Formation of Authigenic Low-Magnesium Calcite from Sites SS296 and GC53 of the Gulf of Mexico

 ,
,
Abstract
1. Introduction
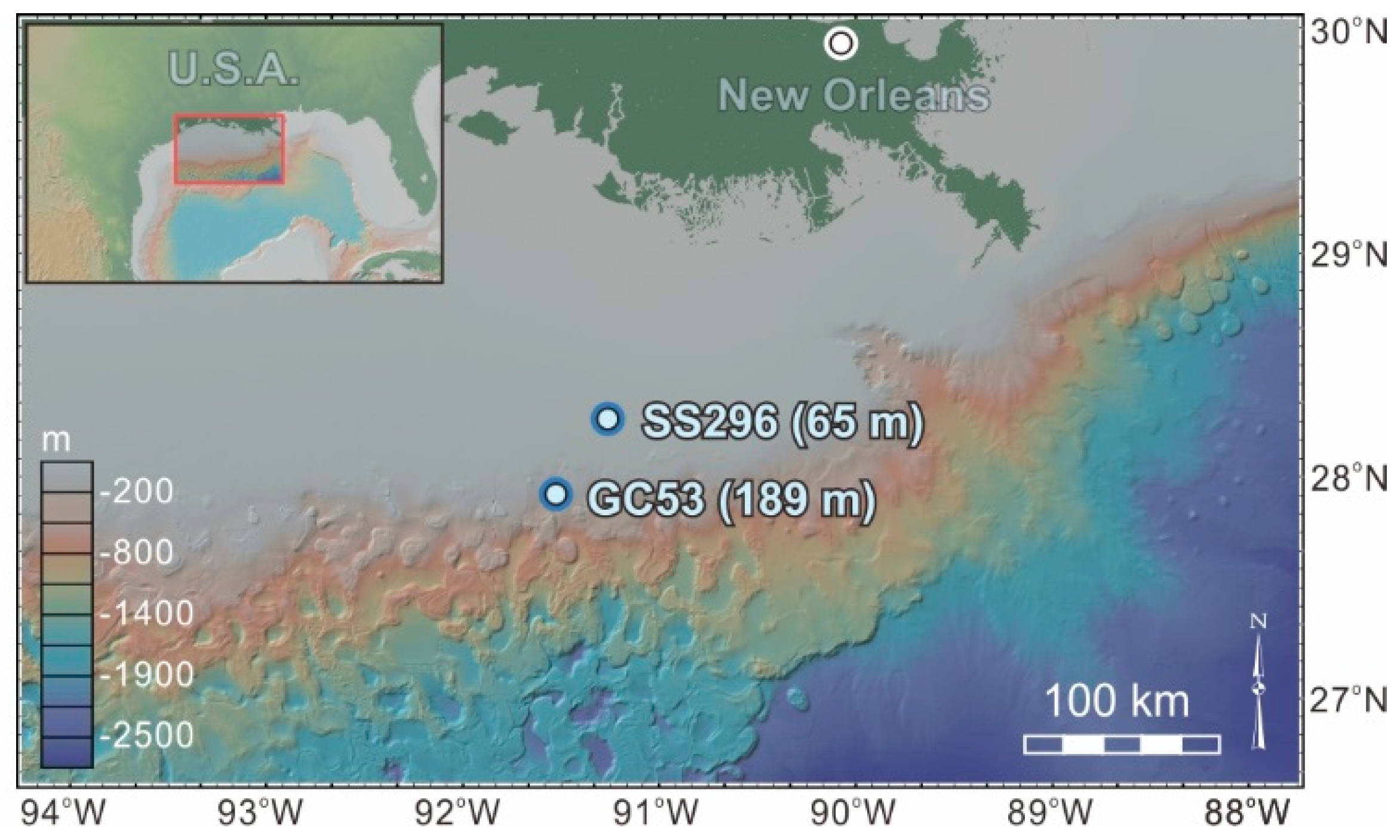
2. Geological Setting and Sampling
3. Methods
3.1. Mineralogy and Petrography
3.2. Major and Trace Element Analyses
3.3. Isotope Analyses
4. Results
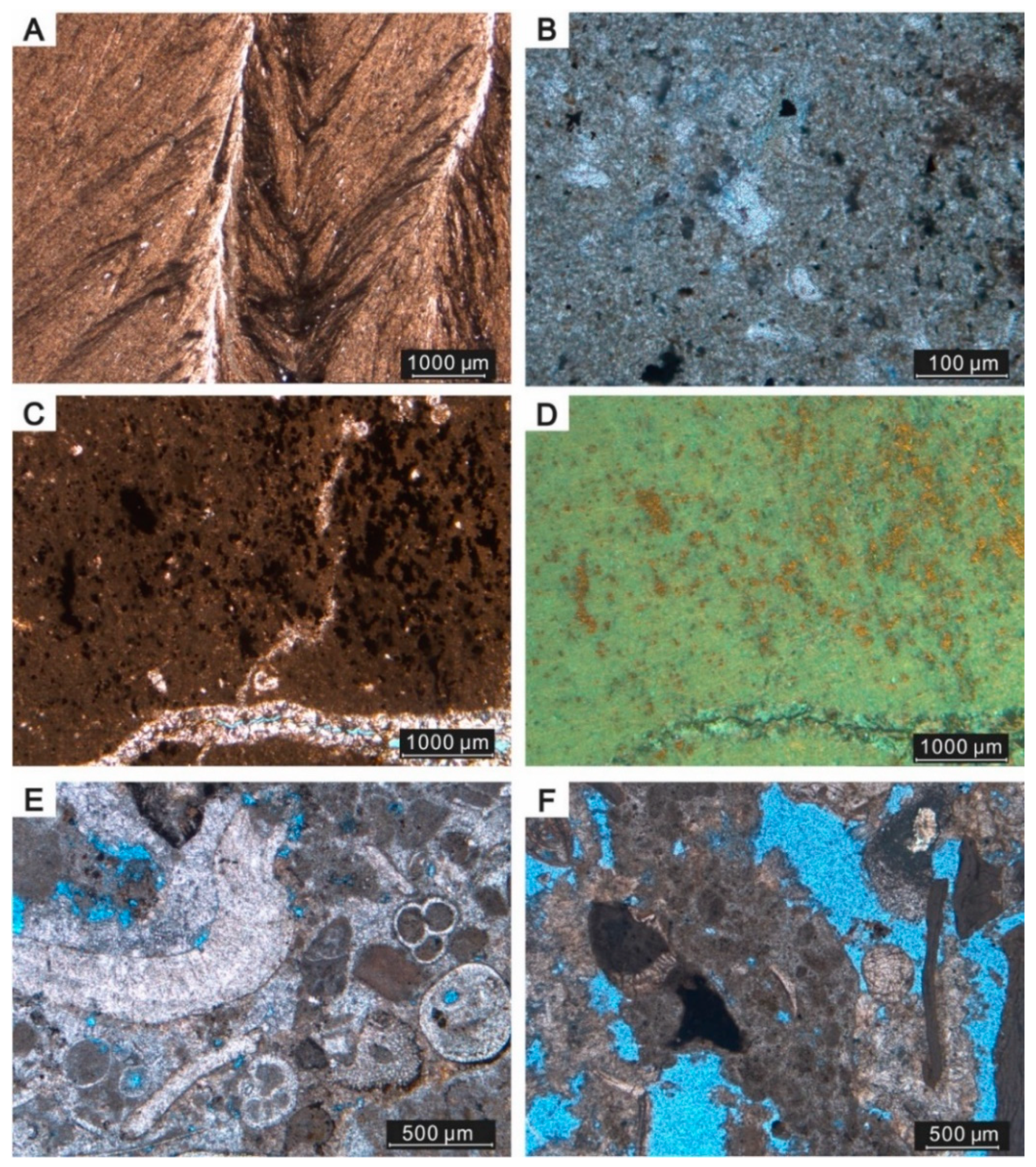
4.1. Samples, Mineralogy, and Petrographic Features
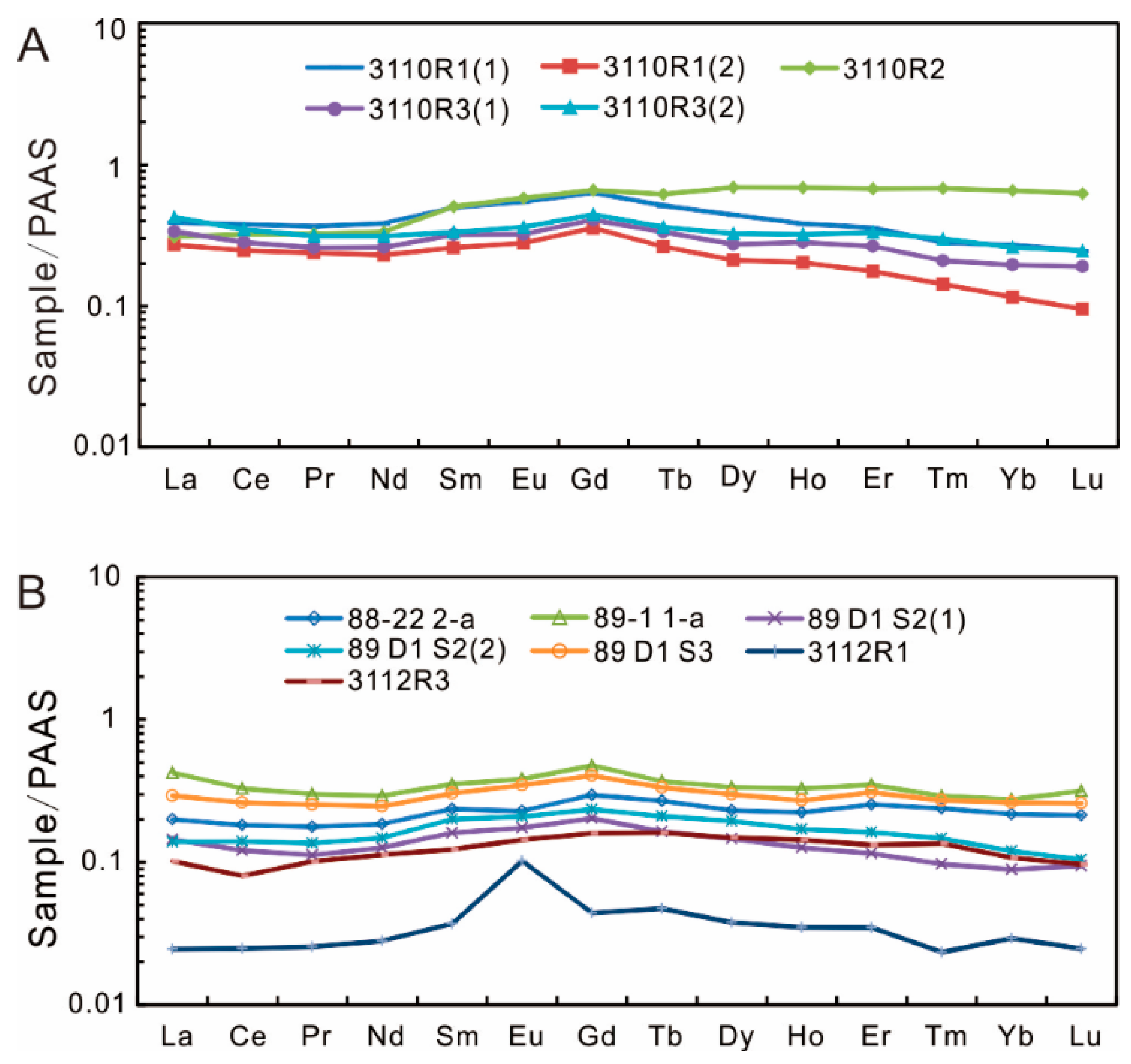
4.2. Major and Trace Elements
4.3. Carbonate Carbon and Oxygen Isotopes
4.4. Organic Carbon and Nitrogen Concentrations and δ13Corg Values
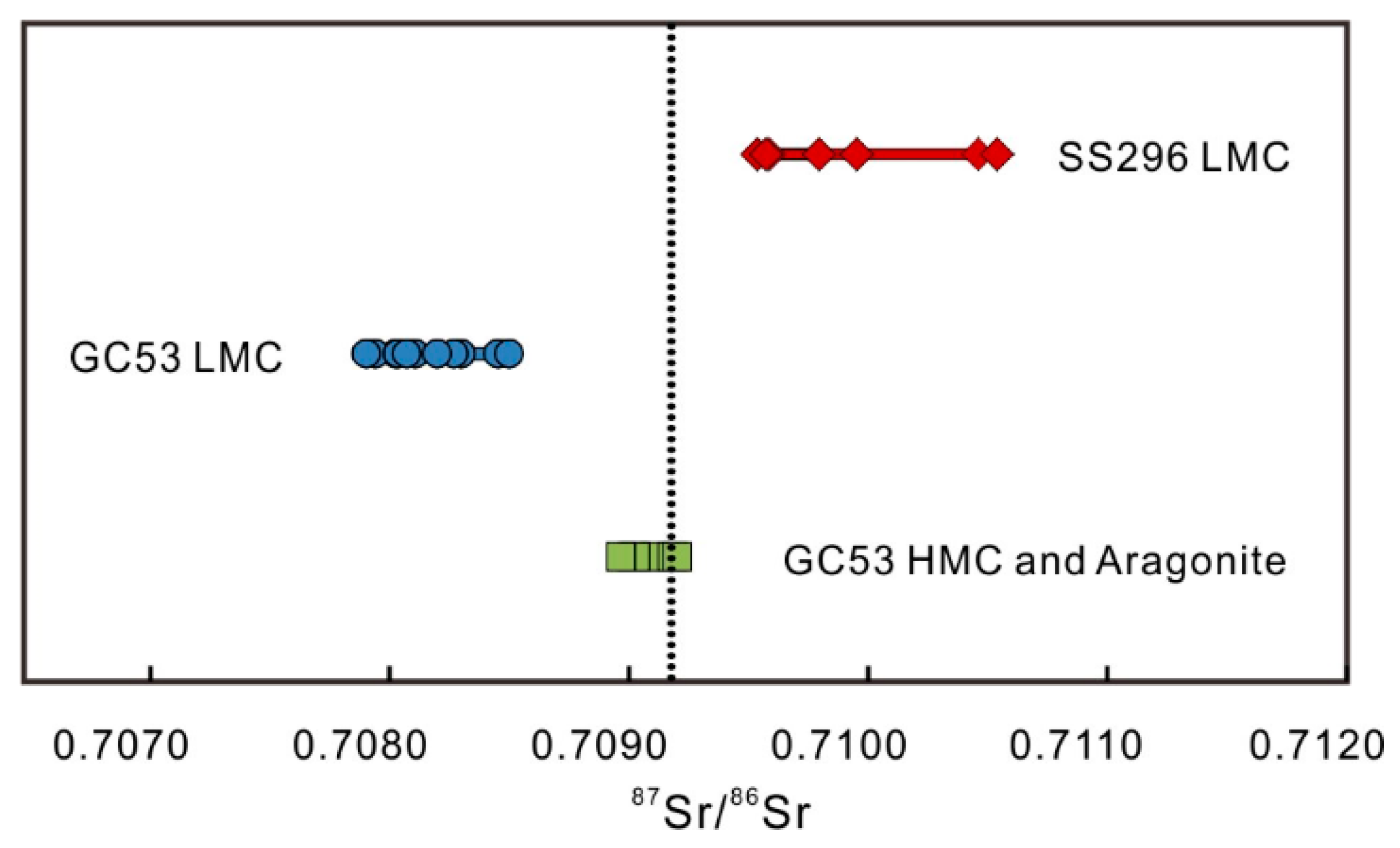
4.5. Sr Isotopes
5. Discussion
5.1. Primary Origin of Low-Mg Calcite
5.2. Constraints on the Formation of Low-Mg Calcite
5.2.1. Site SS296
5.2.2. Site GC53
5.3. Implications of the Occurrence of Low-Mg Calcite in Marine Settings
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardie, L.A. Secular variation in seawater chemistry: An explanation for the coupled secular variation in the mineralogies of marine limestones and potash evaporites over the past 600 m.y. Geology 1996, 24, 279–283. [Google Scholar] [CrossRef]
- Lowenstein, T.K.; Timofeeff, M.N.; Brennan, S.T.; Hardie, L.A.; Demicco, R.V. Oscillations in Phanerozoic seawater chemistry: Evidence from fluid Inclusions. Science 2001, 294, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.M.; Ries, J.B.; Hardie, L.A. Low-magnesium calcite produced by coralline algae in seawater of Late Cretaceous composition. Proc. Natl. Acad. Sci. USA 2002, 99, 15323–15326. [Google Scholar] [CrossRef] [PubMed]
- Burton, E.A.; Walter, L.M. The effects of pCO2 and temperature on magnesium incorporation in calcite in seawater and MgCl2-CaCl2 solutions. Geochim. Cosmochim. Acta 1991, 55, 777–785. [Google Scholar] [CrossRef]
- Feng, D.; Roberts, H.H.; Joye, S.B.; Heydari, E. Formation of low-magnesium calcite at cold seeps in an aragonite sea. Terra Nova 2014, 26, 150–156. [Google Scholar] [CrossRef]
- Zwicker, J.; Smrzka, D.; Himmler, T.; Monien, P.; Gier, S.; Goedert, J.L.; Peckmann, J. Rare earth elements as tracers for microbial activity and early diagenesis: A new perspective from carbonate cements of ancient methane-seep deposits. Chem. Geol. 2018, 501, 77–85. [Google Scholar] [CrossRef]
- Naehr, T.H.; Eichhubl, P.; Orphan, V.J.; Hovland, M.; Paull, C.K.; Ussler, W.; Lorenson, T.D.; Greene, H.G. Authigenic carbonate formation at hydrocarbon seeps in continental margin sediments: A comparative study. Deep Sea Res. Part II 2007, 54, 1268–1291. [Google Scholar] [CrossRef]
- Hu, Y.; Feng, D.; Peckmann, J.; Roberts, H.H.; Chen, D. New insights into cerium anomalies and mechanisms of trace metal enrichment in authigenic carbonate from hydrocarbon seeps. Chem. Geol. 2014, 381, 55–66. [Google Scholar] [CrossRef]
