Enantioselective Catalytic Synthesis of N-alkylated Indoles


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
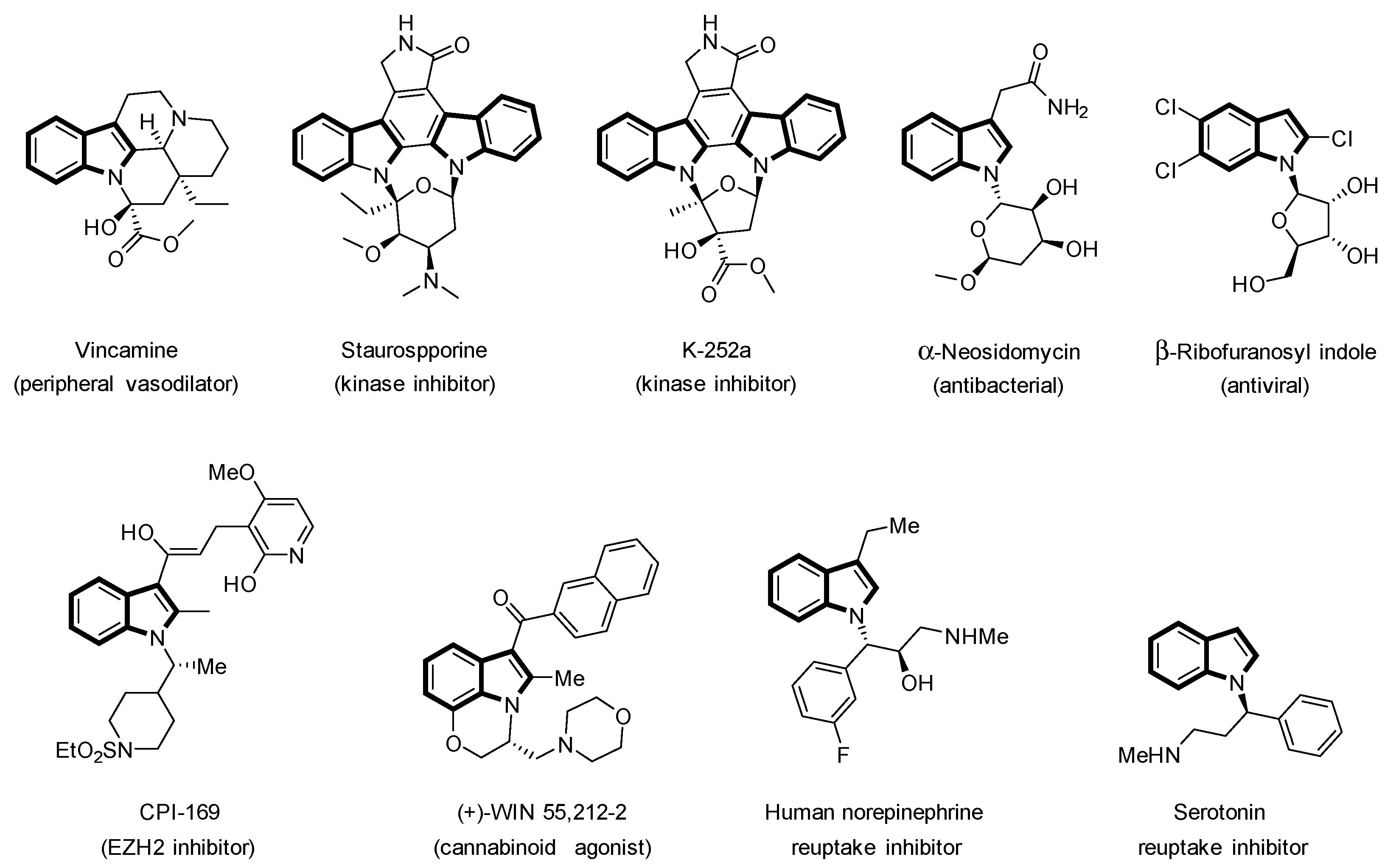
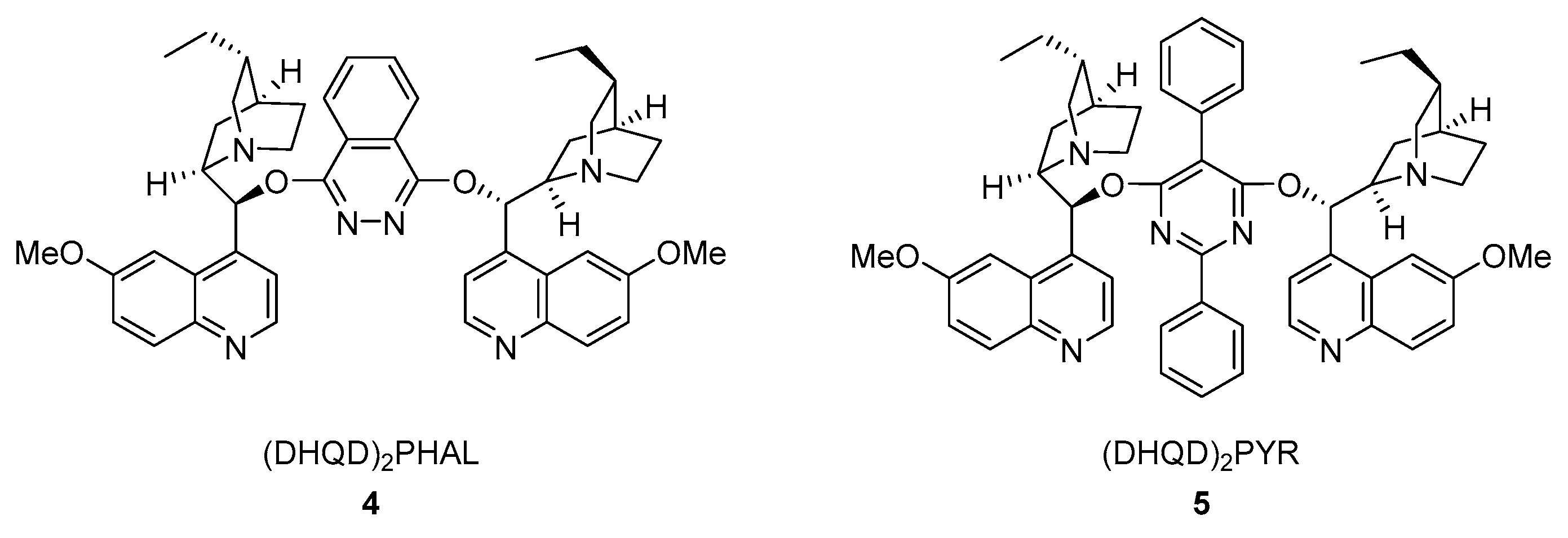
