis of changed proteins revealed an extensive increase in the altered abundance of metabolite interconversion enzymes during aging of the dystrophic mdx-4cv diaphragm muscle. These considerable changes in the expression levels of muscle-associated enzymes included large numbers of hydrolases, isomerases, ligases, lyases, oxidoreductases and transferases. Within the protein class of hydrolases, this included proteins with amylase, deaminase, esterase, glucosidase, lipase, phosphatase, phosphodiesterase and pyrophosphatase activity. Isomerases were represented by epimerases, racemases and mutases. Lyases included aldolases, cyclases, dehydratases and hydratases. Alterations in the abundance of oxidoreductases encompassed dehydrogenases, oxidases, oxygenases, peroxidases and reductases. The protein family of transferases included acetyltransferases, acyltransferases, glycosyltransferases, kinases, methyltransferases and transketolases. A comprehensive biomarker signature of these types of tissue-associated changes in combination with relevant alterations in biofluids, such as saliva, urine and serum/plasma, can be extremely helpful to advance the field of muscular dystrophy research [33,34,35,36,37] and (i) determine the risk for disease prior to the appearance of initial symptoms, (ii) detect the differential nature of specific and potentially sequential pathophysiological processes including progressive fiber degeneration, impaired excitation-contraction coupling, abnormal ion homeostasis, dysregulated cellular signaling, chronic inflammation with immune cell invasion, fat substitution and reactive myofibrosis [28,29,30,31,32,91,92,93], (iii) evaluate disease progression and potential adverse events during treatment, (iv) determine individual sensitivities towards drug treatment, (v) reflect the metabolic/biotransformation responses of detoxifying organs such as the liver and kidneys following exposure to a novel therapeutic agent, (vi) monitor (ideally repeatedly) alterations in the disease status due to therapeutic interventions [51,52,53,54], and (vii) assure the absence of cytotoxic side effects on whole-body physiology [37].
This gives proteomic markers of calcium-dependent membrane repair, tissue regeneration and myofibrosis, as identified in this study, great potential for future usage in disease evaluation and therapeutic-monitoring. The drastic changes in the collagen isoforms COL-V, COL-VI and COL-IV [79] in aged mdx-4cv diaphragm identify these proteins as suitable markers of reactive myofibrosis [91]. Skeletal muscles contain a variety of collagens ranging from COL-I to COL-XXII [79,86]. The comparative proteomic survey of the dystrophic diaphragm identified collagens COL-I, COL-IV, COL-V, COL-VI, COL-XII, COL-XIV, COL-XIV and COL-XVIII, which are located to differing degrees in the basal lamina, endomysium, perimysium, epimysium and myotendinous junctions [85,86,87]. Collagen COL-IV is the basic structural component of the basal lamina and characterized by a helical form [86]. COL-IV functions as the main linker to the dystrophin-glycoprotein complex via laminin-211 and the α/β-dystroglycan subcomplex [16,20,21]. Collagen COL-V forms fibrils and is mostly found in the endomysium where it controls the process of collagen fibrillogenesis [86]. The beaded filaments of collagen COL-VI interact with various cell surface receptors and are intrinsically involved in the maintenance of skeletal muscle integrity [80,81]. COL-XIV links fibrillar collagens to other components of the extracellular matrix and is mostly located in the endomysium and perimysium [87]. The increased levels of these collagens play a crucial role in reactive myofibrosis and are the underlying cause for the loss of tissue elasticity in X-linked muscular dystrophy [91,92,93]. In relation to the progressive nature of X-linked muscular dystrophy, myofibrosis is an excellent indicator of overall muscle deterioration and correlates well with loss in motor functions in Duchenne patients [88,89].
Thus, protein markers of myofibrosis, such as collagen isoform COL-VI, in conjunction with elevated expression levels of other extracellular matrix components, represent excellent indicators of the progression of muscular dystrophy. Additional suitable proteins of the matrisome are the small leucine-rich proteoglycans asporin, biglycan, lumican and mimecan, as well as the proteoglycan decorin, fibronectin, dermatopontin, vitronectin and nidogen-2. The identified increased levels of the tyrosine-rich acidic matrix protein dermatopontin, the dystrophin-associated protein biglycan and the adhesive glycoprotein fibronectin in dystrophic skeletal muscles agree with previous proteomic studies using fluorescence two-dimensional difference in-gel electrophoresis [112,113]. Periostin is a crucial matricellular protein involved in tissue regeneration and cellular signaling events [76,78]. Previous studies indicate that periostin is only temporally expressed in the extracellular matrix during differentiation and regenerative processes [77], which agrees with the findings of the mass spectrometric study presented in this report. Expression levels of periostin are clearly affected by dystrophinopathy-associated changes in the extracellular matrix [91,93,129]. Notably, the deletion of periostin was shown to have a positive effect on X-linked muscular dystrophy by reducing myofibrosis via modulation of the signaling pathway that is associated with transforming growth factor TFG-β [130]. Immunoblotting indicates that periostin exists only at very low levels in normal wild type diaphragm. This makes this component of the extracellular matrix an excellent candidate for evaluating progressive alterations in dystrophin-deficient skeletal muscles.
