A Theoretical Study on the Electronic Structure and Floatability of Rare Earth Elements (La, Ce, Nd and Y) Bearing Fluorapatite




Abstract
:1. Introduction
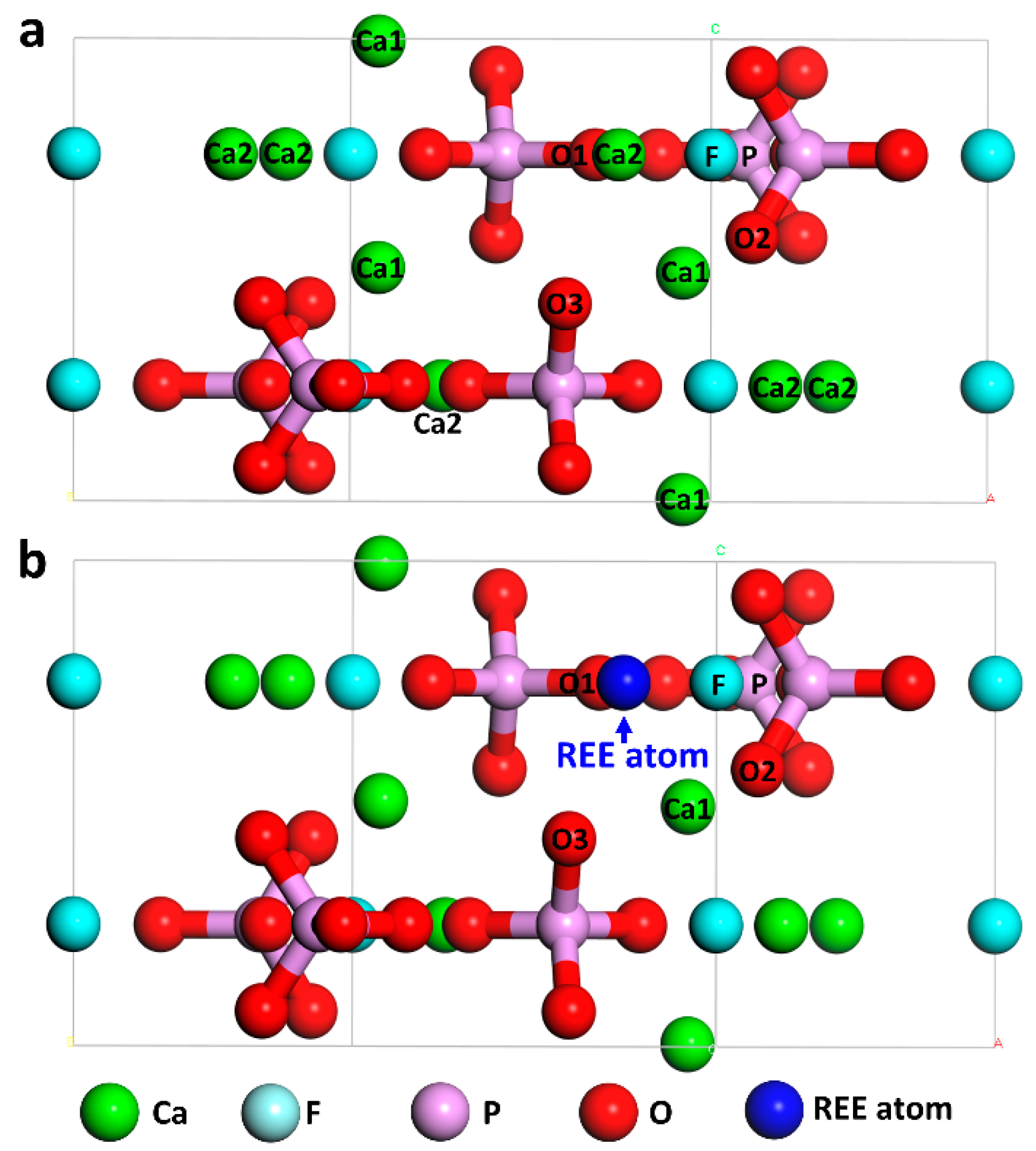
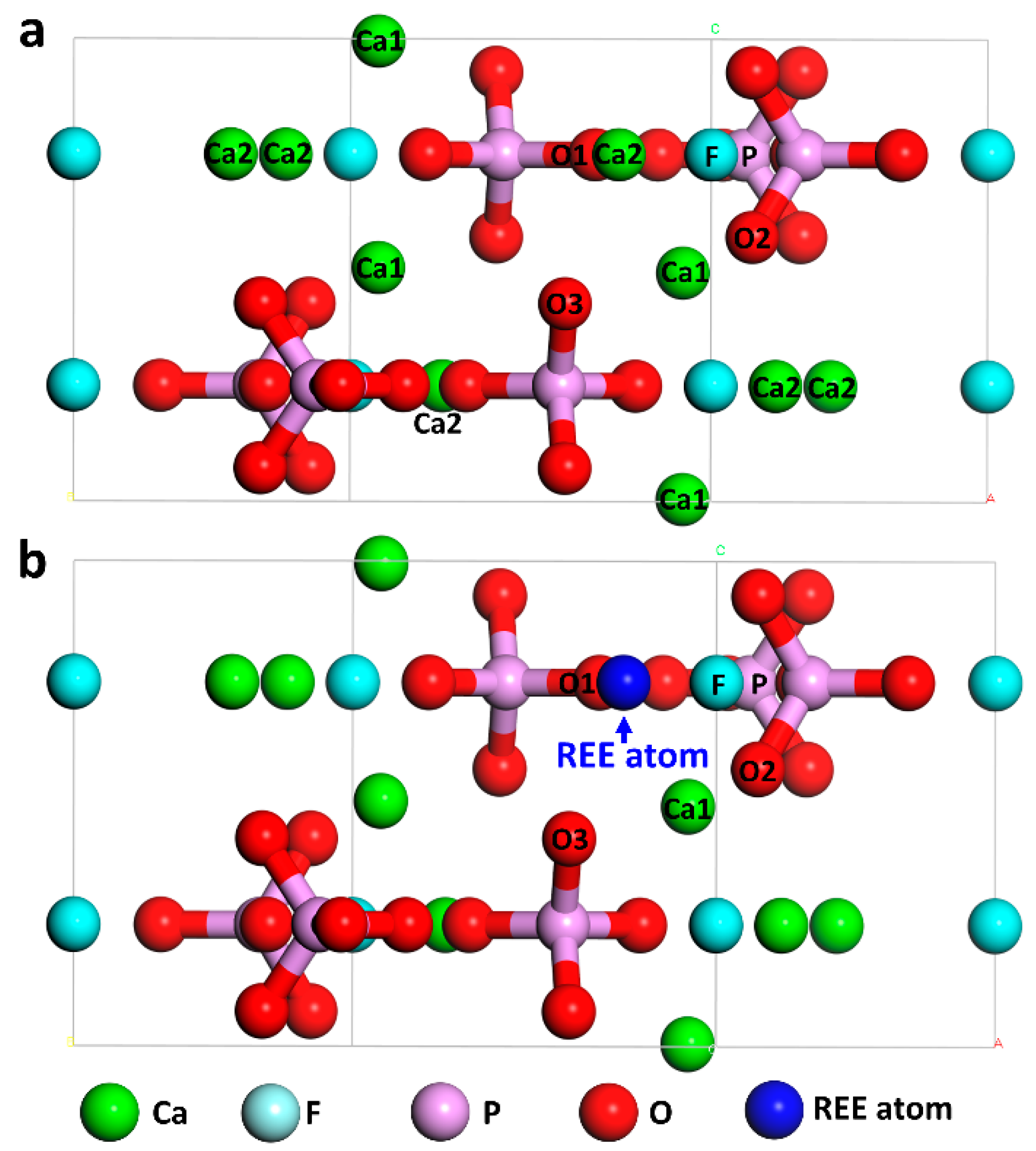
2. Computational Models and Methods
3. Results and Discussion
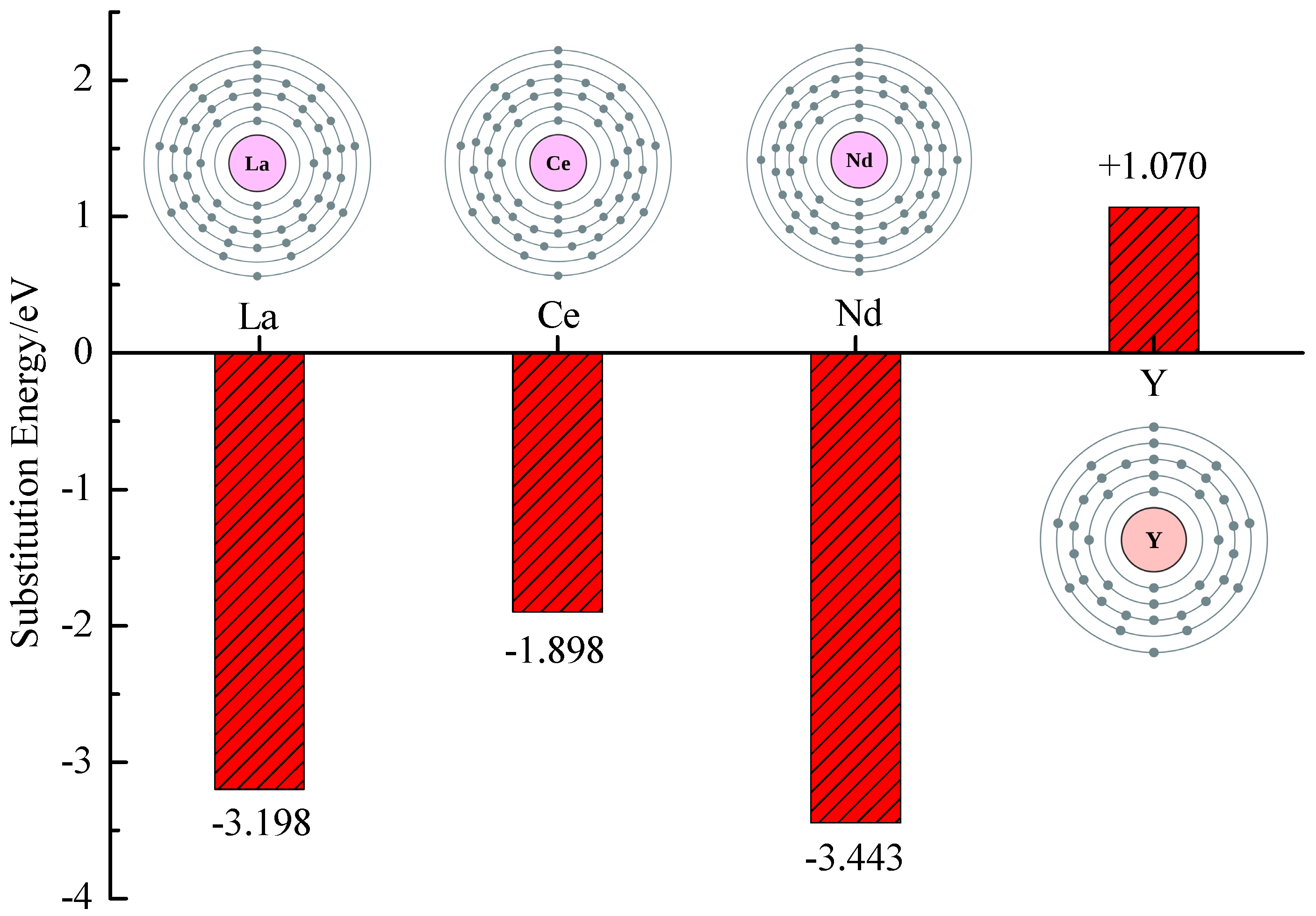
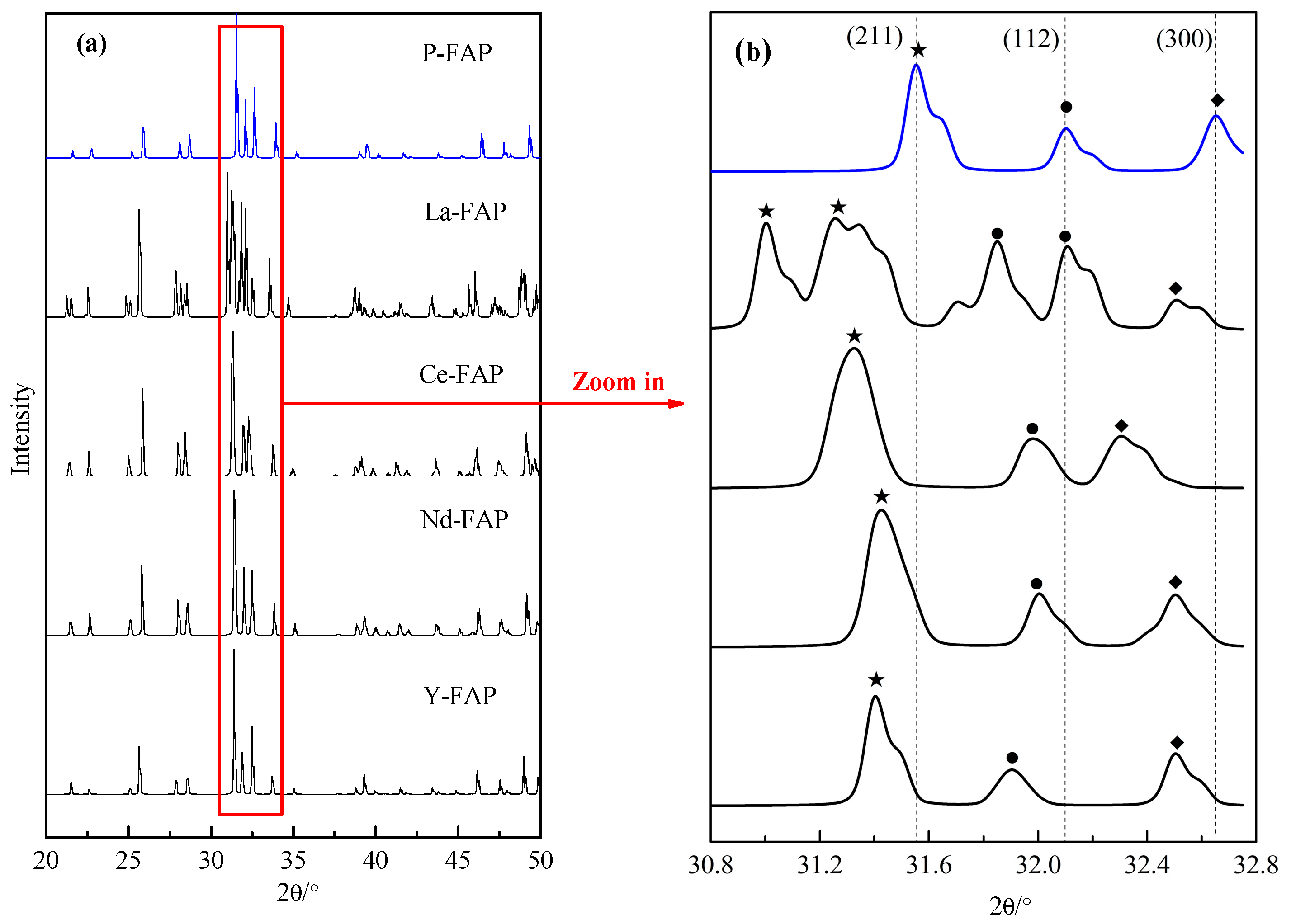
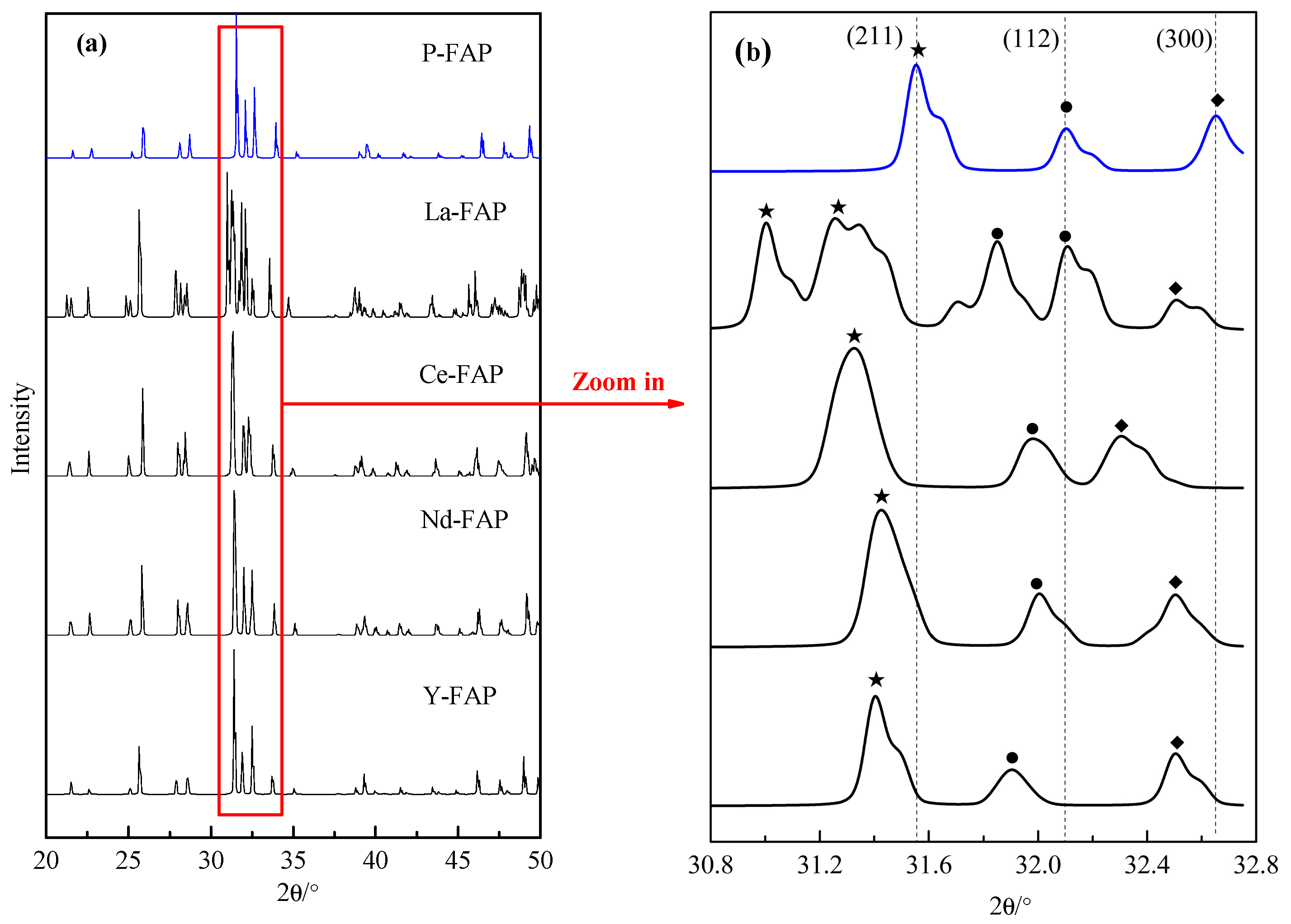
3.1. Substitution Energy and Lattice Parameter
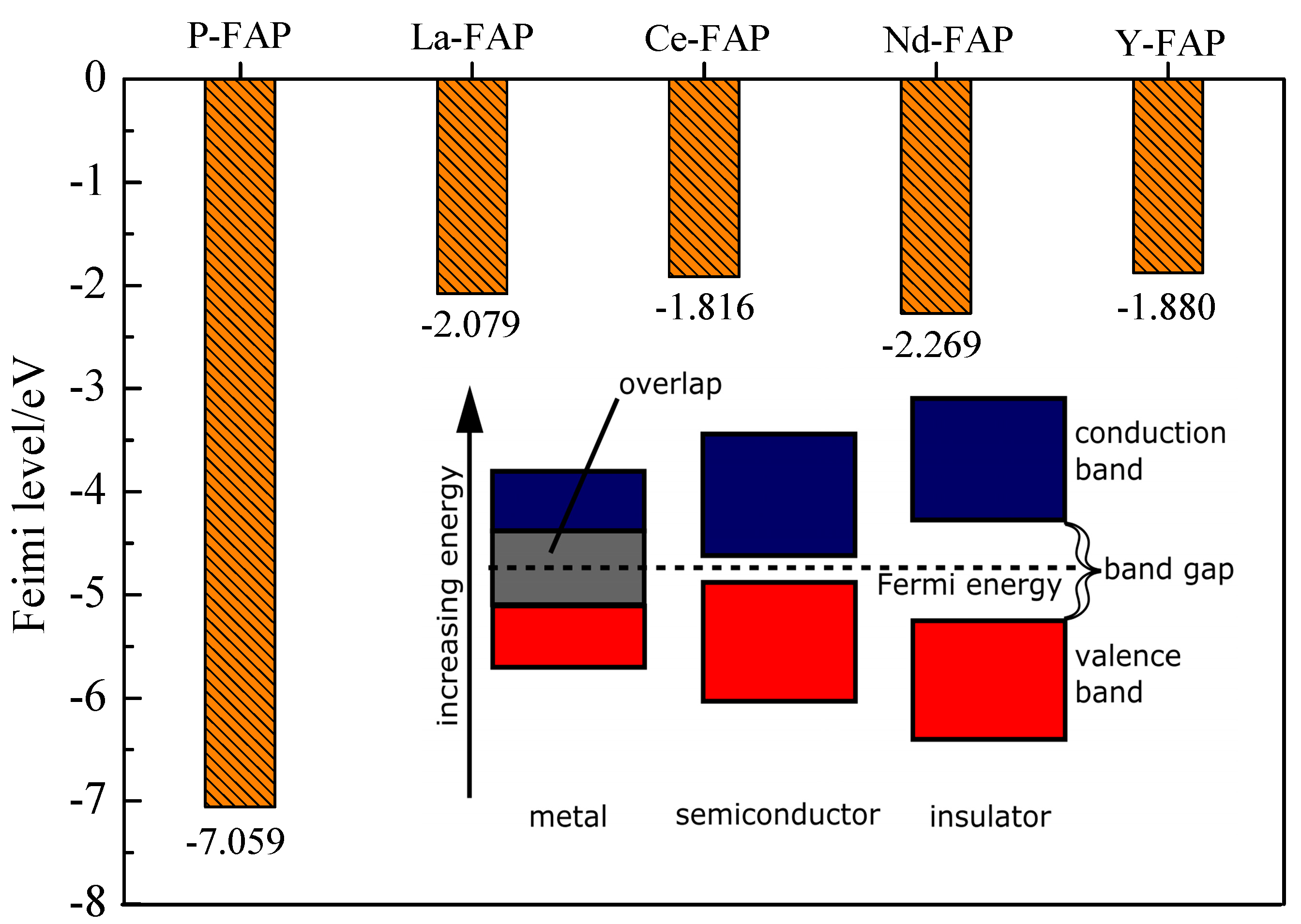
3.2. Fermi Level
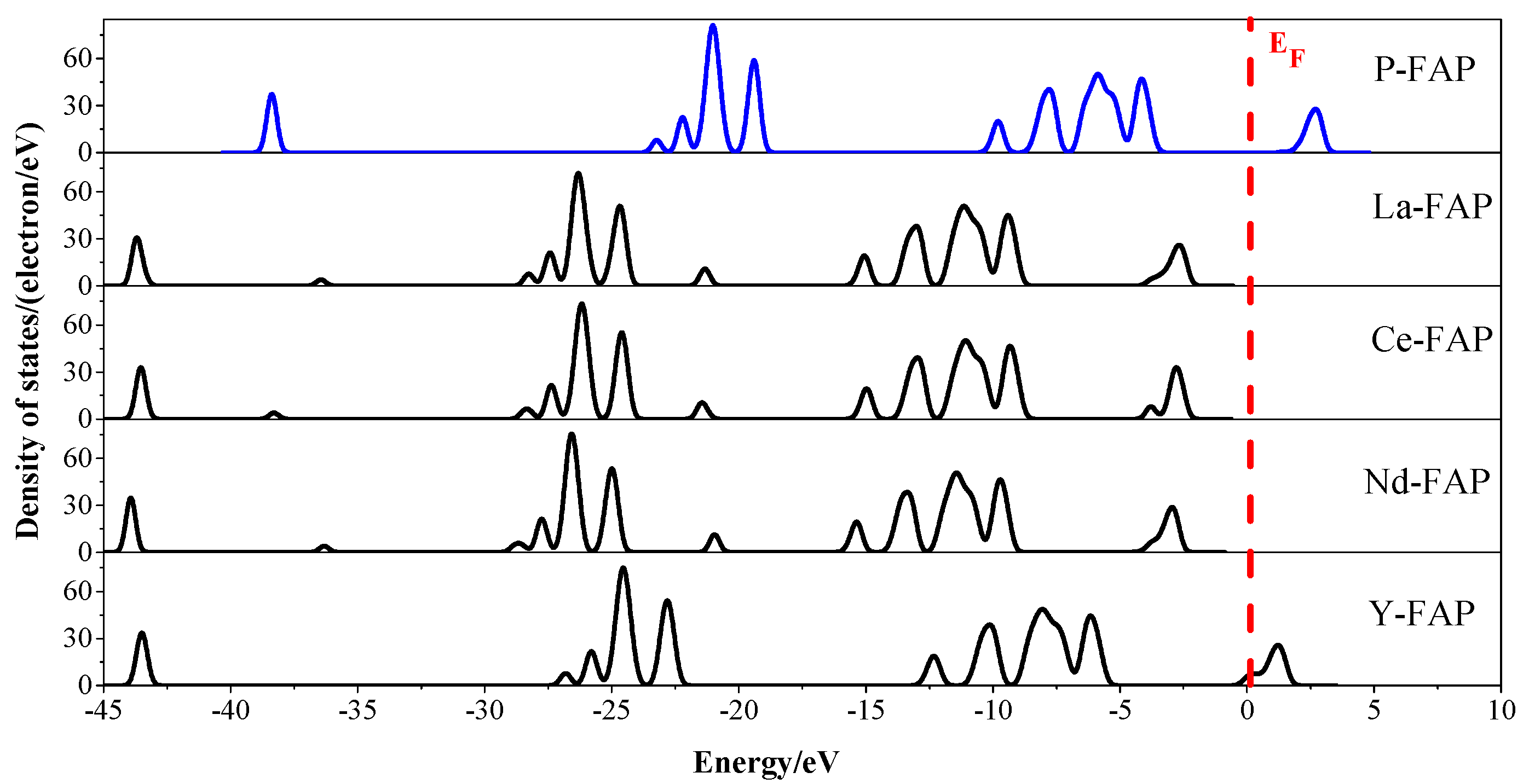
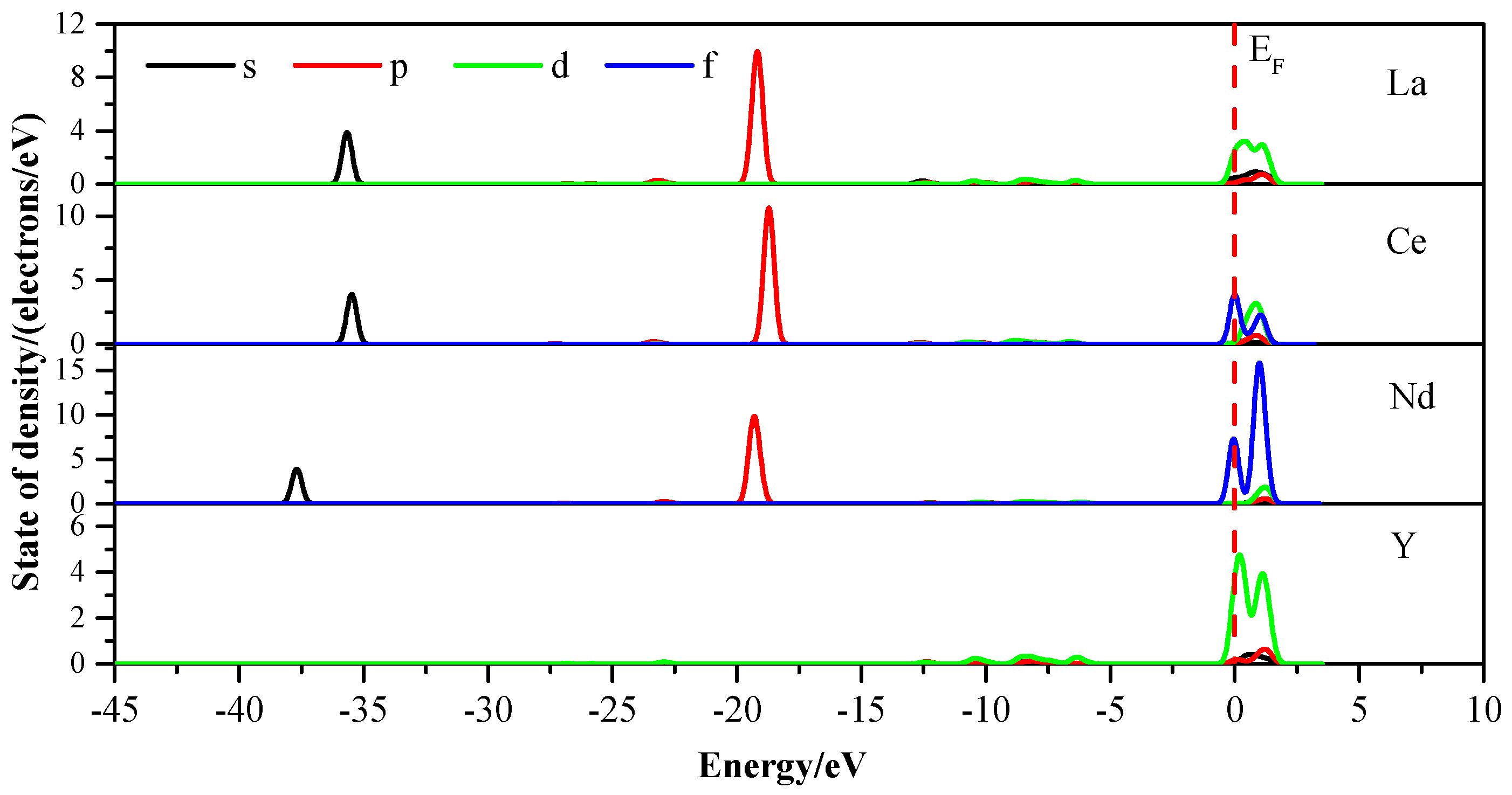
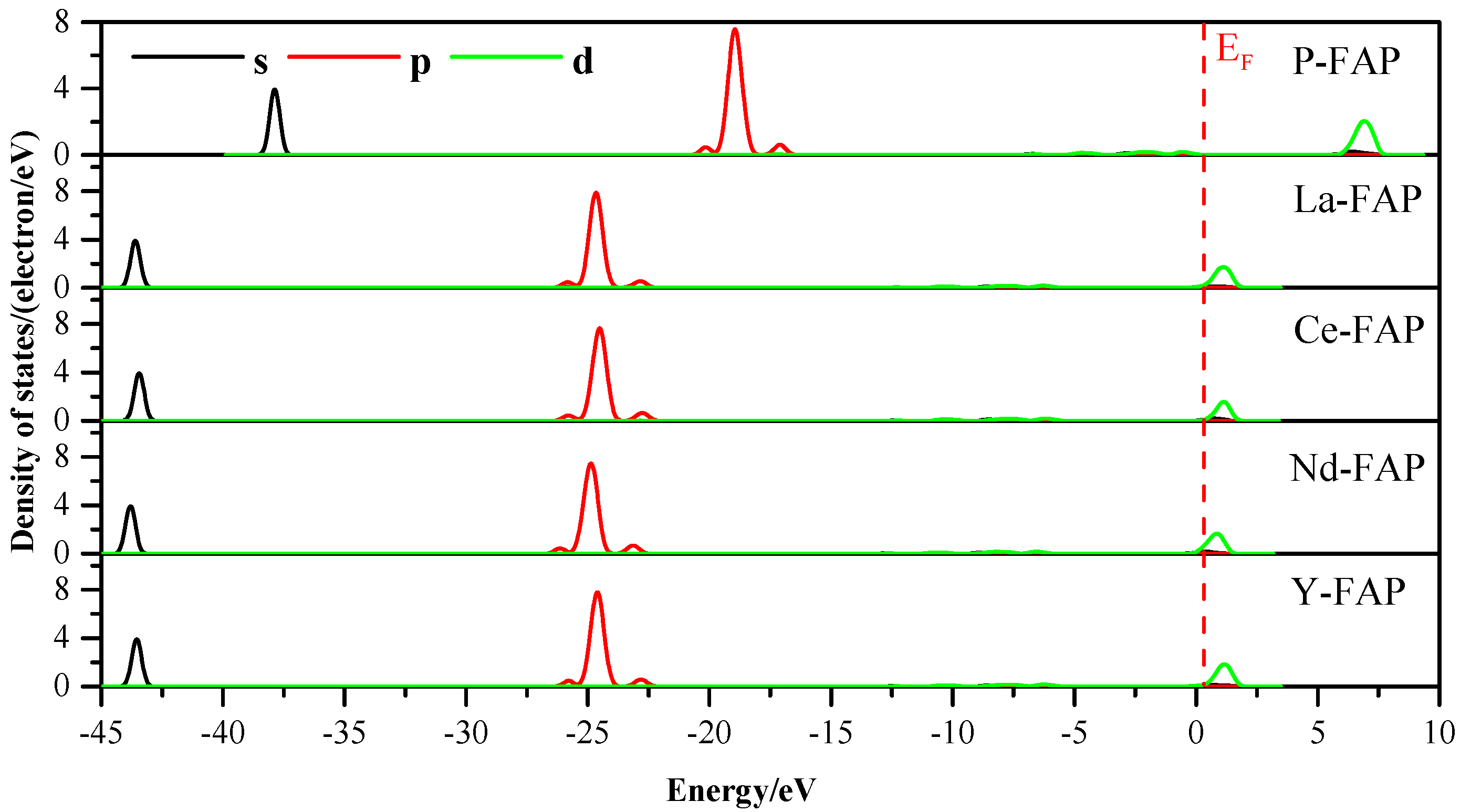
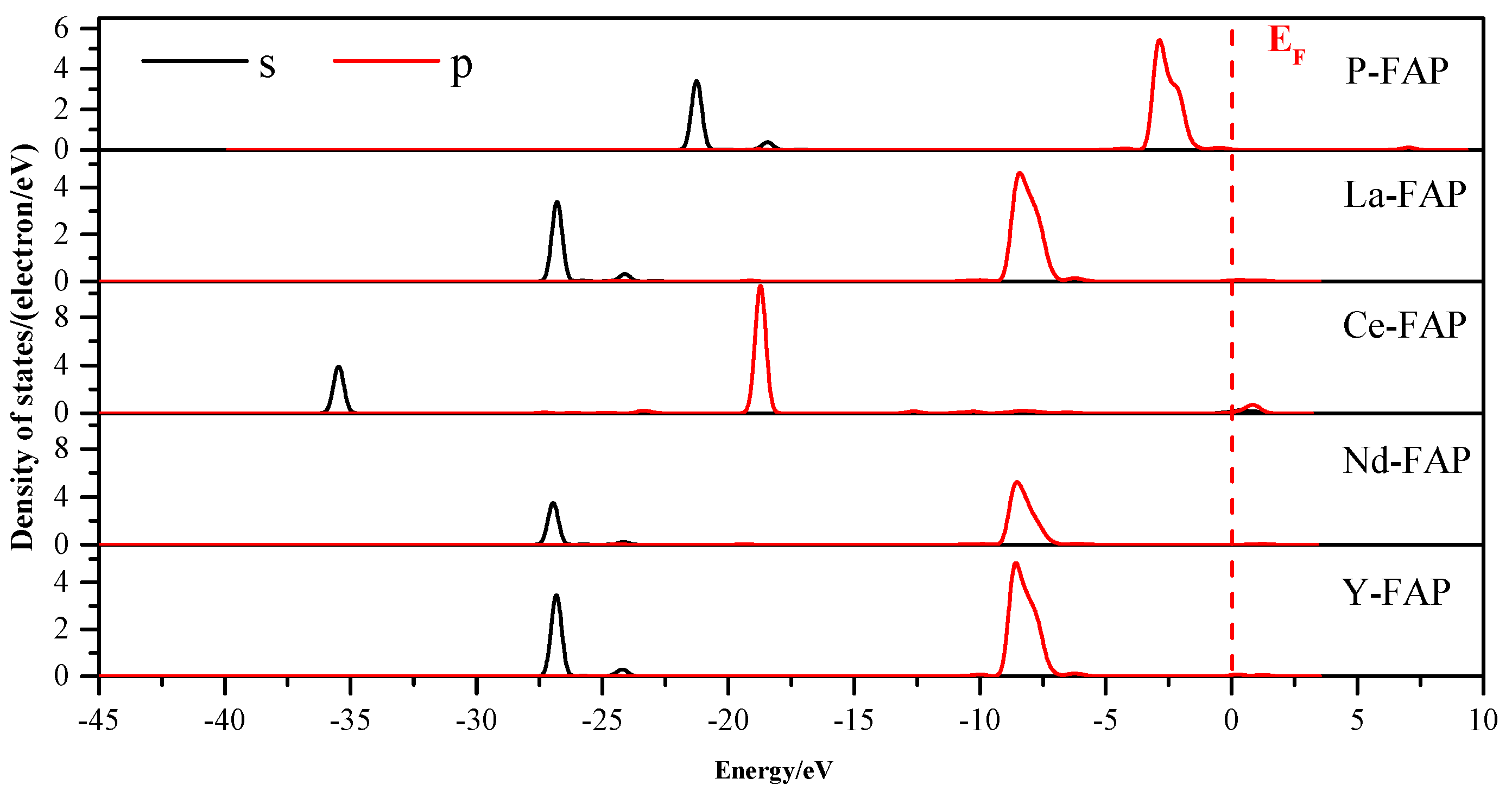
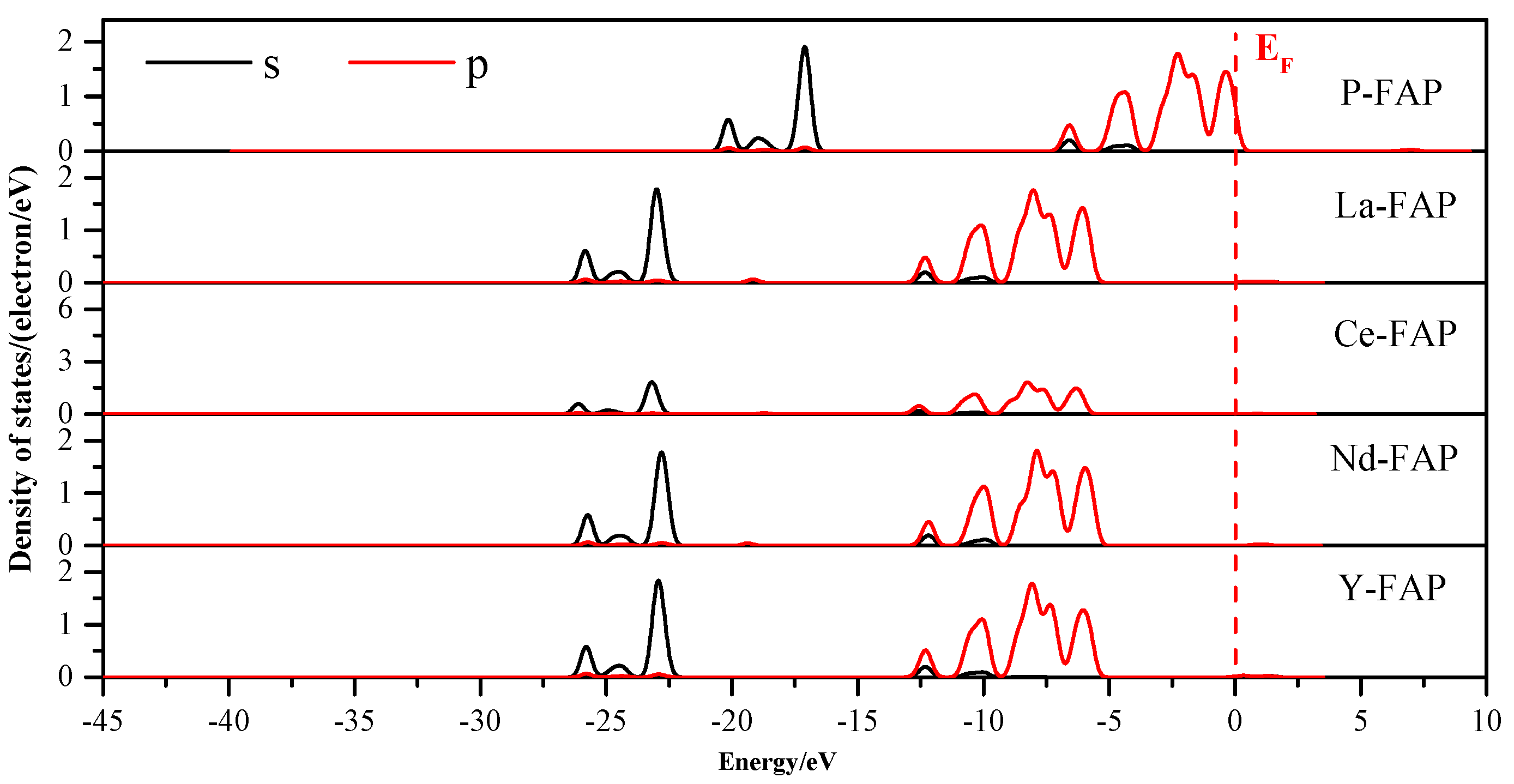
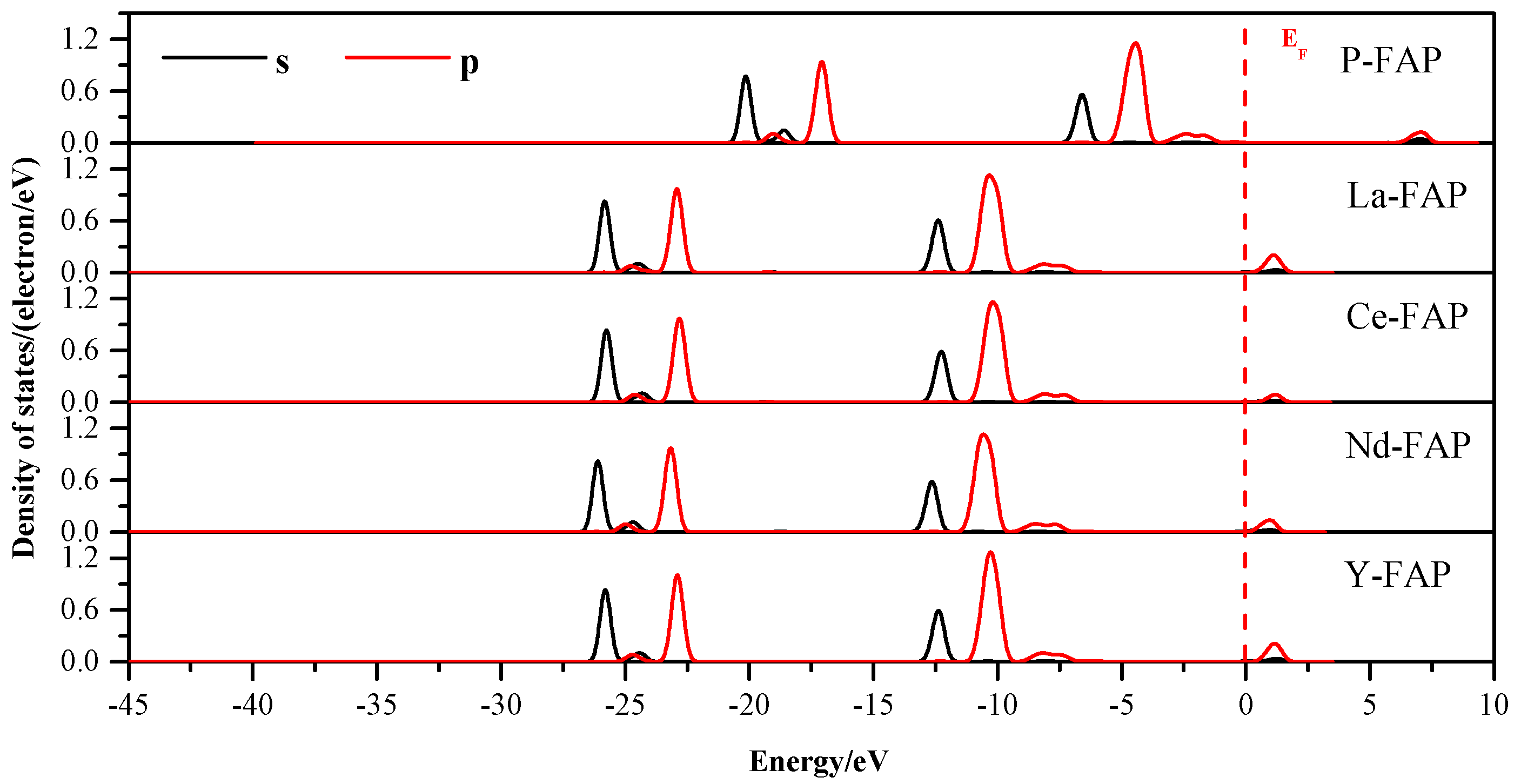
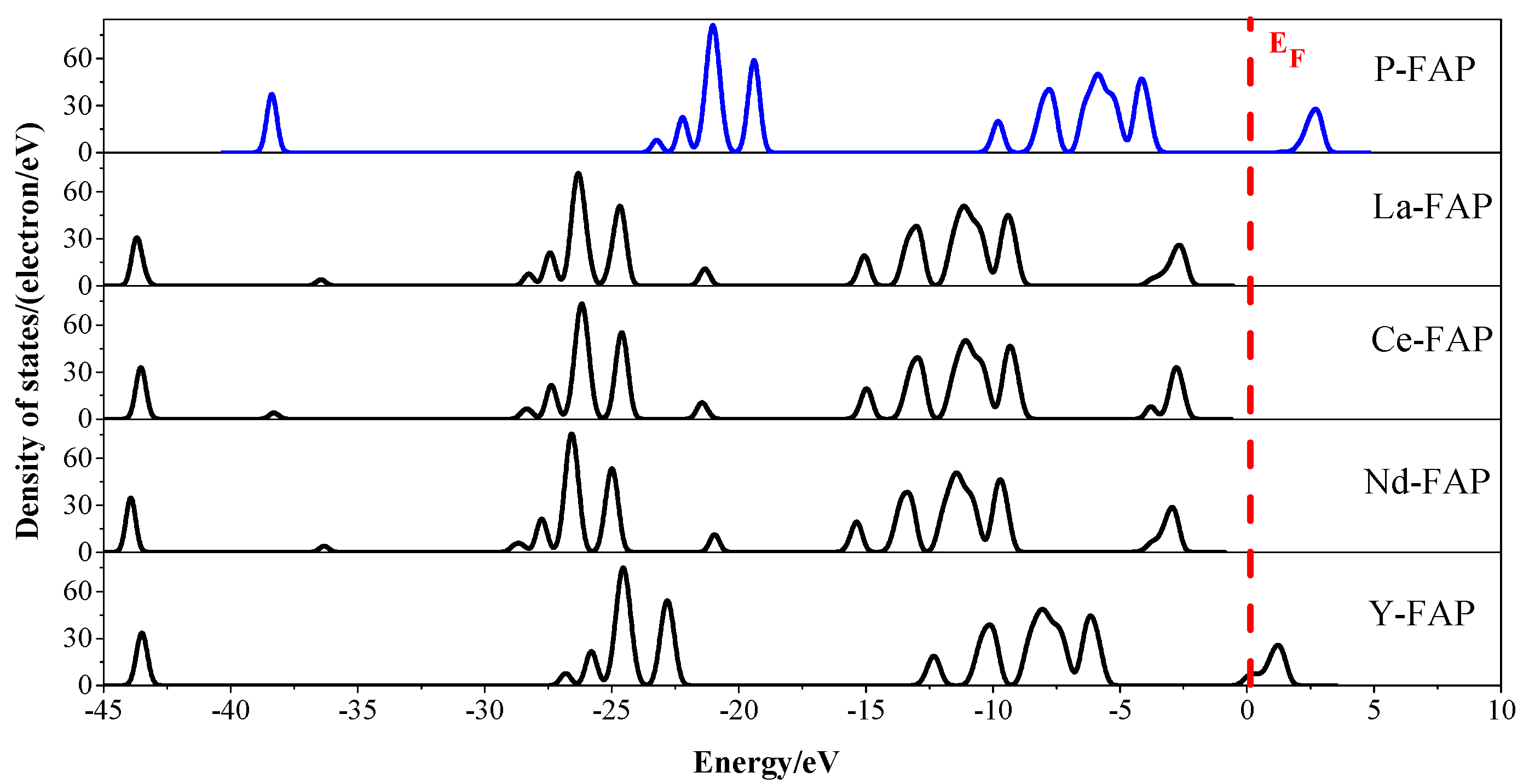
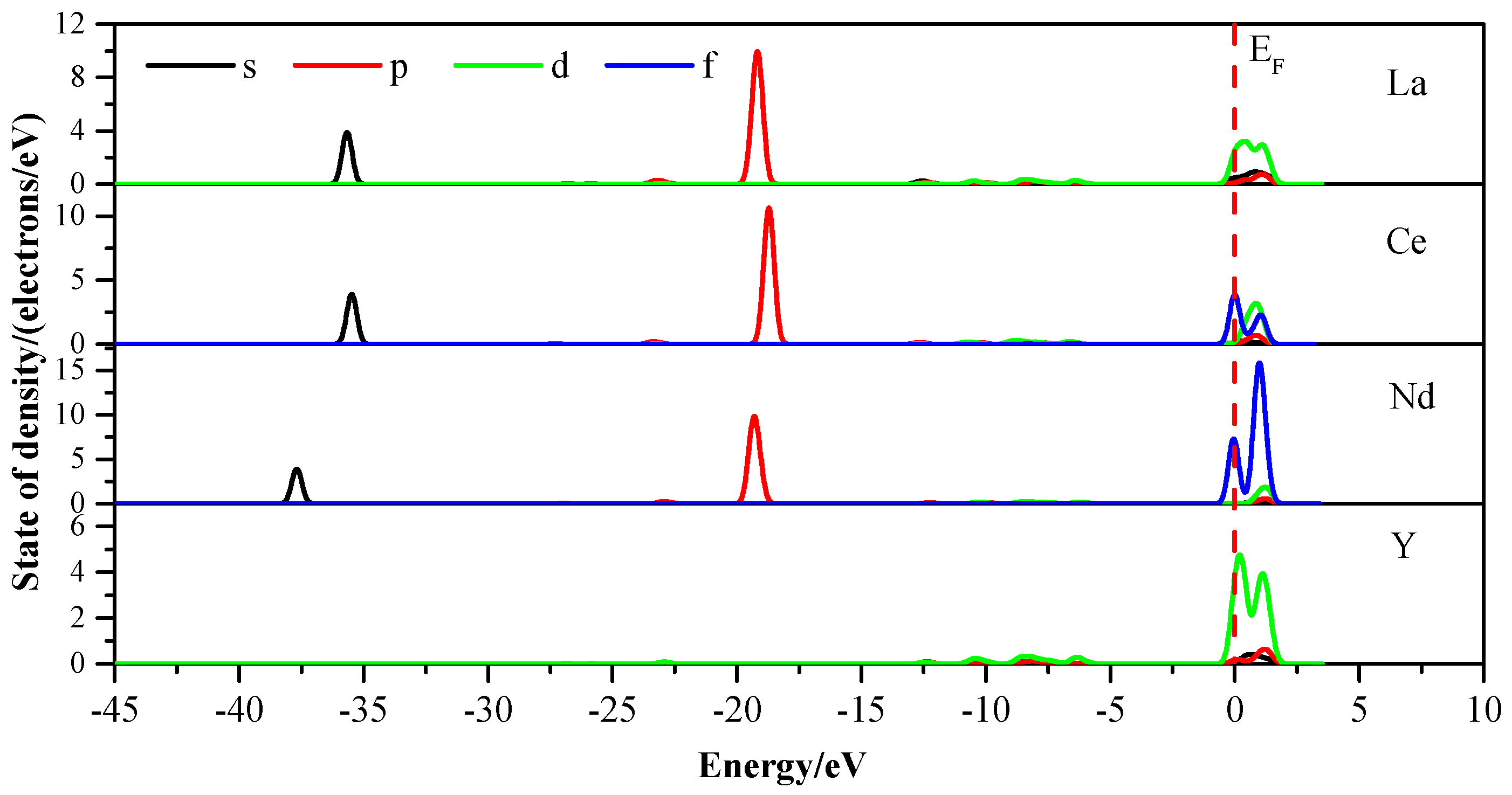
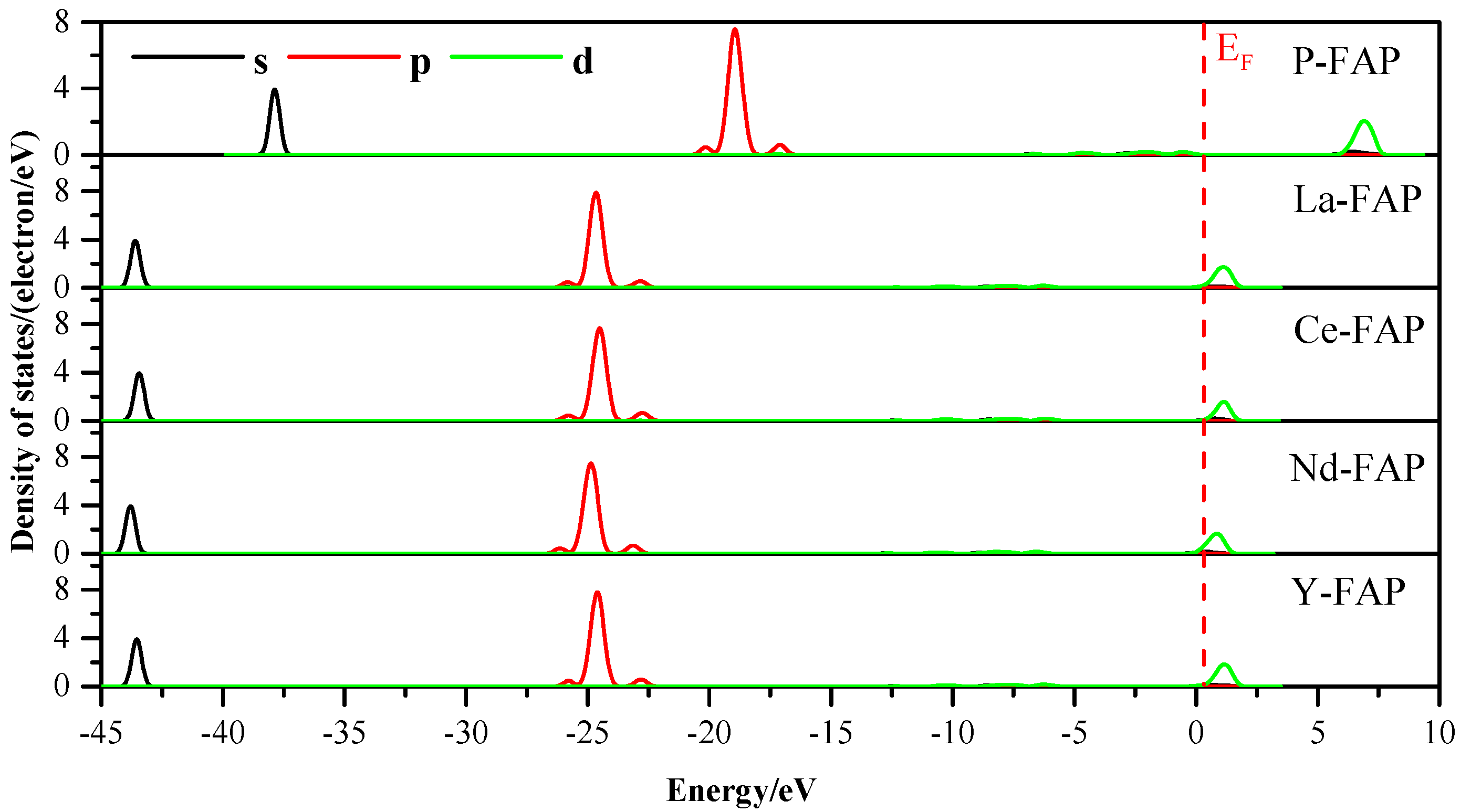
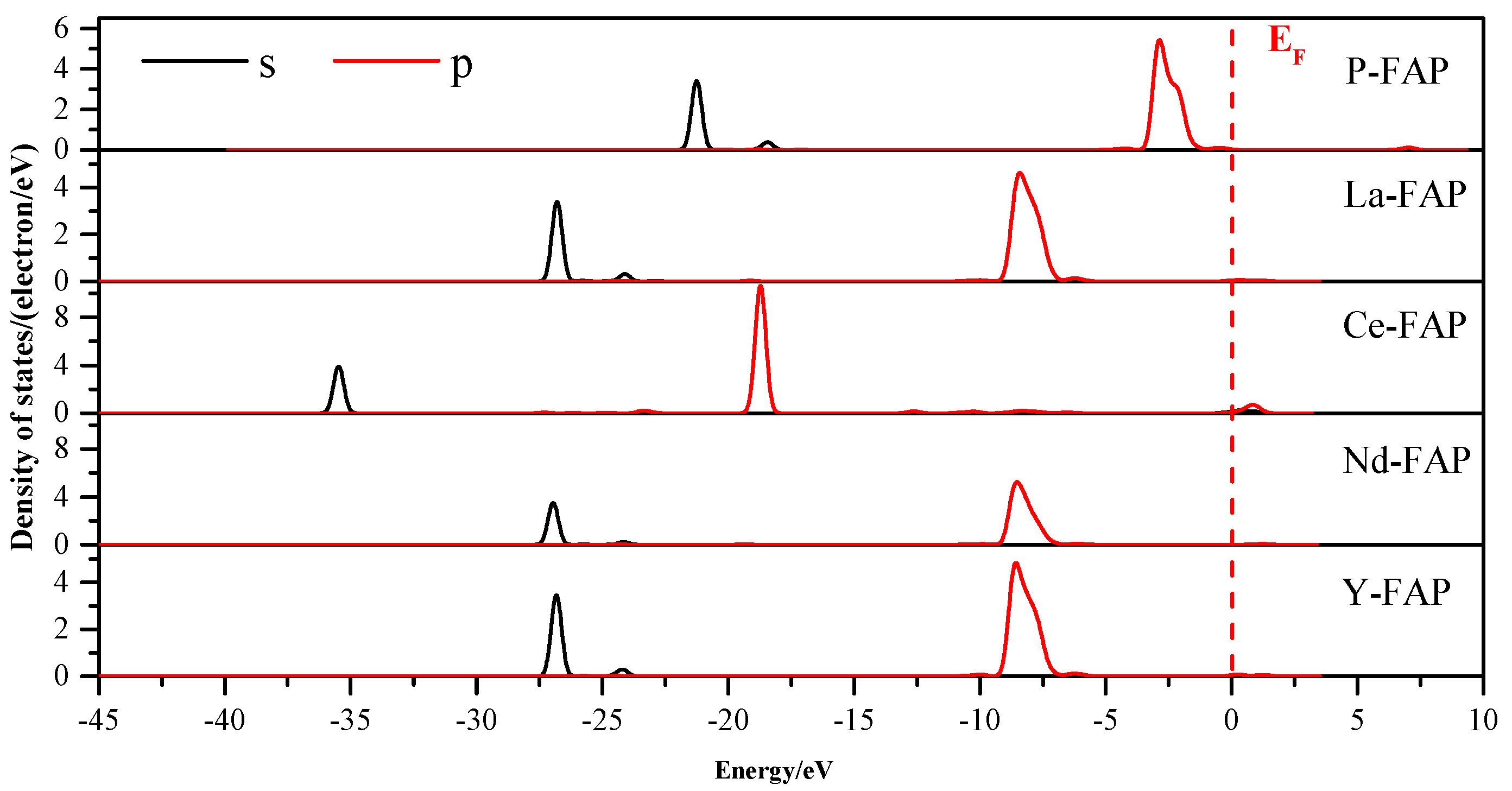
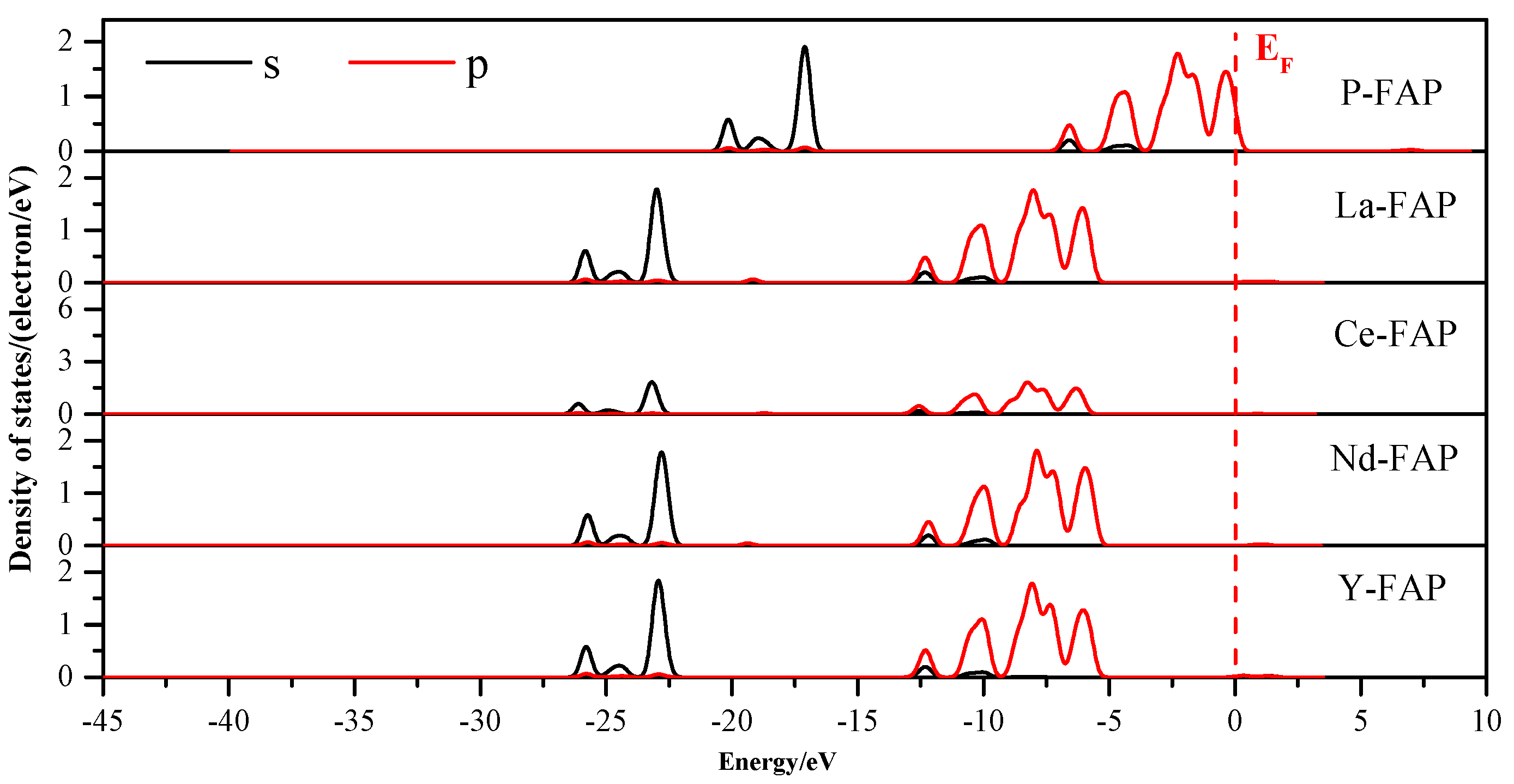
3.3. Density of States
3.4. Analysis of the Mulliken Populationz
3.5. Effect of Impurity on the Reactivity of Fluorapatite
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Connelly, N.G.; Damhus, T.; Hartshorn, R.M.; Hutton, A.T. Nomenclature of Inorganic Chemistry: IUPAC Recommendations 2005; RSC Publishing: Cambridge, UK, 2005. [Google Scholar]
- Chen, Z.H. Global rare earth resources and scenarios of future rare earth industry. J. Rare Earths 2011, 29, 1–6. [Google Scholar] [CrossRef]
- Zhou, B.L.; Li, Z.X.; Chen, C.C. Global potential of rare earth resources and rare earth demand from clean technologies. Minerals 2017, 7, 203. [Google Scholar] [CrossRef]