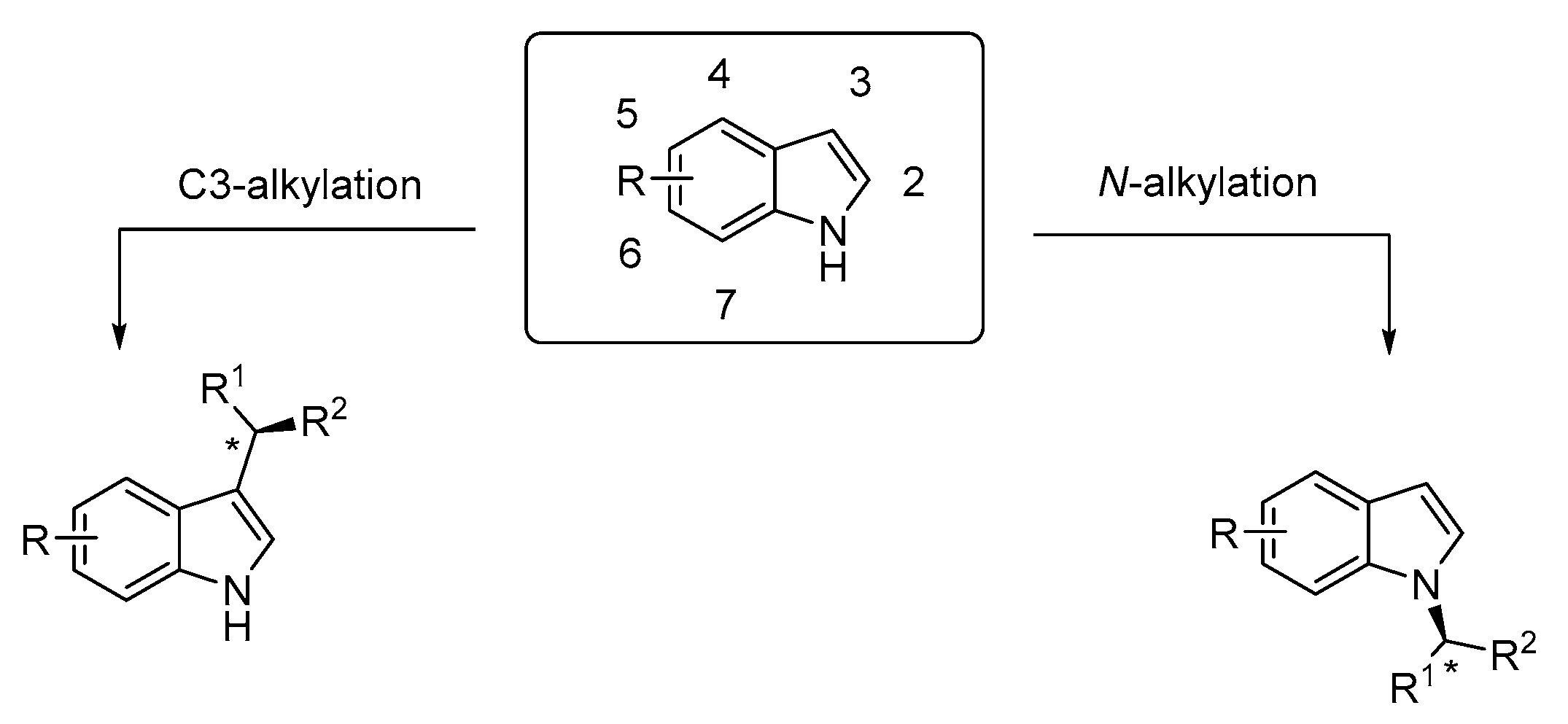
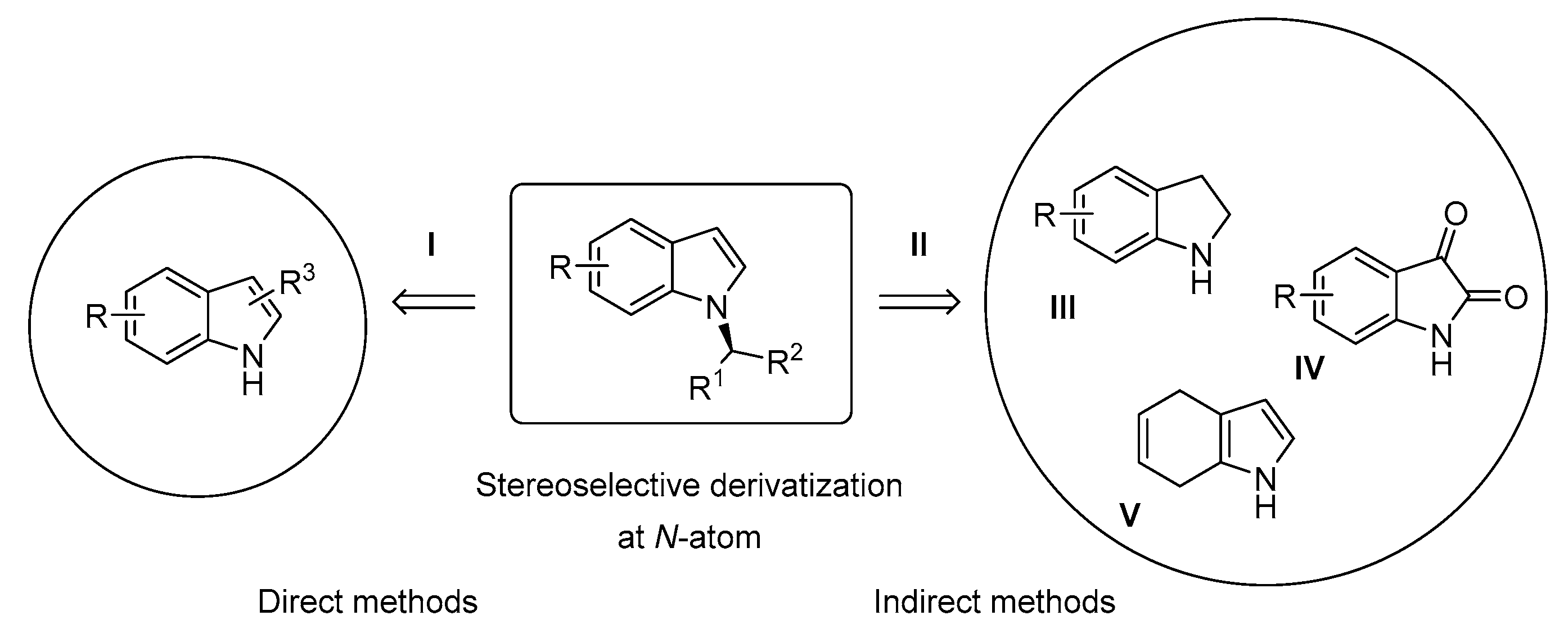
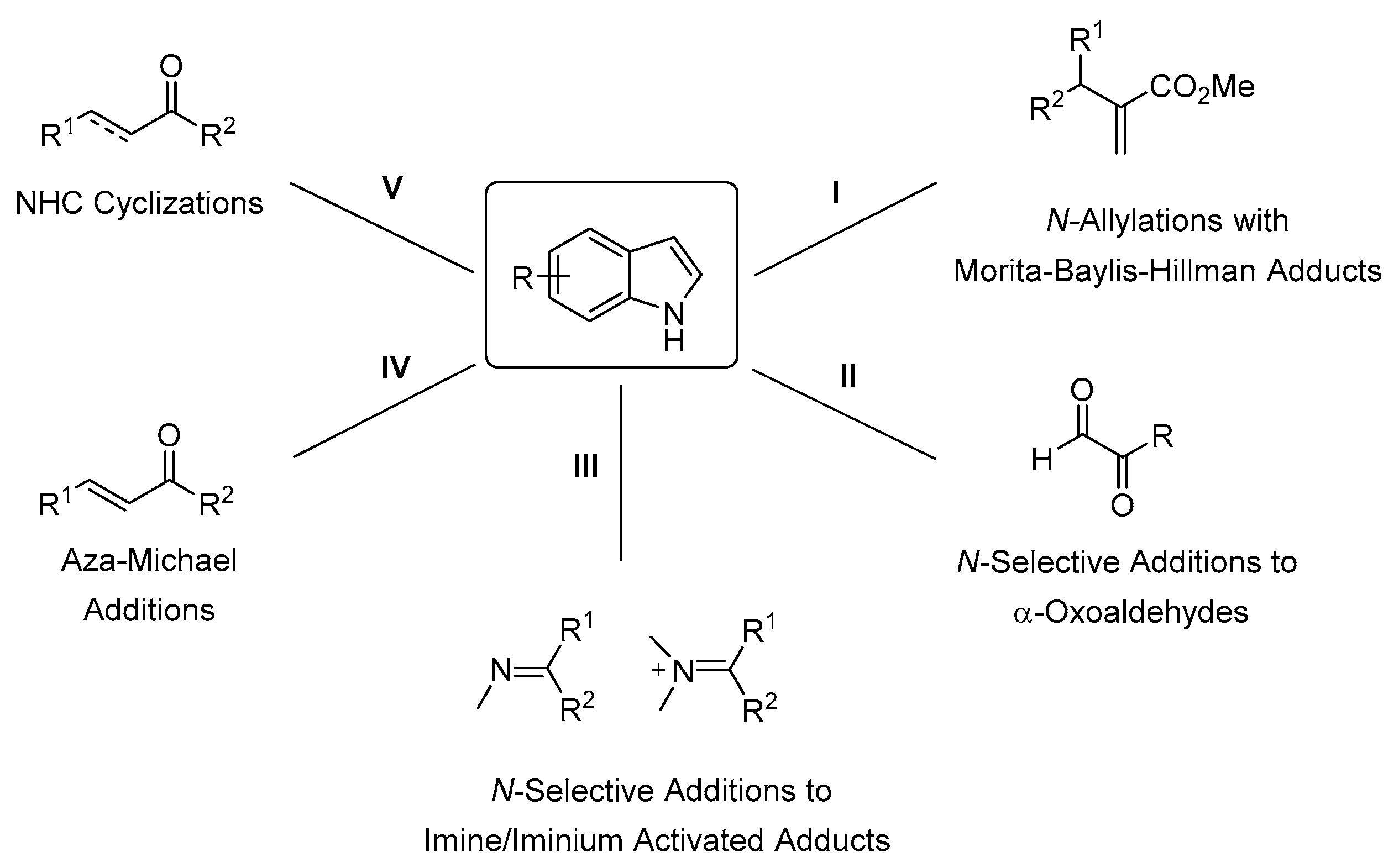
1. Introduction
2. Direct Organocatalytic Methods
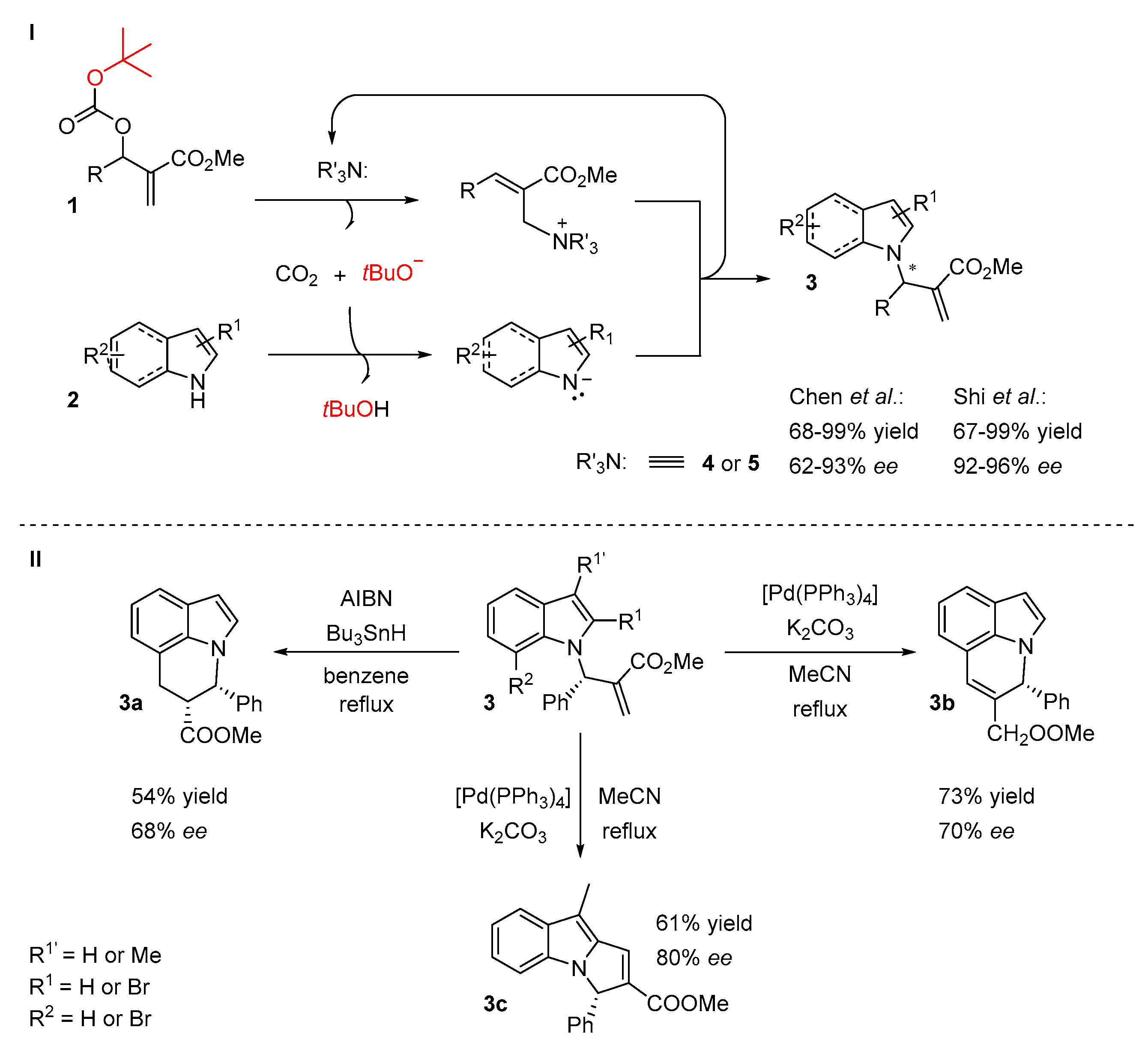