The proteomic evaluation of key proteins involved in the regulation of calcium homeostasis has shown elevated levels of calcium-sensing annexins and a decrease in regulatory components of excitation-contraction coupling. The calcium hypothesis of Duchenne muscular dystrophy assumes that abnormal calcium handling plays a major role in progressive myonecrosis [27]. Full-length dystrophin forms a lattice underneath the sarcolemma in normal muscles, which stabilizes the fiber periphery by linking the actin cytoskeleton to the extracellular matrix via the dystrophin-associated dystroglycan complex [18,20]. The almost complete loss of the Dp427-M isoform in dystrophinopathy weakens this trans-plasmalemmal linkage and renders dystrophic fibers more susceptible to membrane leakage during excitation-contraction-relaxation cycles [28]. The resulting influx of calcium ions, both through membrane tears and calcium leak channels, causes a drastic elevation of the sarcosolic calcium concentration. This in turn activates calcium-dependent proteolysis, which results in the enhanced destruction of muscle proteins [27]. The reduced levels of the voltage-sensing L-type calcium channel of the transverse tubules, the ryanodine receptor calcium release channel and associated junctophilins of the triad junction, the luminal calcium-binding protein calsequestrin and the calcium-pumping ATPase of the sarcoplasmic reticulum agree with the pathophysiological concept of dysregulated calcium fluxes in dystrophin-deficient fibers [28].
The loss of key proteins involved in the temporal and spatial regulation of calcium movements through muscle membranes appears to be the underlying cause for impaired excitation-contraction coupling, decreased calcium buffering and abnormal calcium re-uptake into the lumen of the sarcoplasmic reticulum. These changes in ion-regulatory proteins might represent accumulating abnormalities and/or compensatory mechanisms in dystrophic diaphragm fibers [29]. Another interesting detection of altered expression levels is the mass spectrometric identification of increased levels of calcium-sensing annexins. This large family of proteins is involved in membrane repair [58] and annexin isoform ANXA2 was previously shown to be involved in myofiber repair in conjunction with inflammation and adipogenic replacement in injured contractile tissues [59]. Annexins are crucial facilitators of the accumulation of dysferlin, which is responsible for muscle fiber membrane repair [58,59,60,61,62]. Dysferlin, myoferlin and caveolins closely interact in dystrophic fibres [45,111]. This makes both the ANXA2 isoform of muscle annexin and elevated levels of dysferlin in damaged and dystrophin-deficient fibers [112,131] promising biomarker candidates of calcium-dependent and dysferlin/myoferlin-associated repair processes in X-linked muscular dystrophy.
Another interesting proteomic observation was the increase in integrins [67,68,69]. The α7β1-integrin complex is developmentally regulated and important for sarcolemmal stability and prevention of exercise-induced injury [132] and increased levels of α7β1-integrin were previously shown to counteract muscle degeneration [133,134,135]. Integrins play a key role in the reinforcement of crucial load-bearing structures at costamers and myotendious junctions. Together with the dystrophin-complex, integrins provide structural integrity for lateral and longitudinal force transmission across the sarcolemma [136]. Thus, the increased expression of α7β1-integrin might be part of the repair response in muscular dystrophy and represents a potential marker of cellular proliferation. Since this study was carried out with total extracts from crude muscle preparations, low-abundance markers were not detected. However, increases in the myogenic marker molecules CD34 [73,74,75] and cadherin-13 [71,72,137] were identified in the dystrophic mdx-4cv diaphragm by mass spectrometric analysis. The surface marker CD34 was recently shown to exhibit considerable potential as a satellite cell-linked biomarker of skeletal muscle aging [75]. Cadherin-13 (CDH13), also named heart cadherin (H-cadherin) or truncated cadherin (T-cadherin), attaches to the plasma membrane via a glycosylphosphatidylinositol anchor. Importantly, T-cadherin interacts with integrin-α7 and is linked to signal transduction proteins within caveolae structures [70]. This makes the identified alterations in the abundance of specific isoforms of integrin, cadherin and caveolin in muscular dystrophy an interesting finding with potential for the establishment of a comprehensive biomarker signature of dystrophinopathy.
5. Conclusions
The mass spectrometry-based proteomic survey of 3-month versus 15-month old diaphragm muscles from wild type versus the dystrophic mdx-4cv mouse model of Duchenne muscular dystrophy has identified meaningful biomarker candidates of membrane repair, tissue regeneration and reactive myofibrosis. This included calcium-sensing annexins and caveolins that are involved in dysferlin-related membrane repair and the matricellular protein periostin which plays a crucial role in the extracellular matrix, as well as various isoforms of collagen that indicate the progression of fibrotic changes in the basal lamina, endomysium, perimysium, epimysium and myotendinous junctions of dystrophic skeletal muscles. Although not all of these proteomic candidate markers may be suitable as robust surrogate biomarkers that precisely correlate with a realistic clinical endpoint in therapeutic trials, they can nevertheless be useful for determining the complex pathogenesis of dystrophic muscles and might also be useful to evaluate the varied effects of novel treatments [138]. Biomarker signatures that have been established by omics-type screening processes, and properly verified for their effectiveness and reliability by patient-related analyses, should be able to reflect the pathobiochemical complexity of monogenetic disorders, such as Duchenne muscular dystrophy [57]. In the future, biomarker-guided diagnostics and therapeutic-monitoring will probably play a more central role in pre-clinical studies with animal disease models and patient screening during the main phases of clinical trials.
Author Contributions
Conceptualization, D.S., P.D. and K.O.; methodology, S.G. and P.D.; soft-ware, M.H. and P.M.; validation, S.G.; formal analysis, S.G., P.D., M.Z. and M.H.; resources, M.Z.; data curation, P.D., S.G. and M.H.; writing—original draft preparation, S.G., P.D. and K.O.; writing—review and editing, K.O., D.S., S.G. and P.D.; supervision, K.O., P.D. and P.M.; funding acquisition, K.O. and P.M. All authors have read and agreed to the published version of the manuscript.