- Feng, D.; Peng, Y.; Bao, H.; Peckmann, J.; Roberts, H.H.; Chen, D. A carbonate-based proxy for sulfate-driven anaerobic oxidation of methane. Geology 2016, 44, 999–1002. [Google Scholar] [CrossRef]
- Joye, S.B.; MacDonald, I.R.; Montoya, J.P.; Peccini, M. Geophysical and geochemical signatures of Gulf of Mexico seafloor brines. Biogeosciences 2005, 2, 295–309. [Google Scholar] [CrossRef]
- Greinert, J.; Bohrmann, G.; Suess, E. Gas hydrate-associated carbonates and methane-venting at Hydrate Ridge: Classification, distribution, and origin of authigenic lithologies. Nat. Gas Hydrates Occur. Distrib. Detect. 2001, 124, 99–113. [Google Scholar] [CrossRef]
- Tong, H.; Feng, D.; Cheng, H.; Yang, S.; Wang, H.; Min, A.G.; Edwards, R.L.; Chen, Z.; Chen, D. Authigenic carbonates from seeps on the northern continental slope of the South China Sea: New insights into fluid sources and geochronology. Mar. Pet. Geol. 2013, 43, 260–271. [Google Scholar] [CrossRef]
- Gomberg, D.N.; Bonatti, E. High-magnesian calcite: Leaching of magnesium in the deep sea. Science 1970, 168, 1451–1453. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.J.; Goldstein, R.H. Cambrian sea water preserved as inclusions in marine low-magnesium calcite cement. Nature 1993, 362, 335–337. [Google Scholar] [CrossRef]
- Budd, D.A. Petrographic products of freshwater diagenesis in Holocene ooid sands, Schooner Cays, Bahamas. Carbonates Evaporites 1988, 3, 143–163. [Google Scholar] [CrossRef]
- Frank, T.D.; Lohmann, K.C. Diagenesis of fibrous magnesian calcite marine cement: Implications for the interpretation of δ18O and δ13C values of ancient equivalents. Geochim. Cosmochim. Acta 1996, 60, 2427–2436. [Google Scholar] [CrossRef]
- Salvador, A. Late Triassic- Jurassic paleogeography and origin of Gulf of Mexico basin. AAPG Bull. 1987, 71, 419–451. [Google Scholar] [CrossRef]
- Woodbury, H.O.; Murry, I.B.; Pickford, P.J. Pliocene and Pleistocene depocenters, outer continental shelf, Louisiana and Texas. AAPG Bull. 1973, 57, 2428–2439. [Google Scholar]
- Land, L.S.; Eustice, R.A.; Mack, L.E.; Horita, J. Reactivity of evaporites during burial: An example from the Jurassic of Alabama. Geochim. Cosmochim. Acta 1995, 59, 3765–3778. [Google Scholar] [CrossRef]
- Sassen, R.; Cole, G.A.; Drozd, R.; Roberts, H.H. Oligocene to Holocene hydrocarbon migration and salt-dome carbonates, northern Gulf of Mexico. Mar. Pet. Geol. 1994, 11, 55–65. [Google Scholar] [CrossRef]
- Stueber, A.M.; Walter, L.M. Origin and chemical evolution of formation waters from Silurian-Devonian strata in the Illinois basin, USA. Geochim. Cosmochim. Acta 1991, 55, 309–325. [Google Scholar] [CrossRef]
- Aharon, P.; Roberts, H.H.; Snelling, R. Submarine venting of brines in the deep Gulf of Mexico: observations and geochemistry. Geology 1992, 20, 483–486. [Google Scholar] [CrossRef]