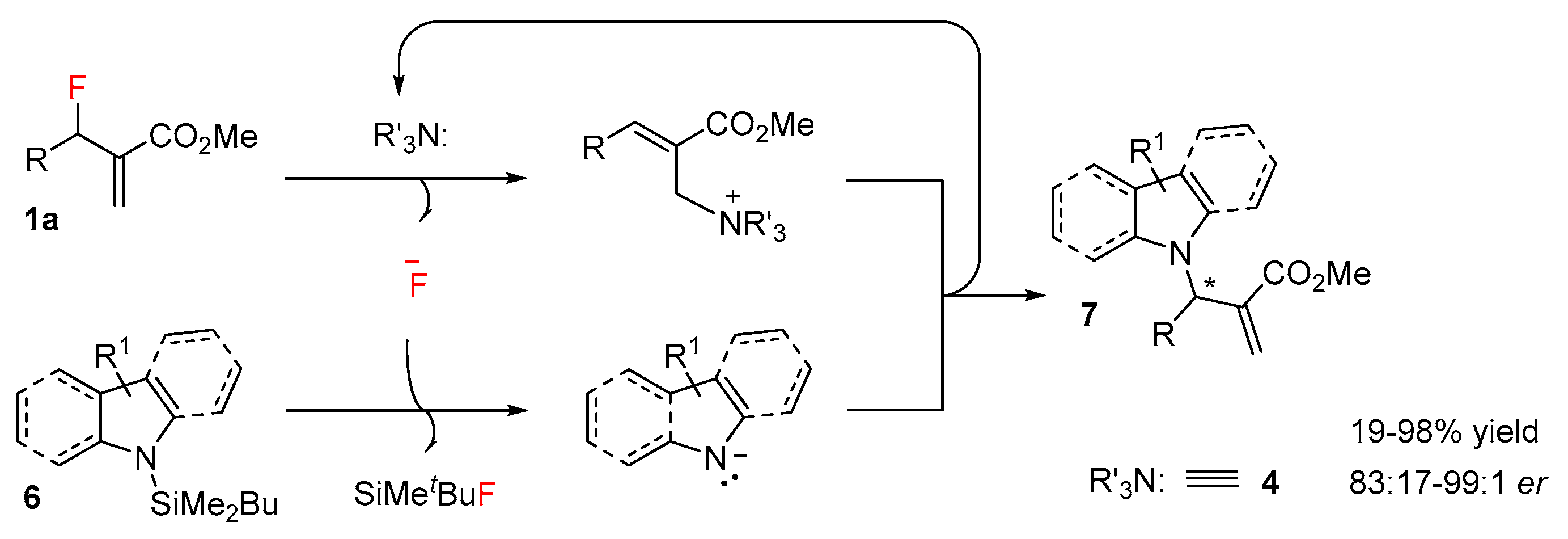
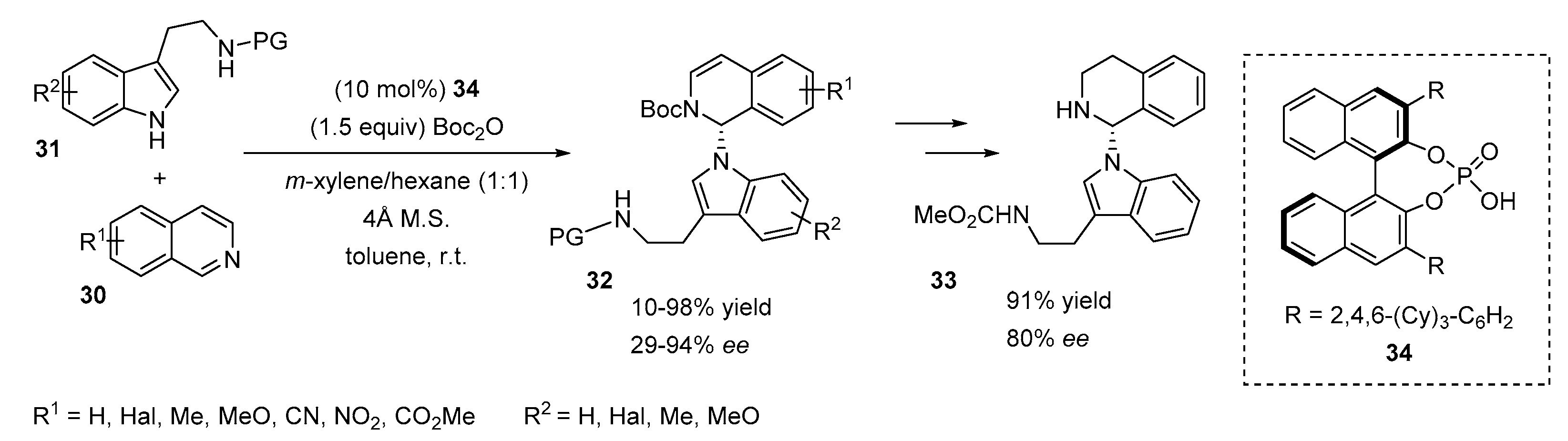
2.1. N-Allylations with Morita-Baylis-Hillman Adducts
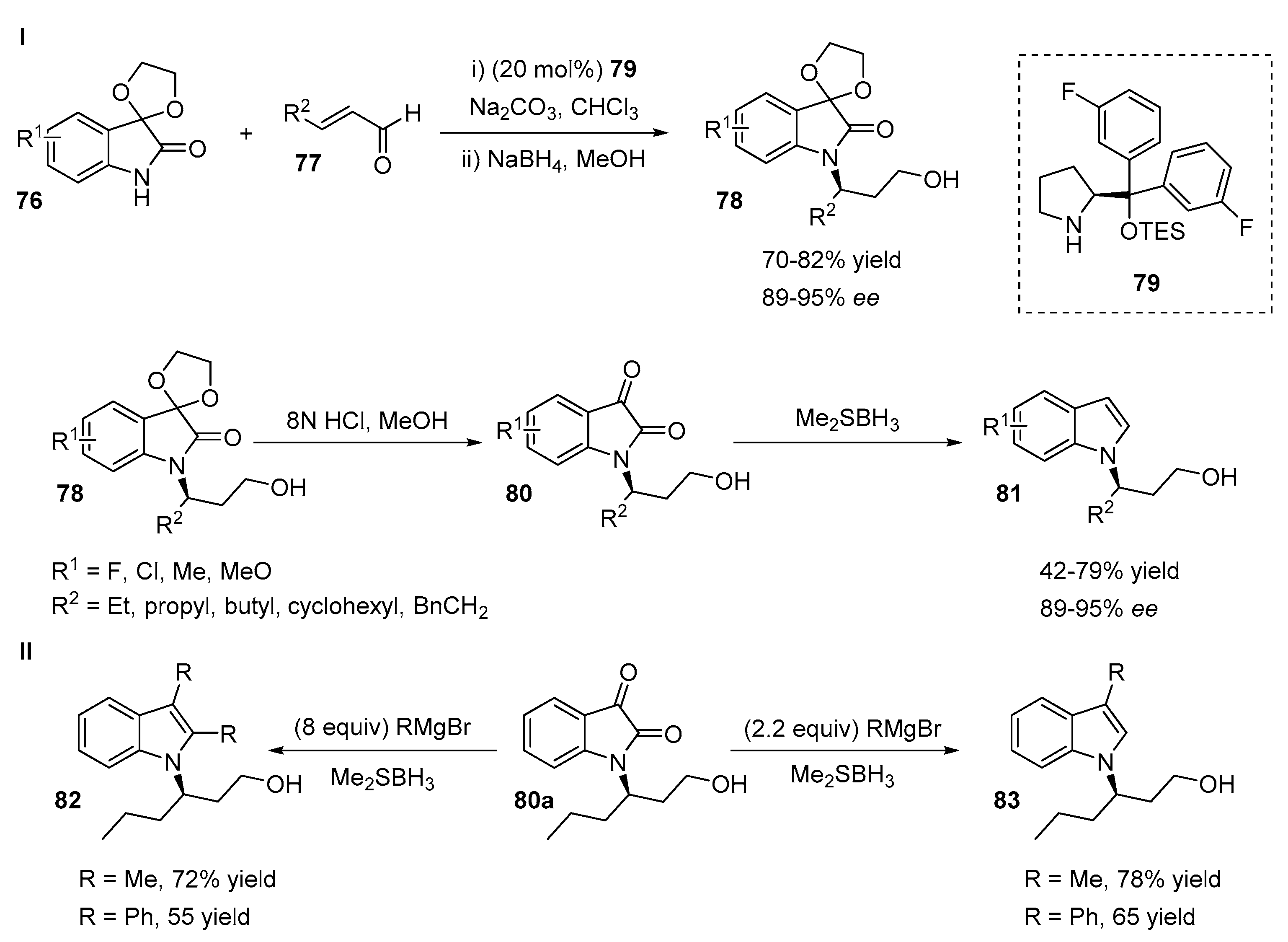
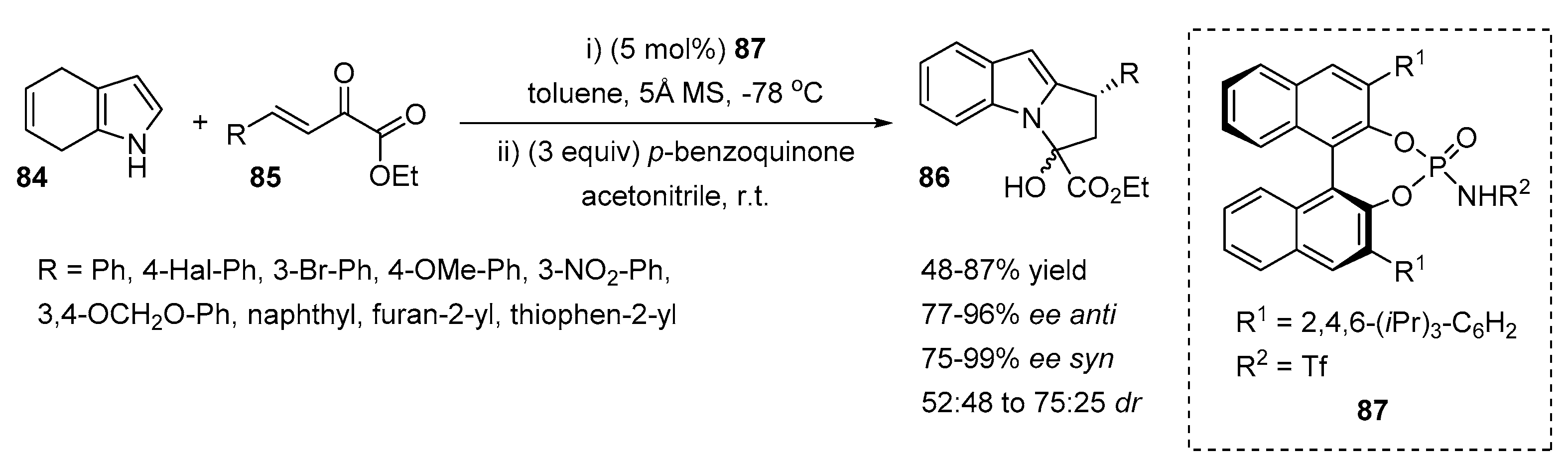