Funding
Research was supported by the Kathleen Lonsdale Institute for Human Health Research at Maynooth University. The Orbitrap Fusion Tribrid mass spectrometer was funded under a Science Foundation Ireland Infrastructure Award to Dublin City University (SFI 16/RI/3701).
Institutional Review Board Statement
Animals were handled in strict adherence to local govern- mental and institutional animal care regulations and were approved by the Institutional Animal Care and Use Committee (Amt für Umwelt, Verbraucherschutz und Lokale Agenda der Stadt Bonn, North Rhine-Westphalia, Germany). Frozen specimens were transported on dry ice to Maynooth University in accordance with the Department of Agriculture (animal by-product register number 2016/16 to the Department of Biology, National University of Ireland, Maynooth).
Data Availability Statement
The multi-consensus MS files and listings of altered proteins in wild type versus dystrophic diaphragm muscle that were generated by the comparative proteomic study shown in this report have been deposited under the title ‘Proteomic analysis of aged mdx-4cv diaphragm’ with the unique identifier ‘qmtre’ to the Open Science Foundation (https://osf.io/qmtre/) (accessed on 15 September 2022).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Benarroch, L.; Bonne, G.; Rivier, F.; Hamroun, D. The 2020 version of the gene table of neuromuscular disorders. Neuromuscul. Disord. 2019, 29, 980–1018. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Spendiff, S.; Roos, A.; Bourque, P.R.; Warman Chardon, J.; Kirschner, J.; Horvath, V.; Lochmüller, H. Advances in the diagnosis of inherited neuromuscular diseases and implications for therapy development. Lancet Neurol. 2020, 19, 522–532. [Google Scholar] [CrossRef]
- Dowling, J.J.; Weihl, C.C.; Spencer, M.J. Molecular and cellular basis of genetically inherited skeletal muscle disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, E.; Samitt, C.E.; Miranda, A.F.; Hays, A.P.; Salviati, G.; DiMauro, S.; Kunkel, L.M.; Hoffman, E.P.; Rowland, L.P. Duchenne muscular dystrophy: Deficiency of dystrophin at the muscle cell surface. Cell 1988, 54, 447–452. [Google Scholar] [CrossRef]
- Hoffman, E.P. Causes of clinical variability in Duchenne and Becker muscular dystrophies and implications for exon skipping therapies. Acta Myol. 2020, 39, 179–186. [Google Scholar] [PubMed]
- Roberts, R.G. Dystrophin, its gene, and the dystrophinopathies. Adv. Genet. 1995, 33, 177–231. [Google Scholar] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Hudecki, M.S.; Rosenberg, P.A.; Pollina, C.M.; Kunkel, L.M. Cell and fiber-type distribution of dystrophin. Neuron 1988, 1, 411–420. [Google Scholar] [CrossRef]
- Delalande, O.; Czogalla, A.; Hubert, J.F.; Sikorski, A.; Le Rumeur, E. Dystrophin and Spectrin, Two Highly Dissimilar Sisters of the Same Family. Subcell. Biochem. 2017, 82, 373–403. [Google Scholar] [PubMed]
- Hoffman, E.P. The discovery of dystrophin, the protein product of the Duchenne muscular dystrophy gene. FEBS J. 2020, 287, 3879–3887. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K.; Campbell, K.P. Dystrophin constitutes 5% of membrane cytoskeleton in skeletal muscle. FEBS Lett. 1991, 283, 230–234. [Google Scholar] [CrossRef]
- Finn, D.M.; Ohlendieck, K. Rabbit brain and muscle isoforms containing the carboxy-terminal domain of 427 kDa skeletal muscle dystrophin exhibit similar biochemical properties. Neurosci. Lett. 1997, 222, 25–28. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. Dystrophin and the membrane skeleton. Curr. Opin. Cell Biol. 1993, 5, 82–87. [Google Scholar] [CrossRef]
- Murphy, S.; Ohlendieck, K. The biochemical and mass spectrometric profiling of the dystrophin complexome from skeletal muscle. Comput. Struct. Biotechnol. J. 2015, 14, 20–27. [Google Scholar] [CrossRef]
- Prins, K.W.; Humston, J.L.; Mehta, A.; Tate, V.; Ralston, E.; Ervasti, J.M. Dystrophin is a microtubule-associated protein. J. Cell Biol. 2009, 186, 363–369. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Murphy, S.; Zweyer, M.; Sabir, H.; Swandulla, D.; Ohlendieck, K. The Dystrophin Node as Integrator of Cytoskeletal Organization, Lateral Force Transmission, Fiber Stability and Cellular Signaling in Skeletal Muscle. Proteomes 2021, 9, 9. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991, 66, 1121–1131. [Google Scholar] [CrossRef]
- Ohlendieck, K. Towards an understanding of the dystrophin-glycoprotein complex: Linkage between the extracellular matrix and the membrane cytoskeleton in muscle fibers. Eur. J. Cell Biol. 1996, 69, 1–10. [Google Scholar] [PubMed]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K.; Campbell, K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J. Cell Biol. 1991, 115, 1685–1694. [Google Scholar] [CrossRef]