- Land, L.S. NaCaCl saline formation waters, Frio Formation (Oligocene), south Texas, USA: Products of diagenesis. Geochim. Cosmochim. Acta 1995, 59, 2163–2174. [Google Scholar] [CrossRef]
- Cohen, D.; Person, M.; Wang, P.; Gable, C.W.; Hutchinson, D.; Marksamer, A.; Dugan, B.; Kooi, H.; Groen, K.; Lizarralde, D.; et al. Origin and extent of fresh paleowaters on the Atlantic continental shelf, USA. Ground Water 2010, 48, 143–158. [Google Scholar] [CrossRef]
- Posey, H.H.; Kyle, J.R. Fluid-rock interactions in the salt dome environment: An introduction and review. Chem. Geol. 1988, 74, 1–24. [Google Scholar] [CrossRef]
- Beckman, J.D.; Williamson, A.K. Salt-dome Locations in the Gulf Coastal Plain, South-Central United States; Center for Integrated Data Analytics Wisconsin Science Center: Austin, TX, USA, 1990. [Google Scholar]
- Roberts, H.H.; Sassen, R.; Carney, R.; Aharon, P. Carbonate buildups on the continental slope off central Louisiana. Offshore Technol. Conf. 1989, 5953, 655–662. [Google Scholar]
- Christopher, K.S.; Joel, W.S. Geometry and development of the salt withdrawal basin in Ship Shoal Block 318 and vicinity. Gulf Coast Assoc. Geol. Soc. Trans. 1998, 48, 173–180. [Google Scholar]
- Cook, D.; D’Onfro, P. Jolliet field thrust fault structure and stratigraphy Green Canyon Block 184, Offshore Louisiana. Gulf Coast Assoc. Geol. Soc. Trans. 1991, 41, 100–121. [Google Scholar]
- Roberts, H.H.; Aharon, P. Hydrocarbon-derived carbonate buildups of the northern Gulf-of-Mexico continental-slope—A review of submersible investigations. Geo-Mar. Lett. 1994, 14, 135–148. [Google Scholar] [CrossRef]
- Sassen, R.; Roberts, H.H.; Aharon, P.; Larkin, J.; Chinn, E.W.; Carney, R. Chemosynthetic bacterial mats at cold hydrocarbon seeps, Gulf of Mexico continental slope. Org. Geochem. 1993, 20, 77–89. [Google Scholar] [CrossRef]
- Goldsmith, J.R.; Graf, D.L.; Heard, H.L. Lattice constants of the calcium-magnesium carbonates. Am. Mineral. 1961, 46, 453–457. [Google Scholar]
- McLennan, S.M. Rare earth elements in sedimentary rocks: Influence of provenance and sedimentary processes. Rev. Mineral. 1989, 21, 169–200. [Google Scholar]
- Lamb, A.L.; Wilson, G.P.; Leng, M.J. A review of coastal palaeoclimate and relative sea-level reconstructions using δ13C and C/N ratios in organic material. Earth Sci. Rev. 2006, 75, 29–57. [Google Scholar] [CrossRef]
- Paytan, A.; Kastner, M.; Martin, E.E.; Macdougall, J.D.; Herbert, T. Marine barite as a monitor of seawater strontium isotope composition. Nature 1993, 366, 445–449. [Google Scholar] [CrossRef]
- Sandberg, P.A. An oscillating trend in Phanerozoic non-skeletal carbonate mineralogy. Nature 1983, 305, 19–22. [Google Scholar] [CrossRef]
- Denison, R.E.; Koepnick, R.B.; Burke, W.H.; Hetherington, E.A. Construction of the Cambrian and Ordovician seawater 87Sr/86Sr curve. Chem. Geol. 1998, 152, 325–340. [Google Scholar] [CrossRef]