2.2. N-Selective Additions to α-Oxoaldehydes
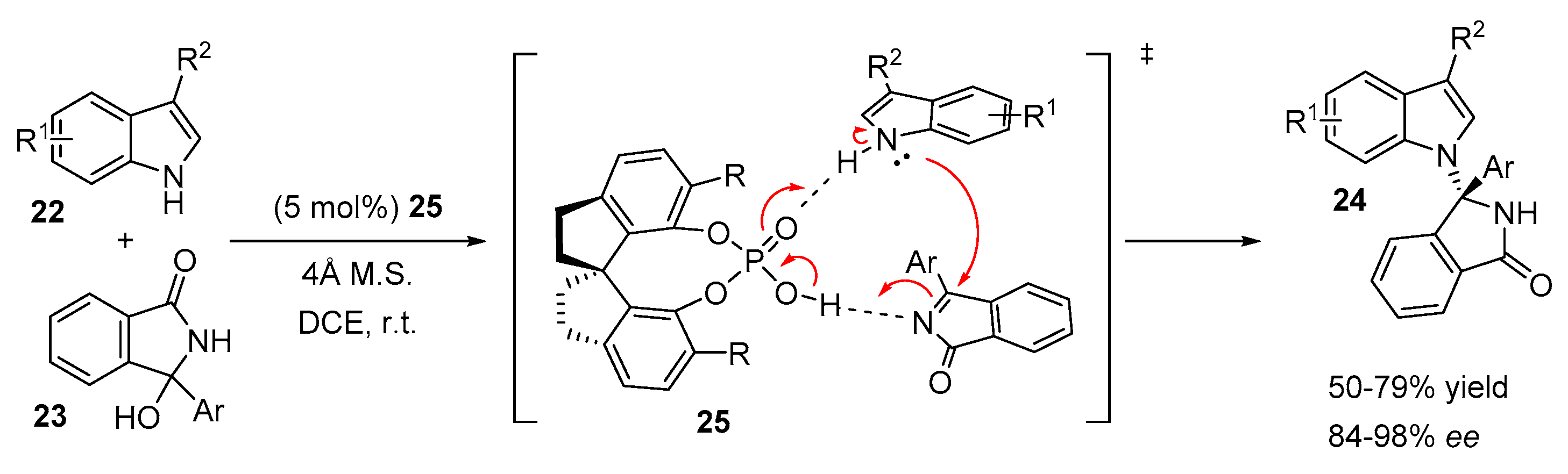
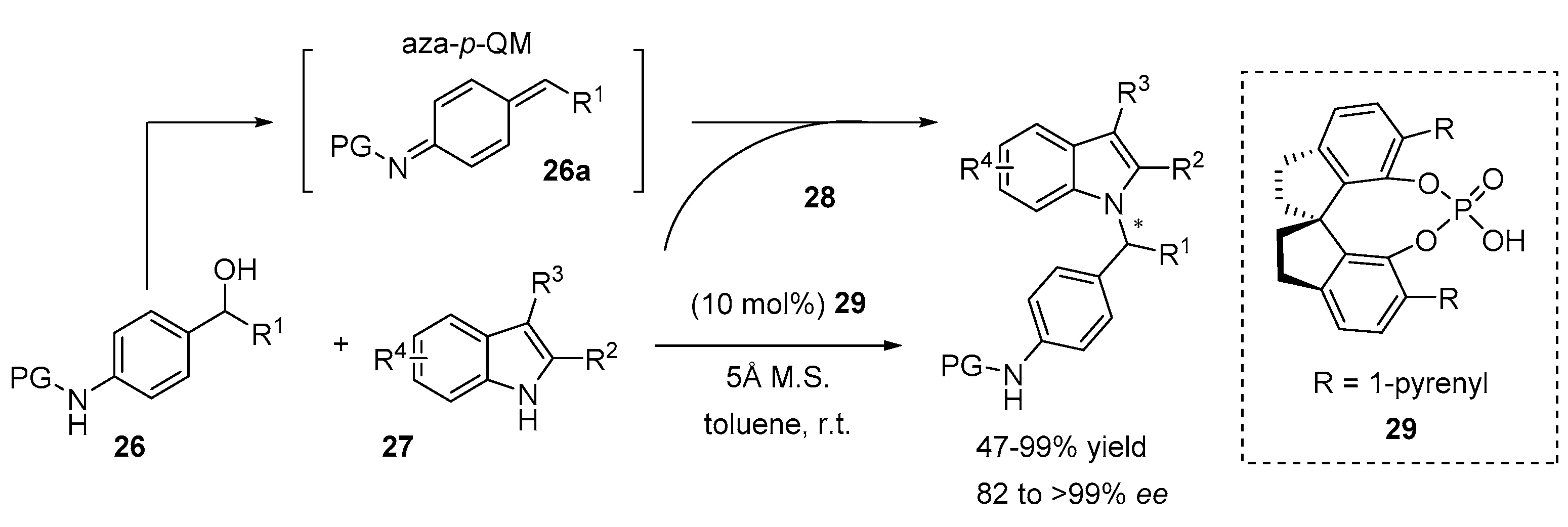
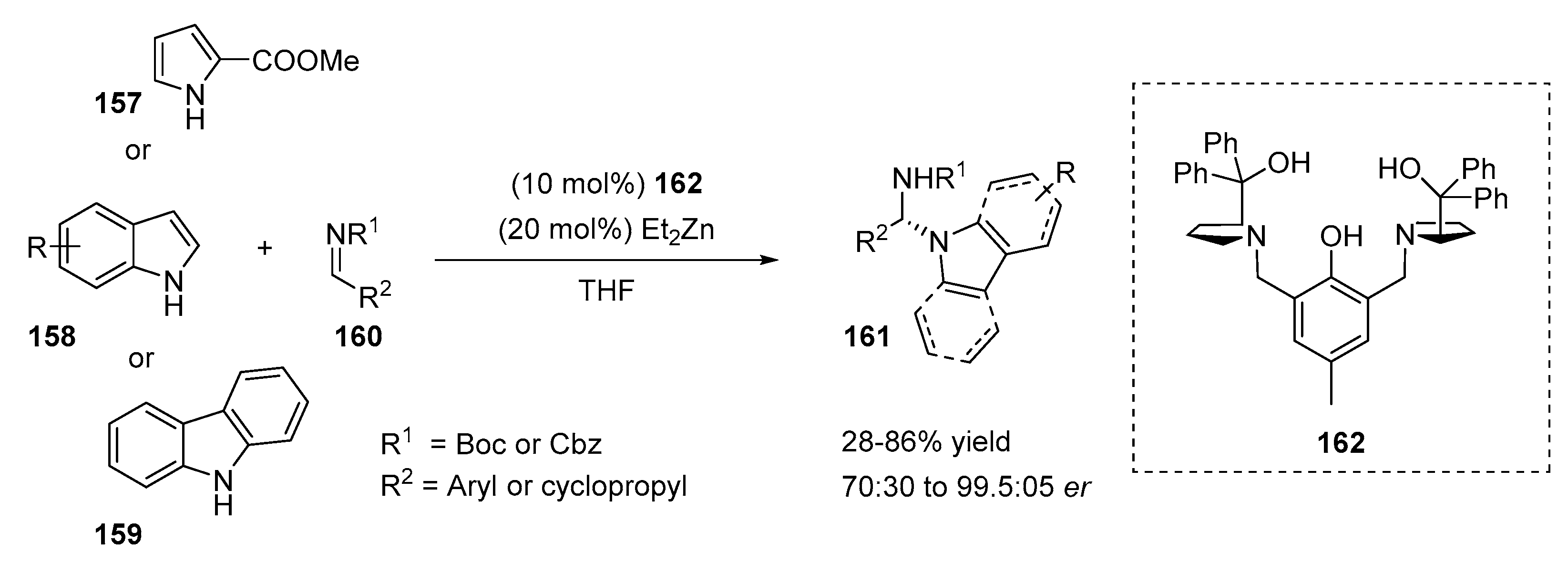
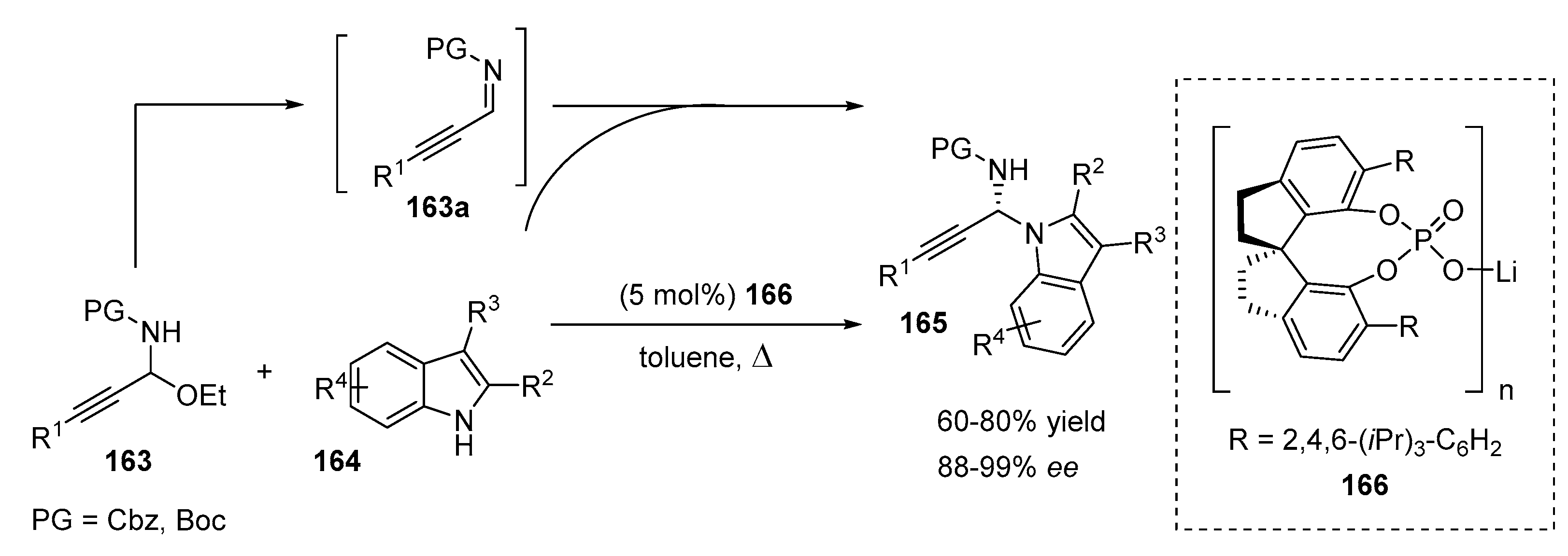