- Ohlendieck, K.; Matsumura, K.; Ionasescu, V.V.; Towbin, J.A.; Bosch, E.P.; Weinstein, S.L.; Sernett, S.W.; Campbell, K.P. Duchenne muscular dystrophy: Deficiency of dystrophin-associated proteins in the sarcolemma. Neurology 1993, 43, 795–800. [Google Scholar] [CrossRef]
- Culligan, K.G.; Mackey, A.J.; Finn, D.M.; Maguire, P.B.; Ohlendieck, K. Role of dystrophin isoforms and associated proteins in muscular dystrophy (review). Int. J. Mol. Med. 1998, 2, 639–648. [Google Scholar] [CrossRef]
- Alderton, J.M.; Steinhardt, R.A. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J. Biol. Chem. 2000, 275, 9452–9460. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of impaired excitation-contraction coupling and abnormal calcium handling in muscular dystrophy. Proteomics 2022, e2200003, in press. [Google Scholar] [CrossRef]
- Ohlendieck, K.; Swandulla, D. Complexity of skeletal muscle degeneration: Multi-systems pathophysiology and organ crosstalk in dystrophinopathy. Pflugers Arch. 2021, 473, 1813–1839. [Google Scholar] [CrossRef]
- Schultz, T.I.; Raucci, F.J., Jr.; Salloum, F.N. Cardiovascular Disease in Duchenne Muscular Dystrophy: Overview and Insight Into Novel Therapeutic Targets. JACC Basic Transl. Sci. 2022, 7, 608–625. [Google Scholar] [CrossRef]
- Tripodi, L.; Villa, C.; Molinaro, D.; Torrente, Y.; Farini, A. The Immune System in Duchenne Muscular Dystrophy Pathogenesis. Biomedicines 2021, 9, 1447. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic serum biomarkers for neuromuscular diseases. Expert Rev. Proteom. 2018, 15, 277–291. [Google Scholar] [CrossRef]
- Spitali, P.; Hettne, K.; Tsonaka, R.; Charrout, M.; van den Bergen, J.; Koeks, Z.; Kan, H.E.; Hooijmans, M.T.; Roos, A.; Straub, V.; et al. Tracking disease progression non-invasively in Duchenne and Becker muscular dystrophies. J. Cachexia Sarcopenia Muscle 2018, 9, 715–726. [Google Scholar] [CrossRef]
- Al-Khalili Szigyarto, C. Duchenne muscular dystrophy: Recent advances in protein biomarkers and the clinical application. Expert Rev. Proteom. 2020, 17, 365–375. [Google Scholar] [CrossRef]
- Carr, S.J.; Zahedi, R.P.; Lochmüller, H.; Roos, A. Mass spectrometry-based protein analysis to unravel the tissue pathophysiology in Duchenne muscular dystrophy. Proteom. Clin. Appl. 2018, 12, 1700071. [Google Scholar] [CrossRef]
- Dowling, P.; Murphy, S.; Zweyer, M.; Raucamp, M.; Swandulla, D.; Ohlendieck, K. Emerging proteomic biomarkers of X- linked muscular dystrophy. Expert Rev. Mol. Diagn. 2019, 19, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Zweyer, M.; Sabir, H.; Dowling, P.; Gargan, S.; Murphy, S.; Swandulla, D.; Ohlendieck, K. Histopathology of Duchenne muscular dystrophy in correlation with changes in proteomic biomarkers. Histol. Histopathol. 2022, 37, 101–116. [Google Scholar] [PubMed]
- Capitanio, D.; Moriggi, M.; Torretta, E.; Barbacini, P.; De Palma, S.; Viganò, A.; Capitanio, D.; Moriggi, M.; Torretta, E.; Barbacini, P.; et al. Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: Changes contributing to preserve muscle function in Becker muscular dystrophy patients. J. Cachexia Sarcopenia Muscle 2020, 11, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, D.; Moriggi, M.; Barbacini, P.; Torretta, E.; Moroni, I.; Blasevich, F.; Morandi, L.; Mora, M.; Gelfi, C. Molecular fingerprint of BMD patients lacking a portion in the rod domain of dystrophin. Int. J. Mol. Sci. 2020, 23, 2624. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Martin, G.; Dowling, P.; Jockusch, H.; Ohlendieck, K. Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics 2006, 6, 4610–4621. [Google Scholar] [CrossRef]
- Guevel, L.; Lavoie, J.R.; Perez-Iratxeta, C.; Rouger, K.; Dubreil, L.; Feron, M.; Talon, S.; Brand, M.; Megeney, L.A. Quantitative proteomic analysis of dystrophic dog muscle. J. Proteome Res. 2011, 10, 2465–2478. [Google Scholar] [CrossRef]
- Rayavarapu, S.; Coley, W.; Cakir, E.; Jahnke, V.; Takeda, S.; Aoki, Y.; Grodish-Dressman, H.; Jaiswal, J.K.; Hoffman, E.P.; Brown, K.J.; et al. Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol. Cell Proteom. 2013, 12, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, T.; Kemter, E.; Flenkenthaler, F.; Klymiuk, N.; Otte, K.A.; Blutke, A.; Krause, S.; Walter, M.C.; Wanke, R.; Wolf, E.; et al. Progressive muscle proteome changes in a clinically relevant pig model of Duchenne muscular dystrophy. Sci. Rep. 2016, 6, 33362. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Comparative gel-based proteomic analysis of chemically crosslinked complexes in dystrophic skeletal muscle. Electrophoresis 2018, 39, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- van Westering, T.L.E.; Johansson, H.J.; Hanson, B.; Coenen-Stass, A.M.L.; Lomonosova, Y.; Tanihata, J.; Motohashi, N.; Yokota, T.; Takeda, S.; Lehtiö, J.; et al. Mutation-independent proteomic signatures of pathological progression in murine models of Duchenne muscular dystrophy. Mol. Cell Proteom. 2020, 19, 2047–2068. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.P.; Kornegay, J.N. Gene therapies in canine models for Duchenne muscular dystrophy. Hum. Genet. 2019, 138, 483–489. [Google Scholar] [CrossRef]