- Massari, S.; Ruberti, M. Rare earth elements as critical raw materials: Focus on international markets and future strategies. Resour. Policy 2013, 38, 36–43. [Google Scholar] [CrossRef]
- Jordens, A.; Cheng, Y.P.; Waters, K.E. A review of the beneficiation of rare earth element bearing minerals. Miner. Eng. 2013, 41, 97–114. [Google Scholar] [CrossRef]
- Edahbi, M.; Plante, B.; Benzaazoua, M.; Kormos, L.; Pelletier, M. Rare earth elements (La, Ce, Pr, Nd, and Sm) from a carbonatite deposit: Mineralogical characterization and geochemical behavior. Minerals 2018, 8, 55. [Google Scholar] [CrossRef]
- Wu, S.X.; Wang, L.S.; Zhao, L.S.; Zhang, P.; El-Shall, H.; Moudgil, B.; Huang, X.W.; Zhang, L.F. Recovery of rare earth elements from phosphate rock by hydrometallurgical processes—A critical review. Chem. Eng. J. 2018, 335, 774–800. [Google Scholar] [CrossRef]
- Jin, H.X.; Wu, F.Z.; Mao, X.H.; Wang, M.L.; Xie, H.Y. Leaching isomorphism rare earths from phosphorite ore by sulfuric acid and phosphoric acid. Rare Met. 2017, 36, 840–850. [Google Scholar] [CrossRef]
- Wang, J.R.; Zhang, J. Study on the selective leaching of Low-Grade phosphate ore for beneficiation of phosphorus and rare earths using citric acid as leaching agent. Russ. J. Appl. Chem. 2016, 89, 1196–1205. [Google Scholar]
- Chen, J.Y.; Yang, R.D.; Wei, H.R.; Gao, J.B. Rare earth element geochemistry of Cambrian phosphorites from the Yangtze Region. J. Rare Earths 2013, 31, 101–112. [Google Scholar] [CrossRef]
- Li, S.; Zhang, J.; Wang, H.F.; Wang, C.L. Geochemical characteristics of dolomitic phosphorite containing rare earth elements and its weathered ore. Minerals 2019, 9, 416. [Google Scholar] [CrossRef]