2.3. N-Selective Additions of Indoles to Imine/Iminium Activated Adducts
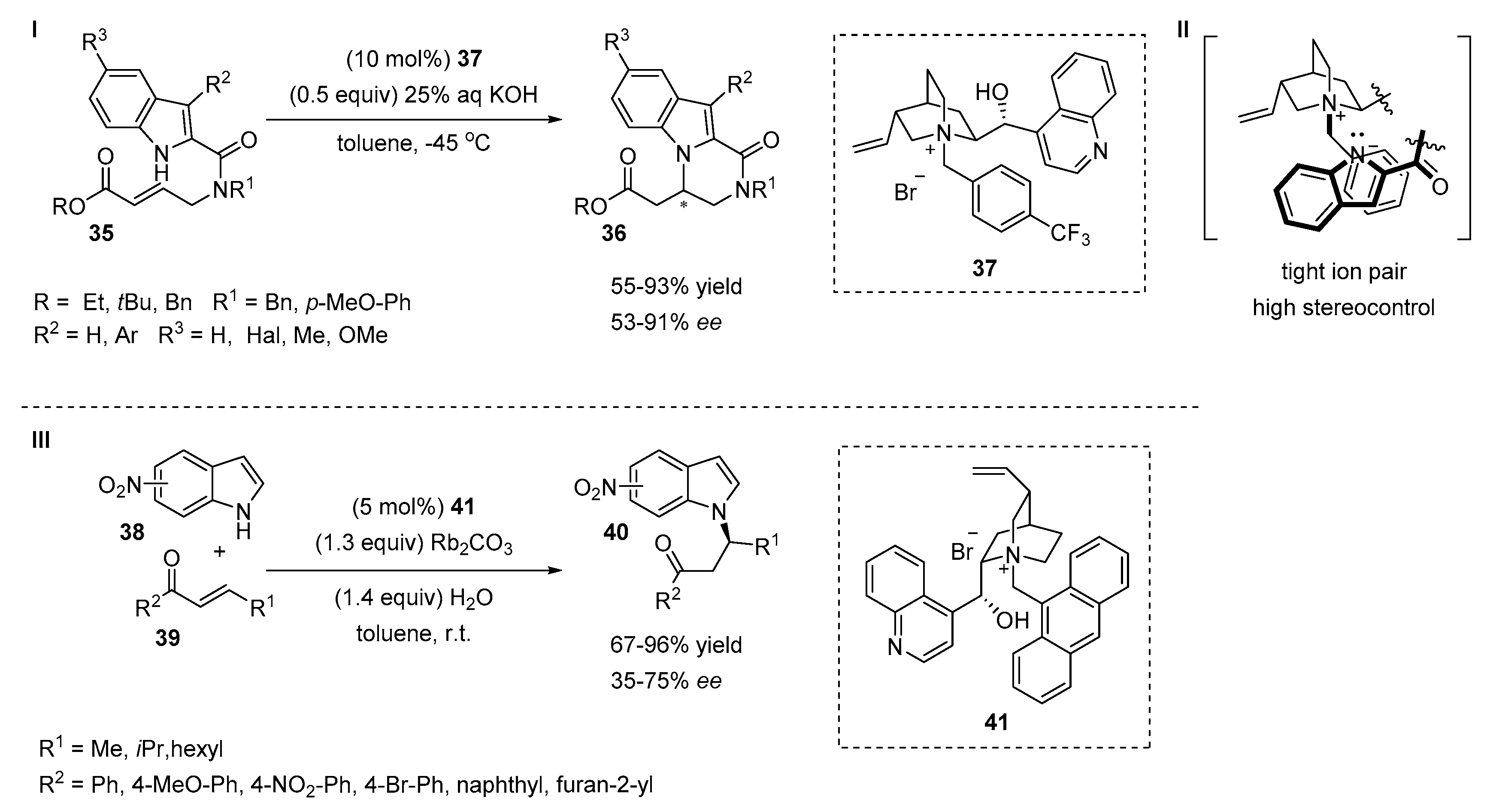
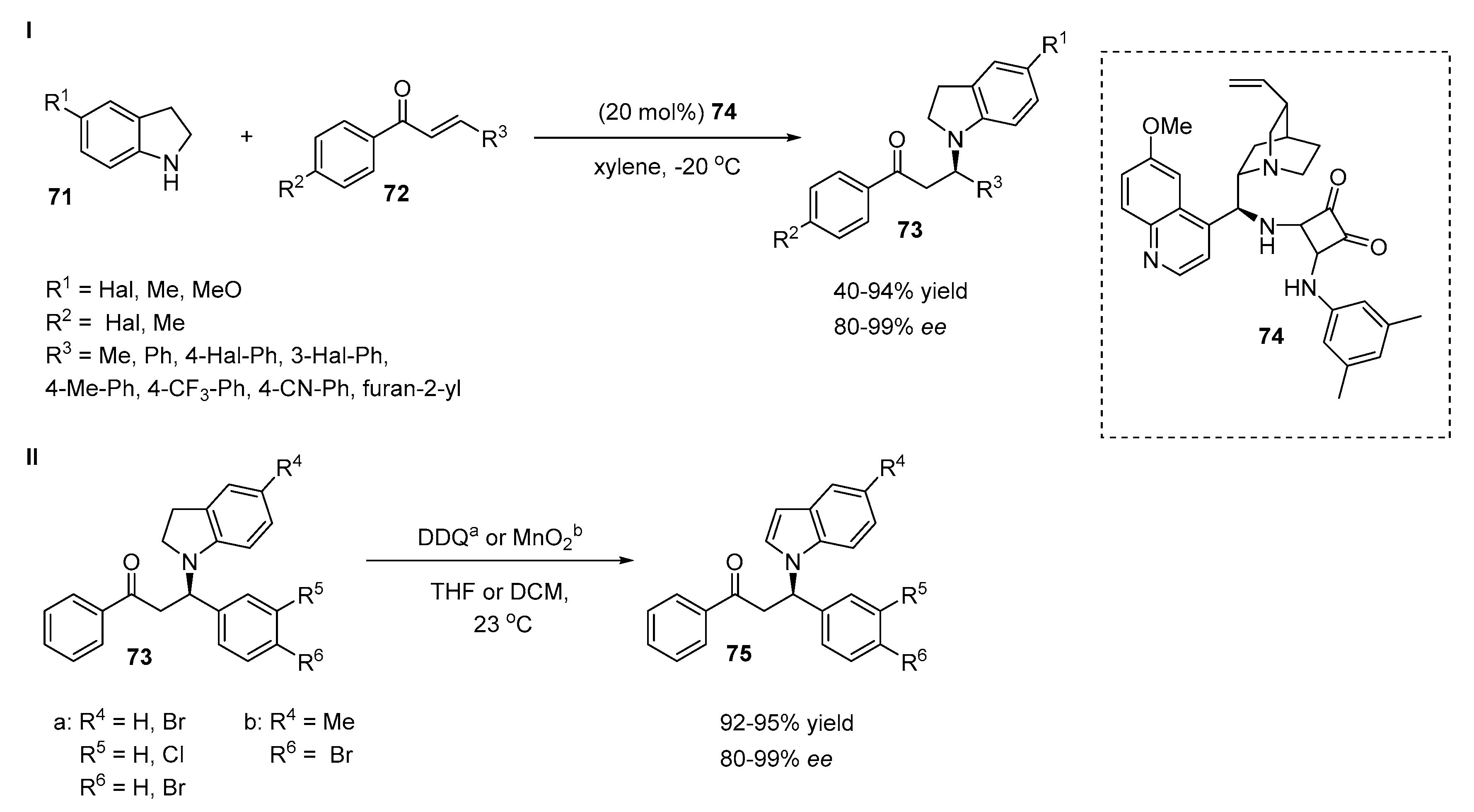
2.4. Aza-Michael Additions