- Zaynitdinova, M.I.; Lavrov, A.V.; Smirnikhina, S.A. Animal models for researching approaches to therapy of Duchenne muscular dystrophy. Transgenic Res. 2021, 30, 709–725. [Google Scholar] [CrossRef]
- Stirm, M.; Fonteyne, L.M.; Shashikadze, B.; Stöckl, J.B.; Kurome, M.; Keßler, B.; Zakhartchenko, V.; Kemter, E.; Blum, H.; Arnold, G.J.; et al. Pig models for Duchenne muscular dystrophy—From disease mechanisms to validation of new diagnostic and therapeutic concepts. Neuromuscul. Disord. 2022, 32, 543–556. [Google Scholar] [CrossRef]
- Markati, T.; Oskoui, M.; Farrar, M.A.; Duong, T.; Goemans, N.; Servais, L. Emerging therapies for Duchenne muscular dystrophy. Lancet Neurol. 2022, 21, 814–829. [Google Scholar] [CrossRef]
- Deng, J.; Zhang, J.; Shi, K.; Liu, Z. Drug development progress in duchenne muscular dystrophy. Front. Pharmacol. 2022, 13, 950651. [Google Scholar] [CrossRef] [PubMed]
- Eser, G.; Topaloğlu, H. Current Outline of Exon Skipping Trials in Duchenne Muscular Dystrophy. Genes 2022, 13, 1241. [Google Scholar] [CrossRef] [PubMed]
- Chung Liang, L.; Sulaiman, N.; Yazid, M.D. A Decade of Progress in Gene Targeted Therapeutic Strategies in Duchenne Muscular Dystrophy: A Systematic Review. Front. Bioeng. Biotechnol. 2022, 10, 833833. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Carberry, S.; Ohlendieck, K. Proteomics of the dystrophin-glycoprotein complex and dystrophinopathy. Curr. Protein Pept. Sci. 2013, 14, 680–697. [Google Scholar] [CrossRef] [PubMed]
- Brinkmeier, H.; Ohlendieck, K. Chaperoning heat shock proteins: Proteomic analysis and relevance for normal and dystrophin-deficient muscle. Proteom. Clin. Appl. 2014, 8, 875–895. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Model Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef]
- Defour, A.; Medikayala, S.; Van der Meulen, J.H.; Hogarth, M.W.; Holdreith, N.; Malatras, A.; Duddy, W.; Boehler, J.; Nagaraju, K.; Jaiswal, J.K. Annexin A2 links poor myofiber repair with inflammation and adipogenic replacement of the injured muscle. Hum. Mol. Genet. 2017, 26, 1979–1991. [Google Scholar] [CrossRef]
- Bittel, D.C.; Chandra, G.; Tirunagri, L.M.S.; Deora, A.B.; Medikayala, S.; Scheffer, L.; Defour, A.; Jaiswal, J.K. Annexin A2 Mediates Dysferlin Accumulation and Muscle Cell Membrane Repair. Cells 2020, 9, 1919. [Google Scholar] [CrossRef]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H., Jr. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar] [CrossRef]
- Han, R.; Campbell, K.P. Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 2007, 19, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; Rossi, A.E.; Alvarez, M.G.; Swanson, K.E.; Deveaux, H.K.; Earley, J.U.; Hadhazy, M.; Vohra, R.; Walter, G.A.; Pytel, P.; et al. Dysferlin and myoferlin regulate transverse tubule formation and glycerol sensitivity. Am. J. Pathol. 2014, 184, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Couchoux, H.; Bichraoui, H.; Chouabe, C.; Altafaj, X.; Bonvallet, R.; Allard, B.; Ronjat, M.; Berthier, C. Caveolin-3 is a direct molecular partner of the Cav1.1 subunit of the skeletal muscle L-type calcium channel. Int. J. Biochem. Cell Biol. 2011, 43, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, B.S.; Prószyński, T.J. A Role for Caveolin-3 in the Pathogenesis of Muscular Dystrophies. Int. J. Mol. Sci. 2020, 21, 8736. [Google Scholar] [CrossRef] [PubMed]
- Matsunobe, M.; Motohashi, N.; Aoki, E.; Tominari, T.; Inada, M.; Aoki, Y. Caveolin-3 regulates the activity of Ca2+/calmodulin-dependent protein kinase II in C2C12 cells. Am. J. Physiol. Cell Physiol. 2022, 323, C1137–C1148. [Google Scholar] [CrossRef]
- Collo, G.; Starr, L.; Quaranta, V. A new isoform of the laminin receptor integrin alpha 7 beta 1 is developmentally regulated in skeletal muscle. J. Biol. Chem. 1993, 268, 19019–19024. [Google Scholar] [CrossRef]
- Burkin, D.J.; Wallace, G.Q.; Milner, D.J.; Chaney, E.J.; Mulligan, J.A.; Kaufman, S.J. Transgenic expression of α7β1 integrin maintains muscle integrity, increases regenerative capacity, promotes hypertrophy, and reduces cardiomyopathy in dystrophic mice. Am. J. Pathol. 2005, 166, 253–263. [Google Scholar] [CrossRef]
- Liu, J.; Burkin, D.J.; Kaufman, S.J. Increasing alpha 7 beta 1-integrin promotes muscle cell proliferation, adhesion, and resistance to apoptosis without changing gene expression. Am. J. Physiol. Cell Physiol. 2008, 294, C627–C640. [Google Scholar] [CrossRef]