- Brand, U. Carbon, oxygen and strontium isotopes in Paleozoic carbonate components: An evaluation of original seawater-chemistry proxies. Chem. Geol. 2004, 204, 23–44. [Google Scholar] [CrossRef]
- Savard, M.M.; Beauchamp, B.; Veizer, J. Significance of aragonite cements around Cretaceous marine methane seeps. J. Sediment. Res. 1996, 66, 430–438. [Google Scholar] [CrossRef]
- Buggisch, W.; Krumm, S. Palaeozoic cold seep carbonates from Europe and North Africa—An integrated isotopic and geochemical approach. Facies 2005, 51, 566–583. [Google Scholar] [CrossRef]
- Beauchamp, B.; Savard, M. Cretaceous chemosynthetic carbonate mounds in the Canadian Arctic. Palaios 1992, 7, 434–450. [Google Scholar] [CrossRef]
- Peckmann, J.; Campbell, K.A.; Walliser, O.H.; Reitner, J. A Late Devonian hydrocarbon-seep deposit dominated by dimerelloid brachiopods, Morocco. Palaios 2007, 22, 114–122. [Google Scholar] [CrossRef]
- Denison, R.E.; Koepnick, R.B.; Fletcher, A.; Howell, M.W.; Callaway, W.S. Criteria for the retention of original seawater 87Sr/86Sr in ancient shelf limestones. Chem. Geol. 1994, 112, 131–143. [Google Scholar] [CrossRef]
- Morse, J.W.; Wang, Q.; Tsio, M.Y. Influences of temperature and Mg:Ca ratio on CaCO3 precipitates from seawater. Geology 1997, 25, 85–87. [Google Scholar] [CrossRef]
- Fu, B.; Aharon, P. Sources of hydrocarbon-rich fluids advecting on the seafloor in the northern Gulf of Mexico. Gulf Coast Assoc. Geol. Soc. Trans. 1998, 48, 73–82. [Google Scholar]
- Kharaka, Y.K.; Hanor, J.S. Deep fluids in sedimentary basins. In Treatise on Geochemistry, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 471–515. [Google Scholar] [CrossRef]
- Bian, Y.; Feng, D.; Roberts, H.H.; Chen, D. Tracing the evolution of seep fluids from authigenic carbonates: Green Canyon, northern Gulf of Mexico. Mar. Pet. Geol. 2013, 44, 71–81. [Google Scholar] [CrossRef]
- Himmler, T.; Birgel, D.; Bayon, G.; Pape, T.; Ge, L.; Bohrmann, G.; Peckmann, J. Formation of seep carbonates along the Makran convergent margin, northern Arabian Sea and a molecular and isotopic approach to constrain the carbon isotopic composition of parent methane. Chem. Geol. 2015, 415, 102–117. [Google Scholar] [CrossRef]
- Birgel, D.; Feng, D.; Roberts, H.H.; Peckmann, J. Changing redox conditions at cold seeps as revealed by authigenic carbonates from Alaminos Canyon, northern Gulf of Mexico. Chem. Geol. 2011, 285, 82–96. [Google Scholar] [CrossRef]
- Kim, S.T.; O’Neil, J.R. Equilibrium and nonequilibrium oxygen isotope effects in synthetic carbonates. Geochim. Cosmochim. Acta 1997, 61, 3461–3475. [Google Scholar] [CrossRef]
- Tarutani, T.; Clayton, R.N.; Mayeda, T.K. The effect of polymorphism and magnesium substitution on oxygen isotope fractionation between calcium carbonate and water. Geochim. Cosmochim. Acta 1969, 33, 987–996. [Google Scholar] [CrossRef]