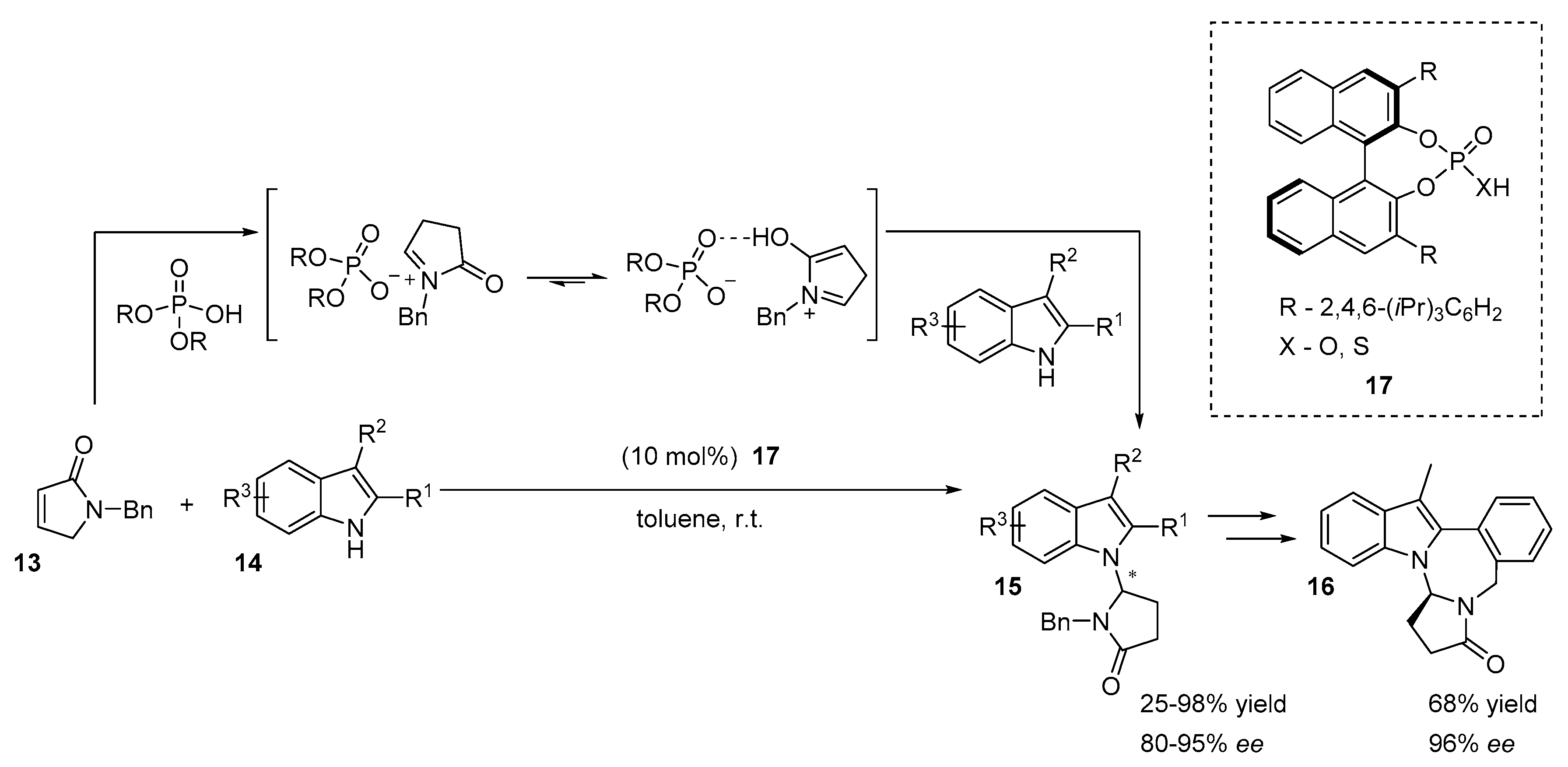
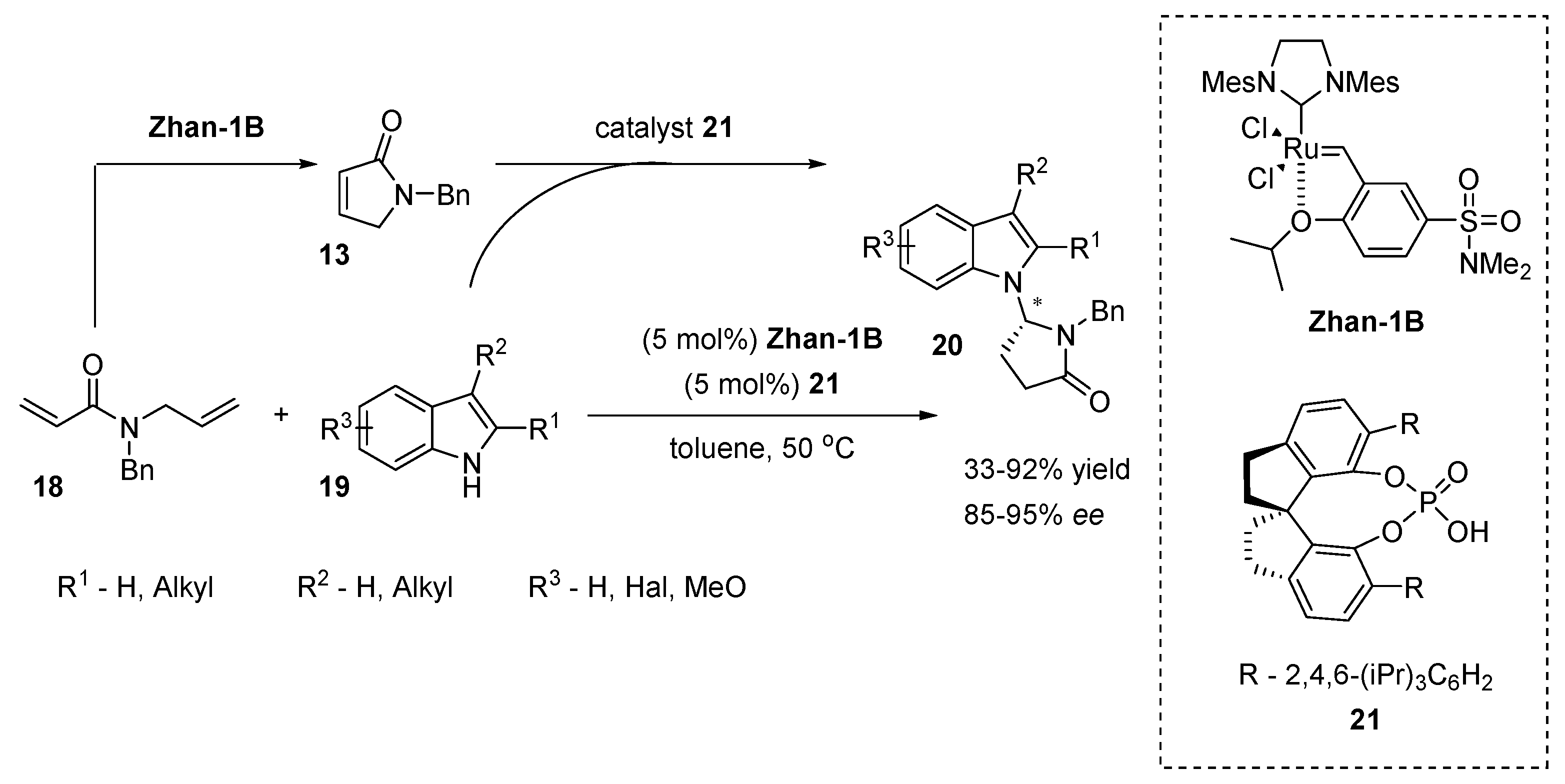
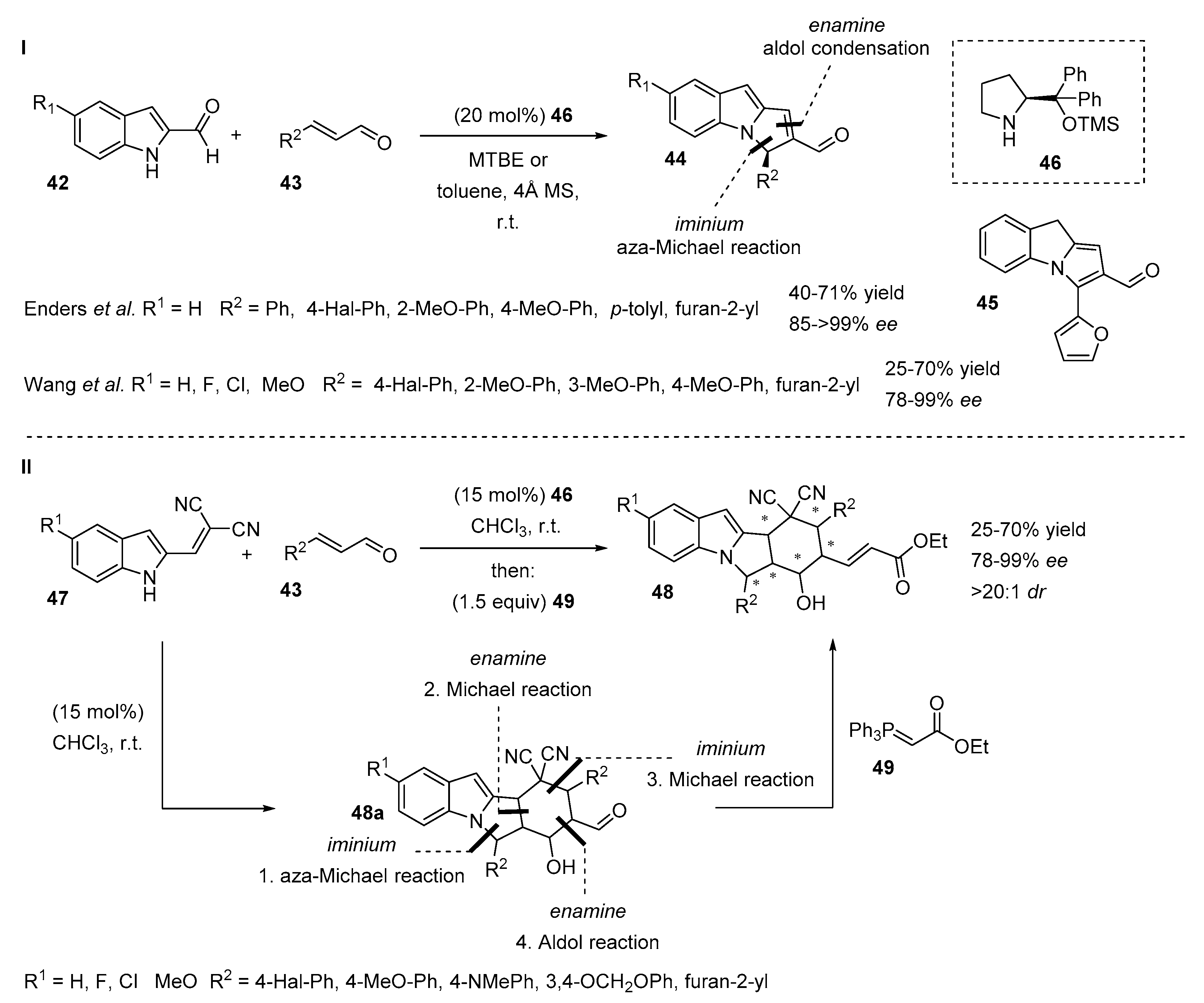
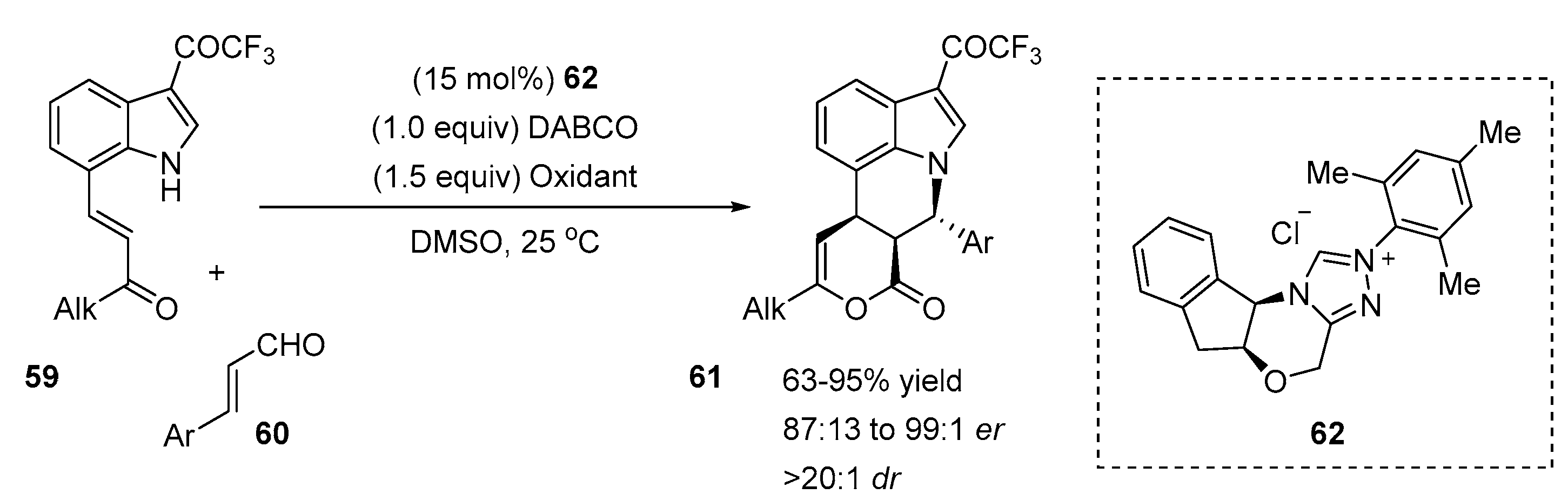
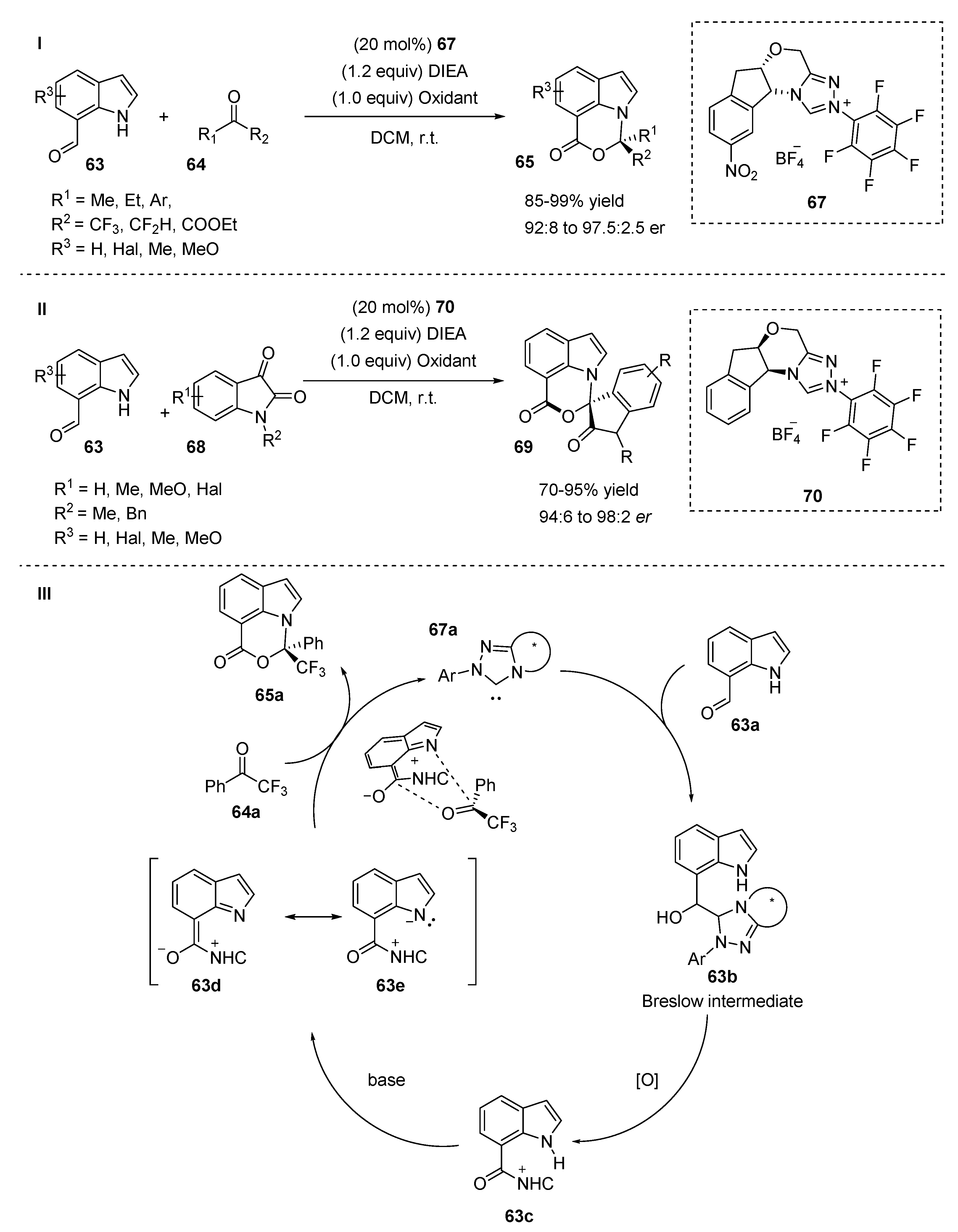