- Philippova, M.P.; Bochkov, V.N.; Stambolsky, D.V.; Tkachuk, V.A.; Resink, T.J. T-cadherin and signal-transducing molecules co-localize in caveolin-rich membrane domains of vascular smooth muscle cells. FEBS Lett. 1998, 429, 207–210. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kita, S.; Nishizawa, H.; Fukuda, S.; Fujishima, Y.; Obata, Y.; Nagao, H.; Masuda, S.; Nakamura, Y.; Shimizu, Y.; et al. Adiponectin promotes muscle regeneration through binding to T-cadherin. Sci. Rep. 2019, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Nalbandian, M.; Zhao, M.; Sasaki-Honda, M.; Jonouchi, T.; Lucena-Cacace, A.; Mizusawa, T.; Yasuda, M.; Yoshida, Y.; Hotta, A.; Sakurai, H. Characterization of hiPSC-Derived Muscle Progenitors Reveals Distinctive Markers for Myogenic Cell Purification Toward Cell Therapy. Stem Cell Rep. 2021, 16, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, J.R.; Heslop, L.; Yu, D.S.; Tajbakhsh, S.; Kelly, R.G.; Wernig, A.; Buckingham, M.E.; Partridge, T.A.; Zammit, P.S. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J. Cell Biol. 2000, 151, 1221–1234. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, R.J.; Deasy, B.M.; Cao, B.; Gates, C.; Huard, J. The role of CD34 expression and cellular fusion in the regeneration capacity of myogenic progenitor cells. J. Cell Sci. 2002, 115, 4361–4374. [Google Scholar] [CrossRef]
- Fernández-Lázaro, D.; Garrosa, E.; Seco-Calvo, J.; Garrosa, M. Potential Satellite Cell-Linked Biomarkers in Aging Skeletal Muscle Tissue: Proteomics and Proteogenomics to Monitor Sarcopenia. Proteomes 2022, 10, 29. [Google Scholar] [CrossRef]
- Ito, N.; Miyagoe-Suzuki, Y.; Takeda, S.; Kudo, A. Periostin Is Required for the Maintenance of Muscle Fibers during Muscle Regeneration. Int. J. Mol. Sci. 2021, 22, 3627. [Google Scholar] [CrossRef]
- Ozdemir, C.; Akpulat, U.; Sharafi, P.; Yıldız, Y.; Onbaşılar, I.; Kocaefe, C. Periostin is temporally expressed as an extracellular matrix component in skeletal muscle regeneration and differentiation. Gene 2014, 553, 130–139. [Google Scholar] [CrossRef]
- Wang, Z.; An, J.; Zhu, D.; Chen, H.; Lin, A.; Kang, J.; Liu, W.; Kang, X. Periostin: An emerging activator of multiple signaling pathways. J. Cell Commun. Signal. 2022. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef]
- Cescon, M.; Gattazzo, F.; Chen, P.; Bonaldo, P. Collagen VI at a glance. J. Cell Sci. 2015, 128, 3525–3531. [Google Scholar] [CrossRef]
- Cescon, M.; Gregorio, I.; Eiber, N.; Borgia, D.; Fusto, A.; Sabatelli, P.; Scorzeto, M.; Megighian, A.; Pegoraro, E.; Hashemolhosseini, S.; et al. Collagen VI is required for the structural and functional integrity of the neuromuscular junction. Acta Neuropathol. 2018, 136, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.; Nylandsted, J. Plasma membrane integrity in health and disease: Significance and therapeutic potential. Cell Discov. 2021, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Croissant, C.; Carmeille, R.; Brévart, C.; Bouter, A. Annexins and Membrane Repair Dysfunctions in Muscular Dystrophies. Int. J. Mol. Sci. 2021, 22, 5276. [Google Scholar] [CrossRef] [PubMed]
- Croissant, C.; Gounou, C.; Bouvet, F.; Tan, S.; Bouter, A. Trafficking of Annexins during Membrane Repair in Human Skeletal Muscle Cells. Membranes 2022, 12, 153. [Google Scholar] [CrossRef]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Csapo, R.; Gumpenberger, M.; Wessner, B. Skeletal Muscle Extracellular Matrix—What Do We Know About Its Composition, Regulation, and Physiological Roles? A Narrative Review. Front. Physiol. 2020, 11, 253. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Zhang, H. Extracellular matrix: An important regulator of cell functions and skeletal muscle development. Cell Biosci. 2021, 11, 65. [Google Scholar] [CrossRef]
- Desguerre, I.; Mayer, M.; Leturcq, F.; Barbet, J.P.; Gherardi, R.K.; Christov, C. Endomysial fibrosis in Duchenne muscular dystrophy: A marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 2009, 68, 762–773. [Google Scholar] [CrossRef]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar]
- Ahmad, K.; Shaikh, S.; Ahmad, S.S.; Lee, E.J.; Choi, I. Cross-Talk Between Extracellular Matrix and Skeletal Muscle: Implications for Myopathies. Front. Pharmacol. 2020, 11, 142. [Google Scholar] [CrossRef]
- Holland, A.; Murphy, S.; Dowling, P.; Ohlendieck, K. Pathoproteomic profiling of the skeletal muscle matrisome in dystrophinopa- thy associated myofibrosis. Proteomics 2016, 16, 345–366. [Google Scholar] [CrossRef]
- Serrano, A.L.; Muñoz-Cánoves, P. Fibrosis development in early-onset muscular dystrophies: Mechanisms and translational implications. Semin. Cell Dev. Biol. 2017, 64, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K.; Swandulla, D. Molecular pathogenesis of Duchenne muscular dystrophy-related fibrosis. Pathologe 2017, 38, 21–29. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Protocol for the Bottom-Up Proteomic Analysis of Mouse Spleen. STAR Protoc. 2020, 1, 100196. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Concurrent Label-Free Mass Spectrometric Analysis of Dystrophin Isoform Dp427 and the Myofibrosis Marker Collagen in Crude Extracts from mdx-4cv Skeletal Muscles. Proteomes 2015, 3, 298–327. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Raucamp, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of the mouse diaphragm and refined mass spectrometric analysis of the dystrophic phenotype. J. Muscle Res. Cell Motil. 2019, 40, 9–28. [Google Scholar] [CrossRef]