- Cobbold, P.R.; Zanella, A.; Rodrigues, N.; Løseth, H. Bedding-parallel fibrous veins (beef and cone-in-cone): Worldwide occurrence and possible significance in terms of fluid overpressure, hydrocarbon generation and mineralization. Mar. Pet. Geol. 2013, 43, 1–20. [Google Scholar] [CrossRef]
- Kershaw, S.; Guo, L. Beef and cone-in-cone calcite fibrous cements associated with the end-Permian and end-Triassic mass extinctions: Reassessment of processes of formation. J. Palaeogeogr. 2016, 5, 28–42. [Google Scholar] [CrossRef]
- Greene, S.E.; Bottjer, D.J.; Corsetti, F.A.; Berelson, W.M.; Zonneveld, J.P. A subseafloor carbonate factory across the Triassic-Jurassic transition. Geology 2012, 40, 1043–1046. [Google Scholar] [CrossRef]
- Omosanya, K.O.; Alves, T.M. Ramps and flats of mass-transport deposits (MTDs) as markers of seafloor strain on the flanks of rising diapirs (Espírito Santo Basin, SE Brazil). Mar. Geol. 2013, 340, 82–97. [Google Scholar] [CrossRef]
- Faure, G. Principles of Isotope Geology, 2nd ed.; Smith and Wyllie Intermediate Geology Series; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Peckmann, J.; Reimer, A.; Luth, U.; Luth, C.; Hansen, B.T.; Heinicke, C.; Hoefs, J.; Reitner, J. Methane-derived carbonates and authigenic pyrite from the northwestern Black Sea. Mar. Geol. 2001, 177, 129–150. [Google Scholar] [CrossRef]
- Osborn, S.G.; McIntosh, J.C.; Hanor, J.S.; Biddulph, D. Iodine-129, 87Sr/ 86Sr, and trace elemental geochemistry of northern Appalachian Basin brines: Evidence for basinal-scale fluid migration and clay mineral diagenesis. Am. J. Sci. 2012, 312, 263–287. [Google Scholar] [CrossRef]
- Kim, J.H.; Torres, M.E.; Haley, B.A.; Ryu, J.S.; Park, M.H.; Hong, W.L.; Choi, J. Marine silicate weathering in the anoxic sediment of the Ulleung Basin: Evidence and consequences. Geochem. Geophy. Geosy. 2016, 17, 3437–3453. [Google Scholar] [CrossRef]
- Li, Y.P.; Jiang, S.Y. Sr isotopic compositions of the interstitial water and carbonate from two basins in the Gulf of Mexico: Implications for fluid flow and origin. Chem. Geol. 2016, 439, 43–51. [Google Scholar] [CrossRef]
- Posey, H.H.; Richard Kyle, J.; Jackson, T.J.; Hurst, S.D.; Price, P.E. Multiple fluid components of salt diapirs and salt dome cap rocks, Gulf Coast, U.S.A. Appl. Geochem. 1987, 2, 523–534. [Google Scholar] [CrossRef]
- Land, L.S.; Kupecz, J.A.; Mack, L.E. Louann salt geochemistry (Gulf of Mexico sedimentary basin, U.S.A.): A preliminary synthesis. Chem. Geol. 1988, 74, 25–35. [Google Scholar] [CrossRef]
- Ge, L.; Jiang, S.Y.; Swennen, R.; Yang, T.; Yang, J.H.; Wu, N.Y.; Liu, J.A.; Chen, D.H. Chemical environment of cold seep carbonate formation on the northern continental slope of South China Sea: Evidence from trace and rare earth element geochemistry. Mar. Geol. 2010, 277, 21–30. [Google Scholar] [CrossRef]