- Roelandts, I. Determination of light rare earth elements in apatite by x-ray fluorescence spectrometry after anion exchange extraction. Anal. Chem. 1981, 53, 676–680. [Google Scholar] [CrossRef]
- Ardanova, L.I.; Get’Man, E.I.; Loboda, S.N.; Prisedsky, V.V.; Tkachenko, T.V.; Marchenko, V.I.; Antonovich, V.P.; Chivireva, N.A.; Chebishev, K.A.; Lyashenko, A.S. Isomorphous substitutions of rare earth elements for calcium in synthetic hydroxyapatites. Inorg. Chem. 2010, 49, 10687–10693. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Peng, M.S. Advances in the researches on structural substitution of apatite. Acta Petrol. Mineral. (Chin.) 2003, 22, 413–415. [Google Scholar]
- Luo, Y.; Rakovan, J.; Tang, Y.; Lupulescu, M.; Hughes, J.M. Crystal chemistry of Th in fluorapatite. Am. Mineral. 2011, 96, 23–33. [Google Scholar] [CrossRef]
- Fleet, E.M.; Pan, Y.M. Site preference of rare earth elements in fluorapatite. Am. Mineral. 1995, 80, 329–335. [Google Scholar] [CrossRef]
- Fleet, E.M.; Pan, Y.M. Site preference of rare earth elements in fluorapatite: Binary (LREE + HREE)-substituted crystals. Am. Mineral. 1997, 82, 870–877. [Google Scholar] [CrossRef]
- Njema, H.; Boughzala, K.; Boughzala, H.; Bouzouita, K. Structural analysis by Rietveld refinement of calcium and lanthanum phosphosilicate apatites. J. Rare Earths 2013, 31, 897–904. [Google Scholar] [CrossRef]
- Hughes, M.J.; Cameron, M. Rare-earth-element ordering and structural variations in natural rare-earth bearing apatites. Am. Mineral. 1991, 76, 1165–1173. [Google Scholar]
- Fleet, M.E.; Pan, Y. Crystal chemistry of rare earth elements in fluorapatite and some calc-silicates. Eur. J. Miner. 1995, 7, 591–605. [Google Scholar] [CrossRef]