- Antharavally, B.S.; Mallia, K.A.; Rangaraj, P.; Haney, P.; Bell, P.A. Quantitation of proteins using a dye-metal-based colorimetric protein assay. Anal. Biochem. 2009, 385, 342–345. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Duddy, W.; Duguez, S.; Johnston, H.; Cohen, T.V.; Phadke, A.; Gordish-Dressman, H.; Nagaraju, K.; Gnocchi, V.; Low, S.; Partridge, T. Muscular dystrophy in the mdx mouse is a severe myopathy compounded by hypotrophy, hypertrophy and hyperplasia. Skelet. Muscle 2015, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Stedman, H.H.; Sweeney, H.L.; Shrager, J.B.; Maguire, H.C.; Panettieri, R.A.; Petrof, B.; Narusawa, M.; Leferovich, J.M.; Sladky, J.T.; Kelly, A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991, 352, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Sahani, R.; Wallace, C.H.; Jones, B.K.; Blemker, S.S. Diaphragm muscle fibrosis involves changes in collagen organization with mechanical implications in Duchenne muscular dystrophy. J. Appl. Physiol. 2022, 132, 653–672. [Google Scholar] [CrossRef] [PubMed]
- Giovarelli, M.; Arnaboldi, F.; Zecchini, S.; Cornaghi, L.B.; Nava, A.; Sommariva, M.; Clementi, E.G.I.; Gagliano, N. Characterisation of Progressive Skeletal Muscle Fibrosis in the Mdx Mouse Model of Duchenne Muscular Dystrophy: An In Vivo and In Vitro Study. Int. J. Mol. Sci. 2022, 23, 8735. [Google Scholar] [CrossRef]
- Im, W.B.; Phelps, S.F.; Copen, E.H.; Adams, E.G.; Slightom, J.L.; Chamberlain, J.S. Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Hum. Mol. Genet. 1996, 5, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Chapman, V.M.; Miller, D.R.; Armstrong, D.; Caskey, C.T. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc. Natl. Acad. Sci. USA 1989, 86, 1292–1296. [Google Scholar] [CrossRef]
- Shin, J.H.; Hakim, C.H.; Zhang, K.; Duan, D. Genotyping mdx, mdx3cv, and mdx4cv mice by primer competition polymerase chain reaction. Muscle Nerve 2011, 43, 283–286. [Google Scholar] [CrossRef]
- Danko, I.; Chapman, V.; Wolff, J.A. The frequency of revertants in mdx mouse genetic models for Duchenne muscular dystrophy. Pediatr. Res. 1992, 32, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Henry, M.; Meleady, P.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Simultaneous Pathoproteomic Evaluation of the Dystrophin-Glycoprotein Complex and Secondary Changes in the mdx-4cv Mouse Model of Duchenne Muscular Dystrophy. Biology 2015, 4, 397–423. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Chemical crosslinking analysis of β-dystroglycan in dystrophin-deficient skeletal muscle. HRB Open Res. 2018, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of the sarcolemma-enriched fraction from dystrophic mdx-4cv skeletal muscle. J. Proteom. 2019, 191, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Dowling, P.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of dystrophin deficiency and associated changes in the aged mdx-4cv heart model of dystrophinopathy-related cardiomyopathy. J. Proteom. 2016, 145, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Gargan, S.; Dowling, P.; Zweyer, M.; Reimann, J.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Mass Spectrometric Profiling of Extraocular Muscle and Proteomic Adaptations in the mdx-4cv Model of Duchenne Muscular Dystrophy. Life 2021, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Dowling, P.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of mdx-4cv serum reveals highly elevated levels of the inflammation-induced plasma marker haptoglobin in muscular dystrophy. Int. J. Mol. Med. 2017, 39, 1357–1370. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic identification of elevated saliva kallikrein levels in the mdx-4cv mouse model of Duchenne muscular dystrophy. Biochem. Biophys. Rep. 2018, 18, 100541. [Google Scholar] [CrossRef]
- Gargan, S.; Dowling, P.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Identification of marker proteins of muscular dystrophy in the urine proteome from the mdx-4cv model of dystrophinopathy. Mol. Omics 2020, 16, 268–278. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-free mass spectrometric analysis reveals complex changes in the brain proteome from the mdx-4cv mouse model of Duchenne muscular dystrophy. Clin. Proteom. 2015, 12, 27. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of liver tissue from the mdx-4cv mouse model of Duchenne muscular dystrophy. Clin. Proteom. 2018, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Gargan, S.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteome-wide Changes in the mdx-4cv Spleen due to Pathophysiological Cross Talk with Dystrophin-Deficient Skeletal Muscle. iScience 2020, 23, 101500. [Google Scholar] [CrossRef]