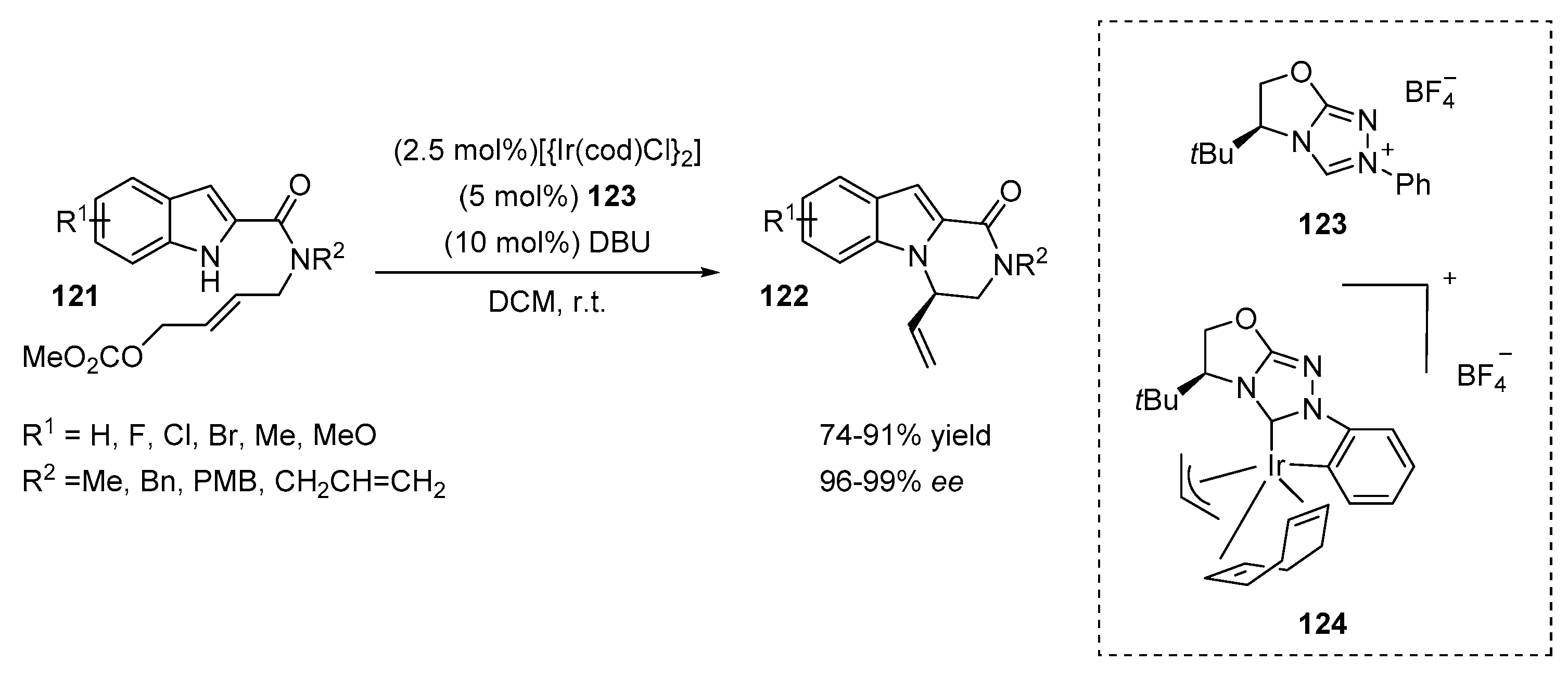
2.5. N-Heterocyclic Carbene-Mediated Cyclizations
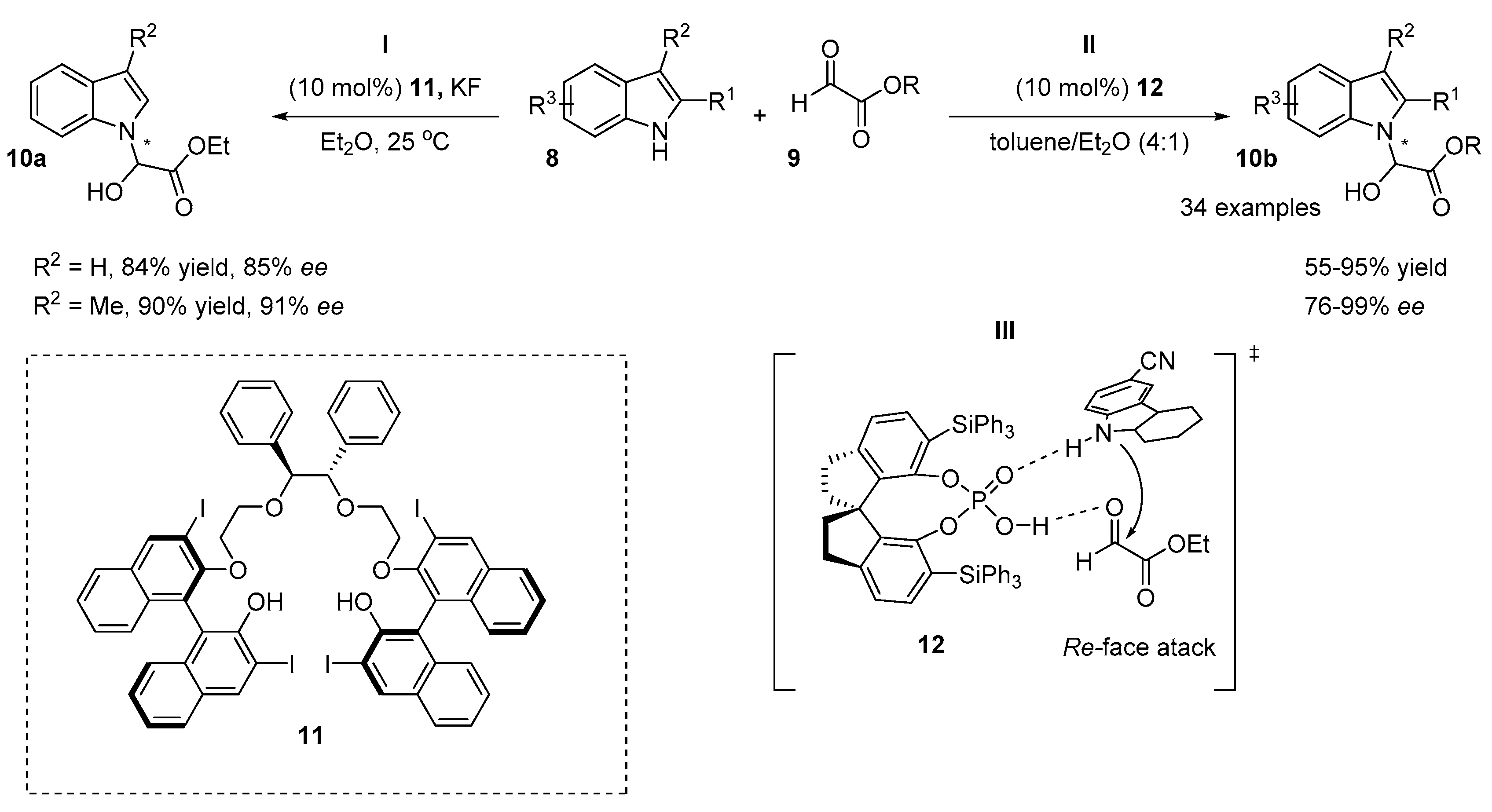
3. Organocatalytic Indirect Methods
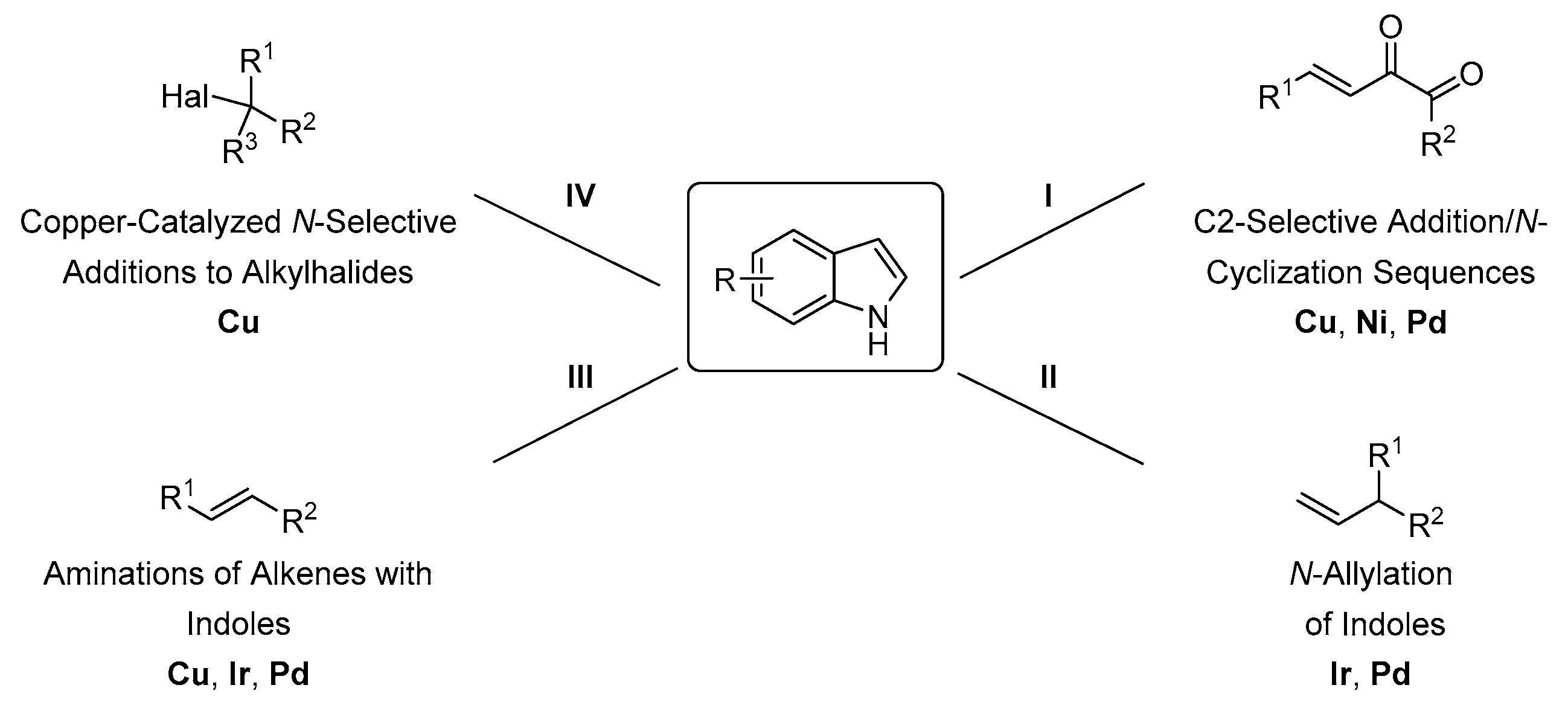