- Kim, J.H.; Torres, M.E.; Haley, B.A.; Kastner, M.; Pohlman, J.W.; Riedel, M.; Lee, Y.J. The effect of diagenesis and fluid migration on rare earth element distribution in pore fluids of the northern Cascadia accretionary margin. Chem. Geol. 2012, 291, 152–165. [Google Scholar] [CrossRef]
- Prikryl, J.D.; Posey, H.H.; Kyle, J.R. A petrographic and geochemical model for the origin of calcite cap rock at Damon Mound Salt. Chem. Geol. 1988, 74, 67–97. [Google Scholar] [CrossRef]
- Anderson, T.F.; Arthur, M.A. Stable isotopes of oxygen and carbon and their application to sedimentologic and paleoenvironmental problems. In Stable Isotopes in Sedimentary Geology; SEPM (Society for Sedimentary Geology): Dallas, TX, USA, 1983; pp. 1–151. [Google Scholar] [CrossRef]
- Himmler, T.; Brinkmann, F.; Bohrmann, G.; Peckmann, J. Corrosion patterns of seep-carbonates from the eastern Mediterranean Sea. Terra Nova 2011, 23, 206–212. [Google Scholar] [CrossRef]
- Feng, D.; Peckmann, J.; Li, N.; Kiel, S.; Qiu, J.W.; Liang, Q.; Carney, R.S.; Peng, Y.; Tao, J.; Chen, D. The stable isotope fingerprint of chemosymbiosis in the shell organic matrix of seep-dwelling bivalves. Chem. Geol. 2018, 479, 241–250. [Google Scholar] [CrossRef]
- Guan, H.; Feng, D.; Birgel, D.; Peckmann, J.; Roberts, H.H.; Wu, N.; Chen, D. Lipid biomarker patterns reflect different formation environments of mussel- and tubeworm-dominated seep carbonates from the Gulf of Mexico (Atwater Valley and Green Canyon). Chem. Geol. 2019, 505, 36–47. [Google Scholar] [CrossRef]
- Lepot, K.; Williford, K.H.; Philippot, P.; Thomazo, C.; Ushikubo, T.; Kitajima, K.; Mostefaoui, S.; Valley, J.W. Extreme 13C-depletions and organic sulfur content argue for S-fueled anaerobic methane oxidation in 2.72 Ga old stromatolites. Geochim. Cosmochim. Acta 2019, 244, 522–547. [Google Scholar] [CrossRef]
- Caesar, K.H.; Kyle, J.R.; Lyons, T.W.; Tripati, A.; Loyd, S.J. Carbonate formation in salt dome cap rocks by microbial anaerobic oxidation of methane. Nat. Commun. 2019, 10, 808. [Google Scholar] [CrossRef]
- Aharon, P.; Graber, E.R.; Roberts, H.H. Dissolved carbon and δ13C anomalies in the water column caused by hydrocarbon seeps on the northwestern Gulf of Mexico slope. Geo-Mar. Lett. 1992, 12, 33–40. [Google Scholar] [CrossRef]
- Welch, S.E. Source(s) of Salinity in the Mississippi River Alluvial Aquifer, Iberville Parish, Louisiana. Master’s Thesis, Louisiana State University, Baton Rouge, LA, USA, 2009. [Google Scholar]
- Kennett, J.P.; Shackleton, N.J. Laurentide ice sheet meltwater recorded in Gulf of Mexico deep-sea cores. Science 1975, 188, 147–150. [Google Scholar] [CrossRef]
- Palmer, M.R.; Edmond, J.M. The strontium isotope budget of the modern ocean. Earth Planet. Sci. Lett. 1989, 92, 11–26. [Google Scholar] [CrossRef]
- Xu, Y.; Marcantonio, F. Strontium isotope variations in the lower Mississippi River and its estuarine mixing zone. Mar. Chem. 2007, 105, 118–128. [Google Scholar] [CrossRef]