- De Leeuw, N.H. Density functional theory calculations of solid solutions of fluor- and chlorapatites. Chem. Mater. 2002, 14, 435–441. [Google Scholar] [CrossRef]
- Qiu, Y.Q.; Cui, W.Y.; Li, L.J.; Ye, J.J.; Wang, J.; Zhang, Q. Structural, electronic properties with different terminations for fluorapatite (0 0 1) surface: A first-principles investigation. Comp. Mater. Sci. 2017, 126, 132–138. [Google Scholar] [CrossRef]
- Rulis, P.; Yao, H.Z.; Ouyang, L.Z.; Ching, W.Y. Electronic structure, bonding, charge distribution, and x-ray absorption spectra of the (0 0 1) surfaces of fluorapatite and hydroxyapatite from first principles. Phys. Rev. B 2007, 76, 245410. [Google Scholar] [CrossRef]
- Wang, X.C.; Zhang, Q.; Li, X.B.; Ye, J.J.; Li, L.J. Structural and electronic properties of different terminations for quartz (001) surfaces as well as water molecule adsorption on it: A First-Principles study. Minerals 2018, 8, 58. [Google Scholar] [CrossRef]
- Xie, J.; Li, X.H.; Mao, S.; Li, L.J.; Ke, B.L.; Zhang, Q. Effects of structure of fatty acid collectors on the adsorption of fluorapatite (0 0 1) surface: A first-principles calculations. Appl. Surf. Sci. 2018, 444, 699–709. [Google Scholar] [CrossRef]
- Chen, J.H. The Solid Physics of Sulphide Minerals Flotation; Central South University Press: Changsha, China, 2015. [Google Scholar]
- Chen, J.H.; Ke, B.L.; Lan, L.H.; Li, Y.Q. DFT and experimental studies of oxygen adsorption on galena surface bearing Ag, Mn, Bi and Cu impurities. Miner. Eng. 2015, 71, 170–179. [Google Scholar]