- Dowling, P.; Zweyer, M.; Raucamp, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic and cell biological profiling of the renal phenotype of the mdx-4cv mouse model of Duchenne muscular dystrophy. Eur. J. Cell Biol. 2020, 99, 151059. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Gargan, S.; Zweyer, M.; Sabir, H.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of the interface between the stomach wall and the pancreas in dystrophinopathy. Eur. J. Transl. Myol. 2021, 31, 9627. [Google Scholar]
- Judge, L.M.; Haraguchiln, M.; Chamberlain, J.S. Dissecting the signaling and mechanical functions of the dystrophin-glycoprotein complex. J. Cell Sci. 2006, 119, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Mitrpant, C.; Fletcher, S.; Iversen, P.L.; Wilton, S.D. By-passing the nonsense mutation in the 4 CV mouse model of muscular dystrophy by induced exon skipping. J. Gene Med. 2009, 11, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Li, S.; Gregorevic, P.; Fall, B.M.; Chamberlain, J.S. Dystrophin delivery to muscles of mdx mice using lentiviral vectors leads to myogenic progenitor targeting and stable gene expression. Mol. Ther. 2010, 18, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Henry, M.; Meleady, P.; Winkler, C.K.; Krautwald, M.; Brinkmeier, H.; Ohlendieck, K. Comparative Label-Free Mass Spectrometric Analysis of Mildly versus Severely Affected mdx Mouse Skeletal Muscles Identifies Annexin, Lamin, and Vimentin as Universal Dystrophic Markers. Molecules 2015, 20, 11317–11344. [Google Scholar] [CrossRef]
- Murphy, S.; Brinkmeier, H.; Krautwald, M.; Henry, M.; Meleady, P.; Ohlendieck, K. Proteomic profiling of the dystrophin complex and membrane fraction from dystrophic mdx muscle reveals decreases in the cytolinker desmoglein and increases in the extracellular matrix stabilizers biglycan and fibronectin. J. Muscle Res. Cell Motil. 2017, 38, 251–268. [Google Scholar] [CrossRef]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Proteomics reveals drastic increase of extracellular matrix proteins collagen and dermatopontin in the aged mdx diaphragm model of Duchenne muscular dystrophy. Int. J. Mol. Med. 2012, 30, 229–234. [Google Scholar] [CrossRef]
- Holland, A.; Dowling, P.; Meleady, P.; Henry, M.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-free mass spectrometric analysis of the mdx-4cv diaphragm identifies the matricellular protein periostin as a potential factor involved in dystrophinopathy-related fibrosis. Proteomics 2015, 15, 2318–2331. [Google Scholar] [CrossRef]
- Lorts, A.; Schwanekamp, J.A.; Baudino, T.A.; McNally, E.M.; Molkentin, J.D. Deletion of periostin reduces muscular dystrophy and fibrosis in mice by modulating the transforming growth factor-β pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 10978–10983. [Google Scholar] [CrossRef]
- Vontzalidis, A.; Terzis, G.; Manta, P. Increased dysferlin expression in Duchenne muscular dystrophy. Anal. Quant. Cytopathol. Histpathol. 2014, 36, 15–22. [Google Scholar] [PubMed]
- Boppart, M.D.; Mahmassani, Z.S. Integrin signaling: Linking mechanical stimulation to skeletal muscle hypertrophy. Am. J. Physiol. Cell Physiol. 2019, 317, C629–C641. [Google Scholar] [CrossRef] [PubMed]
- Burkin, D.J.; Wallace, G.Q.; Nicol, K.J.; Kaufman, D.J.; Kaufman, S.J. Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J. Cell Biol. 2001, 152, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Rooney, J.E.; Gurpur, P.B.; Burkin, D.J. Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 7991–7996. [Google Scholar] [CrossRef] [PubMed]
- Heller, K.N.; Montgomery, C.L.; Janssen, P.M.; Clark, K.R.; Mendell, J.R.; Rodino-Klapac, L.R. AAV-mediated overexpression of human α7 integrin leads to histological and functional improvement in dystrophic mice. Mol. Ther. 2013, 21, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.L.; Oh, J.; Chou, E.; Lee, J.A.; Holmberg, J.; Burkin, D.J.; Crosbie-Watson, R.H. Sarcospan integration into laminin-binding adhesion complexes that ameliorate muscular dystrophy requires utrophin and α7 integrin. Hum. Mol. Genet. 2015, 24, 2011–2022. [Google Scholar] [CrossRef]
- Kim, H.; Perlingeiro, R.C.R. Generation of human myogenic progenitors from pluripotent stem cells for in vivo regeneration. Cell. Mol. Life Sci. 2022, 79, 406. [Google Scholar] [CrossRef]
- Szigyarto, C.A.; Spitali, P. Biomarkers of Duchenne muscular dystrophy: Current findings. Degener. Neurol. Neuromuscul. Dis. 2018, 8, 1–13. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).