- McManus, K.M.; Hanor, J.S. Calcite and iron sulfide cementation of Miocene sediments flanking the West Hackberry salt dome, southwest Louisiana, U.S.A. Chem. Geol. 1988, 74, 99–112. [Google Scholar] [CrossRef]
- Reilly, J.F., Jr.; MacDonald, I.R.; Biegert, E.K.; Brooks, J.M. Geologic controls on the distribution of chemosynthetic communities in the Gulf of Mexico. In Hydrocarbon Migration and Its Near-Surface Expression. AAPG Memoir No. 66; AAPG: Tulsa, OK, USA, 1996. [Google Scholar]
- Lu, Y.; Liu, Y.; Sun, X.; Lin, Z.; Xu, L.; Lu, H.; Hao, X.; Peckmann, J. Intensity of methane seepage reflected by relative enrichment of heavy magnesium isotopes in authigenic carbonates: A case study from the South China Sea. Deep Sea Res. Part I 2017, 129, 10–21. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
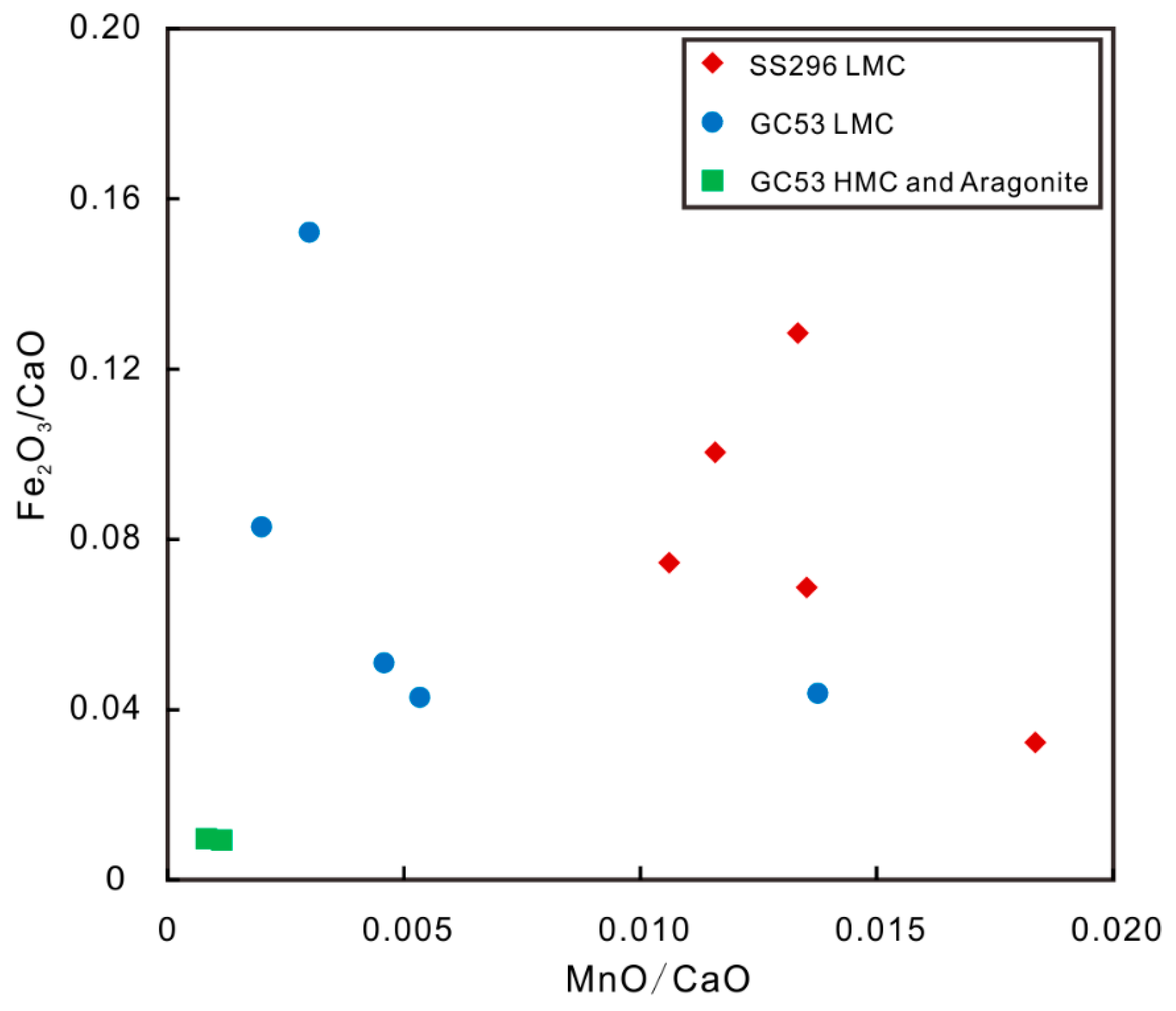{kind=link}
| Sample No. | Qtz | LMC | MgCO3 (%) | HMC | MgCO3 (%) | Arag. | Dolo. | Sd |
|---|---|---|---|---|---|---|---|---|
| SS296 | ||||||||
| 3110R1 | 16 | 82 | 4 | 18 | ||||
| 3110R2 | 12 | 100 | 2 | |||||
| 3110R3 | 14 | 93 | 5 | 7 | ||||
| GC53 | ||||||||
| 88–22 2-a | 6 | 89 | 1 | 11 | ||||
| 89-1 1-a | 12 | 98 | 1 | 2 | ||||
| 89 D1 S2 | 5 | 61 | 5 | 39 | ||||
| 89 D1 S3 | 7 | 100 | 2 | |||||
| 3112R1 | 90 | 10 | 10 | |||||
| 3112R3 | 10 | 45 | 16 | 55 | ||||
| Sample | SS296 | GC53 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3110R1(1) | 3110R1(2) | 3110R2 | 3110R3(1) | 3110R3(2) | 88-22 2-a | 89-1 1-a | 89 D1 S2(1) | 89 D1 S2(2) | 89 D1 S3 | 3112R1 | 3112R3 | |
| Na2O | 0.66 | 0.93 | 0.65 | 0.77 | 0.78 | 0.63 | 0.71 | 0.60 | 0.69 | 0.58 | 0.75 | 0.77 |
| MgO | 2.23 | 1.87 | 1.38 | 1.43 | 1.56 | 1.23 | 1.32 | 3.90 | 1.66 | 1.15 | 2.59 | 2.98 |
| Al2O3 | 5.40 | 4.65 | 5.01 | 5.47 | 4.90 | 4.39 | 5.90 | 3.45 | 4.78 | 3.98 | 0.58 | 1.71 |
| P2O5 | 0.19 | 0.37 | 0.16 | 0.13 | 0.12 | 3.15 | 0.61 | 0.62 | 0.17 | 0.50 | 0.16 | 0.11 |
| K2O | 1.27 | 1.20 | 1.14 | 1.28 | 1.21 | 0.97 | 1.27 | 0.76 | 1.04 | 0.91 | 0.15 | 0.39 |
| CaO | 26.93 | 20.81 | 35.96 | 32.62 | 34.86 | 37.52 | 32.55 | 23.39 | 35.59 | 38.38 | 41.53 | 43.91 |
| TiO2 | 0.25 | 0.23 | 0.22 | 0.25 | 0.22 | 0.18 | 0.24 | 0.17 | 0.22 | 0.16 | 0.03 | 0.07 |
| MnO | 0.34 | 0.26 | 0.44 | 0.34 | 0.29 | 0.16 | 0.13 | 0.31 | 0.09 | 0.41 | 0.05 | 0.07 |
| Fe2O3 | 8.87 | 8.84 | 1.90 | 3.90 | 3.78 | 5.06 | 5.15 | 23.72 | 5.91 | 2.71 | 5.96 | 1.07 |
| Sample | SS296 | GC53 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3110R1(1) | 3110R1(2) | 3110R2 | 3110R3(1) | 3110R3(2) | 88-22 2-a | 89-1 1-a | 89 D1 S2(1) | 89 D1 S2(2) | 89 D1 S3 | 3112R1 | 3112R3 | |
| La | 14.90 | 10.30 | 11.80 | 12.80 | 16.20 | 7.66 | 16.20 | 5.51 | 5.31 | 11.20 | 0.94 | 3.88 |
| Ce | 30.10 | 19.70 | 25.50 | 22.50 | 27.60 | 14.50 | 26.10 | 9.62 | 11.10 | 20.90 | 1.98 | 6.36 |