- Chen, J.H.; Ke, B.L.; Lan, L.H.; Li, Y.Q. Influence of Ag, Sb, Bi and Zn impurities on electrochemical and flotation behaviour of galena. Miner. Eng. 2015, 72, 10–16. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.H.; Guo, J. A DFT study on the effect of lattice impurities on the electronic structures and floatability of sphalerite. Miner. Eng. 2010, 23, 1120–1130. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.H.; Lan, L.H.; Yang, M.J. The influence of the impurities on the flotation behaviors of synthetic ZnS. Miner. Eng. 2012, 27, 65–71. [Google Scholar] [CrossRef]
- Li, Y.Q.; Chen, J.H.; Chen, Y. Electronic structures and flotation behavior of pyrite containing vacancy defects. Acta Phys. Chim. Sin. 2010, 26, 1435–1441. [Google Scholar]
- Chen, J.H.; Wang, L.; Chen, Y.; Guo, Y. Density functional theory of effects of vacancy defects on electronic structure and flotation of galena. Chin. J. Nonferr. Met. 2010, 20, 1815–1821. [Google Scholar]
- Gao, Z.; Sun, W.; Gao, J.; Hu, Y. A Density Functional Theory Study on the Effect of Lattice Impurities on the Electronic Structures and Reactivity of Fluorite. Minerals 2017, 7, 160. [Google Scholar]
- Filippova, I.V.; Filippov, L.O.; Lafhaj, Z.; Barres, O.; Fornasiero, D. Effect of calcium minerals reactivity on fatty acids adsorption and flotation. Colloids Surf. A Phys. Eng. Asp. 2018, 545, 157–166. [Google Scholar] [CrossRef]
- Owens, C.L.; Nash, G.R.; Hadler, K.; Fitzpatrick, R.S.; Anderson, C.G.; Wall, F. Apatite enrichment by rare earth elements: A review of the effects of surface properties. Adv. Colloid Interface Sci. 2019, 265, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.; Tao, D.; Xu, G. Fatty acid collectors for phosphate flotation and their adsorption behavior using QCM-D. Int. J. Miner. Process. 2010, 95, 1–9. [Google Scholar] [CrossRef]
- Paiva, P.R.P.; Monte, M.B.M.; Sim, O.R.A.; Gaspar, J.C. In Situ AFM study of potassium oleate adsorption and calcium precipitate formation on an apatite surface. Miner. Eng. 2011, 24, 387–395. [Google Scholar] [CrossRef]
- Santos, E.P.; Dutra, A.; Oliveira, J.F. The effect of jojoba oil on the surface properties of calcite and apatite aiming at their selective flotation. Int. J. Miner. Process. 2015, 143, 34–38. [Google Scholar] [CrossRef]
- Ye, J.J.; Zhang, Q.; Li, X.B.; Wang, X.C.; Ke, B.L.; Li, X.H.; Shen, Z.H. Effect of the morphology of adsorbed oleate on the wettability of a collophane surface. Appl. Surf. Sci. 2018, 444, 87–96. [Google Scholar] [CrossRef]
- Hughes, M.J.; Cameron, M.; Crowley, K.D. Structural variations in natural F, OH, and Cl apatites. Am. Mineral. 1989, 74, 870–876. [Google Scholar]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.J.; De La Heras, C.; Sanchez, C. The effect of Ni impurities on some structural properties of pyrite thin films. J. Phys. Condens. Matter 1995, 7, 2115–2121. [Google Scholar] [CrossRef]
- Imai, Y.; Mukaida, M.; Tsunoda, T. Calculation of electronic energy and density of state of iron-disilicides using a total-energy pseudopotential method, CASTEP. Thin Solid Films 2001, 381, 176–182. [Google Scholar] [CrossRef]
- Yamabe, S.; Minato, T. Frontier orbital theory. J. Chem. Educ. 1992, 40, 450–454. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluorapatite Type | Lattice Parameter/Å | |||||
|---|---|---|---|---|---|---|
| a | Δa/% | b | Δb/% | c | Δc/% | |