| Pr | 3.22 | 2.10 | 2.85 | 2.28 | 2.76 | 1.56 | 2.66 | 0.99 | 1.20 | 2.24 | 0.23 | 0.89 |
| Nd | 13.00 | 7.78 | 11.30 | 8.79 | 10.60 | 6.28 | 9.95 | 4.29 | 5.00 | 8.34 | 0.95 | 3.82 |
| Sm | 2.75 | 1.43 | 2.80 | 1.77 | 1.84 | 1.31 | 1.96 | 0.89 | 1.11 | 1.69 | 0.21 | 0.68 |
| Eu | 0.59 | 0.30 | 0.63 | 0.35 | 0.39 | 0.25 | 0.41 | 0.19 | 0.23 | 0.38 | 0.11 | 0.16 |
| Gd | 2.94 | 1.65 | 3.08 | 1.89 | 2.07 | 1.38 | 2.22 | 0.95 | 1.10 | 1.89 | 0.21 | 0.75 |
| Tb | 0.40 | 0.20 | 0.48 | 0.26 | 0.28 | 0.21 | 0.29 | 0.13 | 0.16 | 0.26 | 0.04 | 0.13 |
| Dy | 2.07 | 0.99 | 3.23 | 1.28 | 1.52 | 1.08 | 1.57 | 0.68 | 0.91 | 1.40 | 0.18 | 0.69 |
| Ho | 0.38 | 0.20 | 0.68 | 0.28 | 0.32 | 0.22 | 0.33 | 0.13 | 0.17 | 0.27 | 0.03 | 0.14 |
| Er | 1.02 | 0.50 | 1.93 | 0.75 | 0.94 | 0.72 | 1.00 | 0.33 | 0.46 | 0.88 | 0.10 | 0.38 |
| Tm | 0.11 | 0.06 | 0.28 | 0.08 | 0.12 | 0.10 | 0.12 | 0.04 | 0.06 | 0.11 | 0.01 | 0.05 |
| Yb | 0.76 | 0.33 | 1.85 | 0.55 | 0.73 | 0.61 | 0.78 | 0.25 | 0.34 | 0.74 | 0.08 | 0.30 |
| Lu | 0.11 | 0.04 | 0.27 | 0.08 | 0.11 | 0.09 | 0.14 | 0.04 | 0.05 | 0.11 | 0.01 | 0.04 |
| Y | 13.60 | 7.36 | 21.10 | 9.74 | 12.50 | 9.11 | 14.80 | 5.00 | 5.43 | 10.90 | 1.33 | 4.85 |
| ∑REE | 72.35 | 45.58 | 66.67 | 53.66 | 65.48 | 35.97 | 63.71 | 24.03 | 27.19 | 50.41 | 5.06 | 18.27 |
| MnO | 0.36 | 0.24 | 0.66 | 0.44 | 0.37 | 0.20 | 0.15 | 0.07 | 0.07 | 0.53 | 0.03 | 0.05 |
| Fe2O3 | 3.46 | 2.09 | 1.16 | 2.24 | 2.60 | 1.61 | 1.66 | 3.56 | 2.95 | 1.68 | 0.40 | 0.41 |
| Sr | 288 | 219 | 379 | 252 | 278 | 512 | 350 | 185 | 245 | 373 | 3300 | 4010 |
| Ba | 133 | 59 | 6 | 8 | 8 | 100 | 53 | 69 | 76 | 22 | 693 | 166 |
| Sr/Mn | 0.10 | 0.12 | 0.07 | 0.07 | 0.10 | 0.33 | 0.30 | 0.34 | 0.45 | 0.09 | 12.60 | 10.31 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.; Gong, S.; Li, N.; Birgel, D.; Peckmann, J.; Jin, M.; Cheng, M.; Roberts, H.H.; Chen, D.; Feng, D. Formation of Authigenic Low-Magnesium Calcite from Sites SS296 and GC53 of the Gulf of Mexico. Minerals 2019, 9, 251. https://doi.org/10.3390/min9040251
Huang H, Gong S, Li N, Birgel D, Peckmann J, Jin M, Cheng M, Roberts HH, Chen D, Feng D. Formation of Authigenic Low-Magnesium Calcite from Sites SS296 and GC53 of the Gulf of Mexico. Minerals. 2019; 9(4):251. https://doi.org/10.3390/min9040251
Chicago/Turabian StyleHuang, Huiwen, Shanggui Gong, Niu Li, Daniel Birgel, Jörn Peckmann, Meng Jin, Ming Cheng, Harry H. Roberts, Duofu Chen, and Dong Feng. 2019. "Formation of Authigenic Low-Magnesium Calcite from Sites SS296 and GC53 of the Gulf of Mexico" Minerals 9, no. 4: 251. https://doi.org/10.3390/min9040251
APA StyleHuang, H., Gong, S., Li, N., Birgel, D., Peckmann, J., Jin, M., Cheng, M., Roberts, H. H., Chen, D., & Feng, D. (2019). Formation of Authigenic Low-Magnesium Calcite from Sites SS296 and GC53 of the Gulf of Mexico. Minerals, 9(4), 251. https://doi.org/10.3390/min9040251