| P-FAP | 9.455 | 0 | 9.455 | 0 | 6.888 | 0 |
| La-FAP | 9.493 | 0.40 | 9.651 | 2.10 | 6.927 | 0.57 |
| Ce-FAP | 9.570 | 1.22 | 9.530 | 0.80 | 6.920 | 0.47 |
| Nd-FAP | 9.543 | 0.93 | 9.509 | 0.57 | 6.919 | 0.45 |
| Y-FAP | 9.506 | 0.54 | 9.515 | 0.64 | 6.929 | 0.60 |
| Defect Type | Species | Population | Total | Charge/e | |||
|---|---|---|---|---|---|---|---|
| s | p | d | f | ||||
| P-FAP | Ca2 | 2.13 (−0.13) | 6.00 (0) | 0.51 (1.49) | 0 | 8.64 | 1.36 |
| F | 1.96 (0.04) | 5.74 (−0.74) | 0 | 0 | 7.70 | −0.70 | |
| O1 | 1.86 (0.14) | 5.21 (−1.21) | 0 | 0 | 7.07 | −1.07 | |
| La-FAP | La | 2.09 (−0.09) | 6.10 (−0.10) | 1.40 (0.60) | 0 | 9.60 | 1.21 |
| F | 1.96 (0.04) | 5.72 (−0.72) | 0 | 0 | 7.68 | −0.68 | |
| O1 | 1.86 (0.14) | 5.16 (−1.16) | 0 | 0 | 7.02 | −1.02 | |
| Ce-FAP | Ce | 2.15 (−0.15) | 6.09 (−0.09) | 0.79 (1.21) | 1.65 (0.35) | 10.67 | 1.32 |
| F | 1.95 (0.05) | 5.72 (−0.72) | 0 | 0 | 7.67 | −0.67 | |
| O1 | 1.86 (0.14) | 5.16 (−1.16) | 0 | 0 | 7.02 | −1.02 | |
| Nd-FAP | Nd | 2.09 (−0.09) | 6.06 (−0.06) | 0.77 (1.23) | 3.86 (0.14) | 12.85 | 1.22 |
| F | 1.95 (0.05) | 5.71 (−0.71) | 0 | 0 | 7.66 | −0.66 | |
| O1 | 1.86 (0.14) | 5.16 (−1.16) | 0 | 0 | 7.02 | −1.02 | |
| Y-FAP | Y | 0.16 (1.84) | 0.09 (−0.09) | 1.57 (−0.43) | 0 | 1.82 | 1.32 |
| F | 1.96 (0.04) | 5.72 (−0.72) | 0 | 0 | 7.68 | −0.68 | |
| O1 | 1.86 (0.14) | 5.15 (−1.15) | 0 | 0 | 7.01 | −1.01 | |
| Defect Type | Bond | Population | Bond Length/nm |
|---|---|---|---|
| P-FAP | Ca2–F | 0.10 | 0.2327 |
| Ca2–O1 | 0.13 | 0.2363 | |
| Ca2–O2 | 0.11 | 0.2385 | |
| Ca2–O3 | 0.08 | 0.2478 | |
| La-FAP | La–F | 0.11 | 0.2514 |
| La–O1 | 0.23 | 0.2447 | |
| La–O2 | 0.19 | 0.2563 | |
| La–O3 | 0.08 | 0.2590 | |
| Ce-FAP | Ce–F | 0.13 | 0.2577 |
| Ce–O1 | 0.17 | 0.2461 | |
| Ce–O2 | 0.14 | 0.2560 | |
| Ce–O3 | 0.09 | 0.2709 | |
| Nd-FAP | Nd–F | 0.15 | 0.2502 |
| Nd–O1 | 0.22 | 0.2474 | |
| Nd–O2 | 0.16 | 0.2613 | |
| Nd–O3 | 0.11 | 0.2703 | |
| Y-FAP | Y–F | 0.17 | 0.2507 |
| Y–O1 | 0.30 | 0.2416 | |
| Y–O2 | 0.23 | 0.2502 | |
| Y–O3 | 0.15 | 0.2576 |
| Defect Type | EHOMO/eV | ELUMO/eV | |∆E|/eV |
|---|---|---|---|
| P-FAP | −7.059 | −1.518 | 4.096 |
| La-FAP | −7.233 | −2.050 | 3.566 |
| Ce-FAP | −1.987 | −1.731 | 3.883 |
| Nd-FAP | −2.898 | −1.688 | 3.856 |
| Y-FAP | −7.074 | −1.639 | 3.975 |
| OA | −5.614 | −0.942 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhang, Q.; Mao, S.; Cheng, W. A Theoretical Study on the Electronic Structure and Floatability of Rare Earth Elements (La, Ce, Nd and Y) Bearing Fluorapatite. Minerals 2019, 9, 500. https://doi.org/10.3390/min9080500
Wang X, Zhang Q, Mao S, Cheng W. A Theoretical Study on the Electronic Structure and Floatability of Rare Earth Elements (La, Ce, Nd and Y) Bearing Fluorapatite. Minerals. 2019; 9(8):500. https://doi.org/10.3390/min9080500
Chicago/Turabian StyleWang, Xianchen, Qin Zhang, Song Mao, and Wei Cheng. 2019. "A Theoretical Study on the Electronic Structure and Floatability of Rare Earth Elements (La, Ce, Nd and Y) Bearing Fluorapatite" Minerals 9, no. 8: 500. https://doi.org/10.3390/min9080500
APA StyleWang, X., Zhang, Q., Mao, S., & Cheng, W. (2019). A Theoretical Study on the Electronic Structure and Floatability of Rare Earth Elements (La, Ce, Nd and Y) Bearing Fluorapatite. Minerals, 9(8), 500. https://doi.org/10.3390/min9080500