4. Direct Organometallic Methods
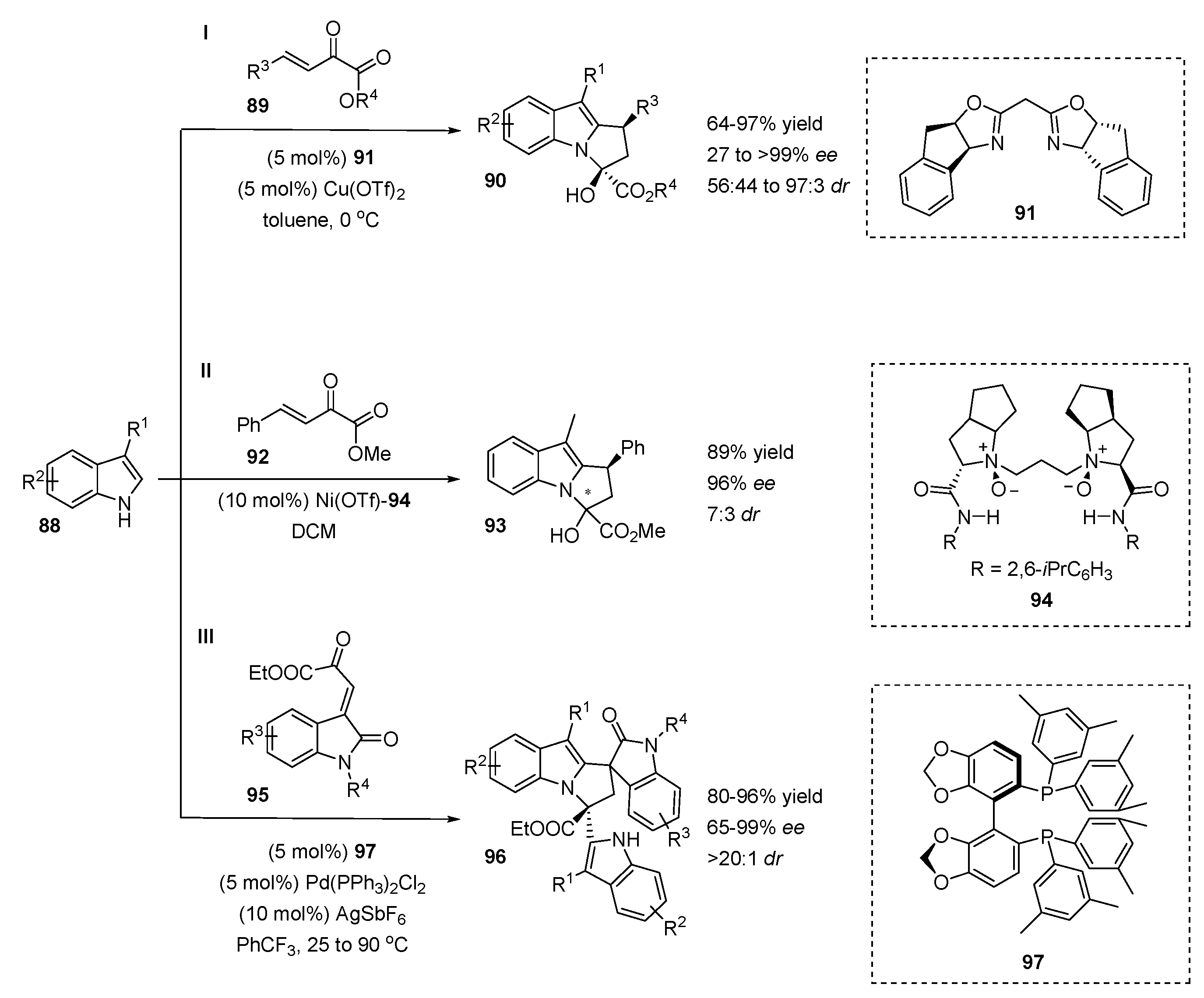
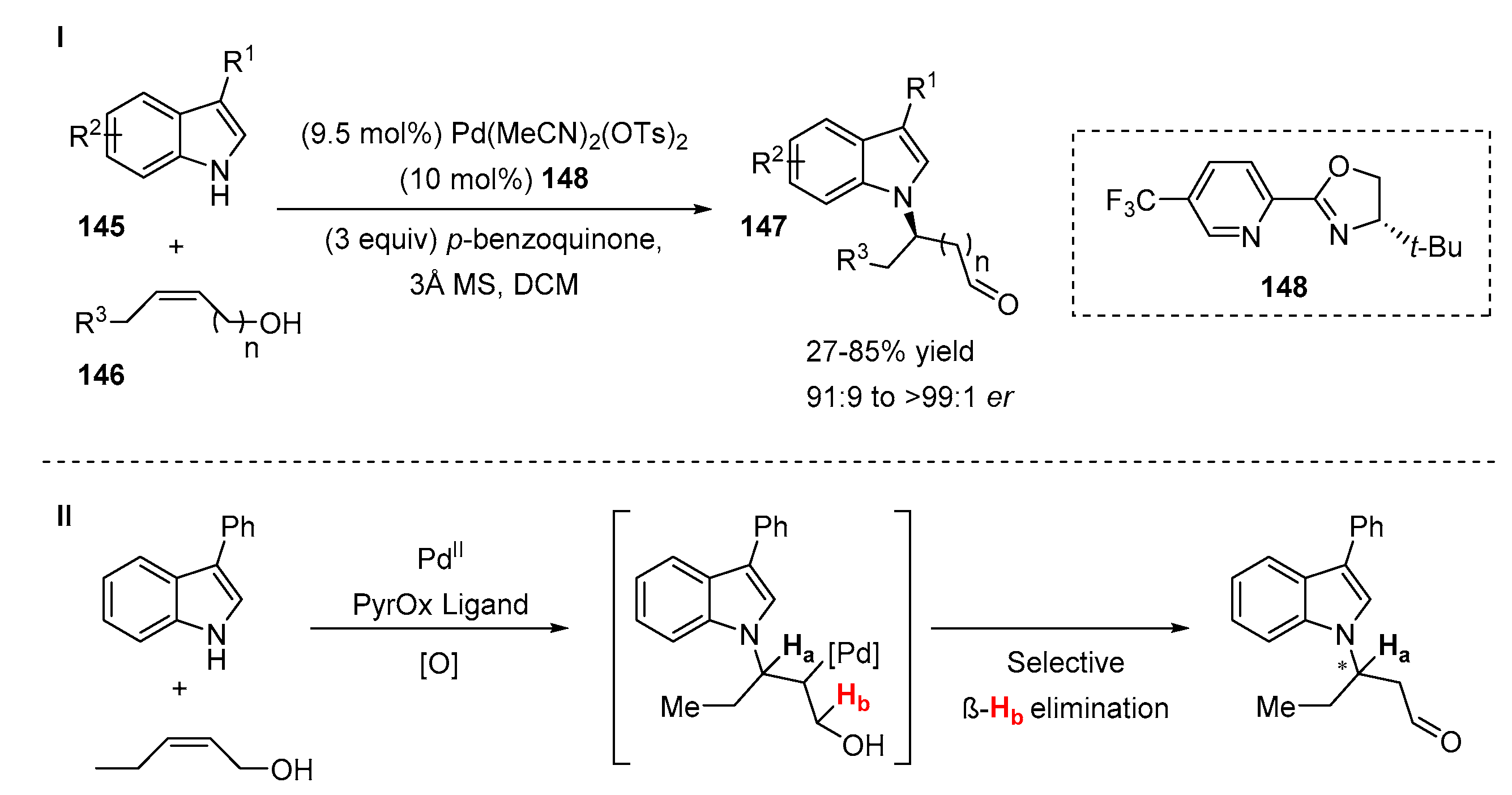
4.1. C2-Selective Addition/N-Cyclization Sequences
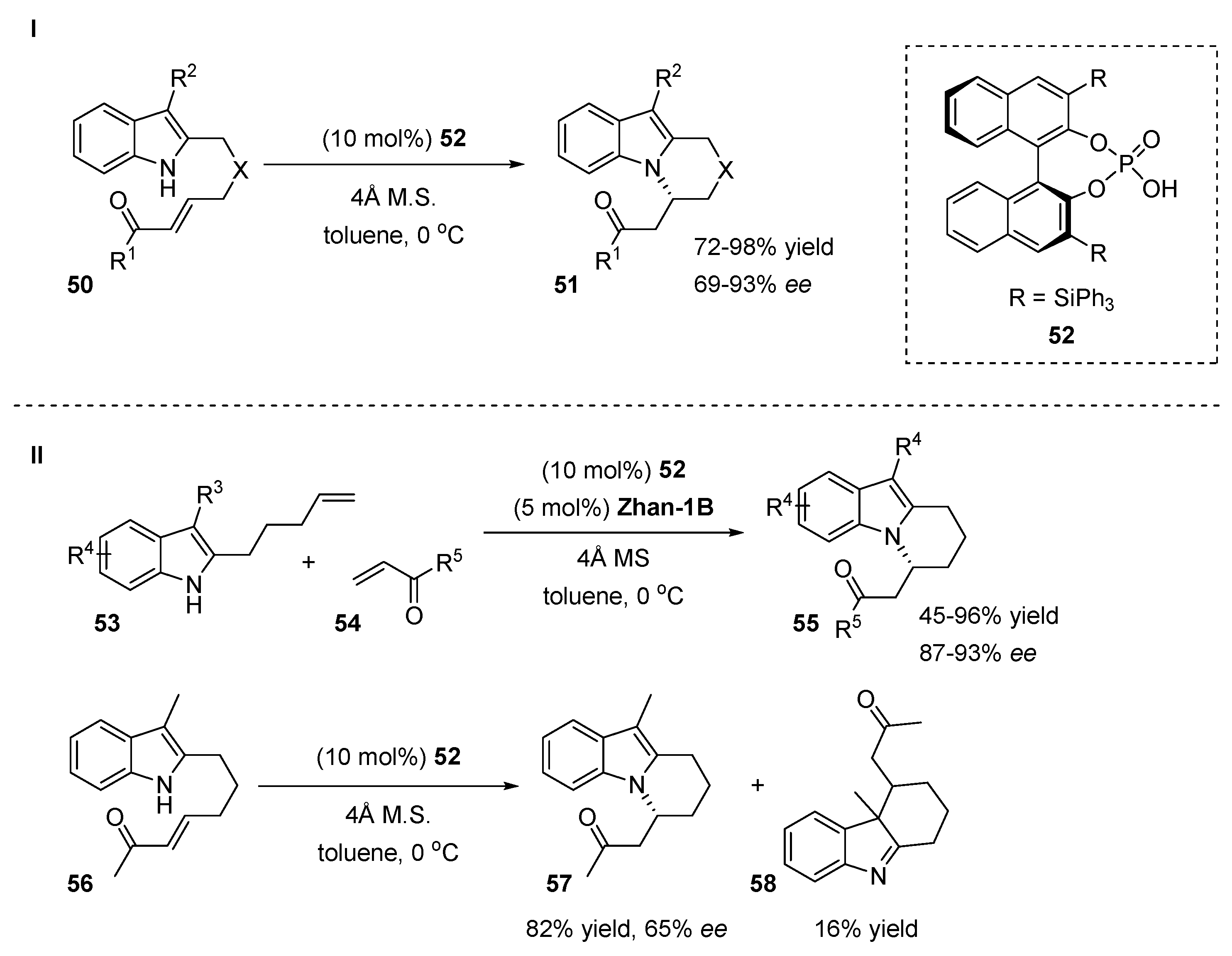
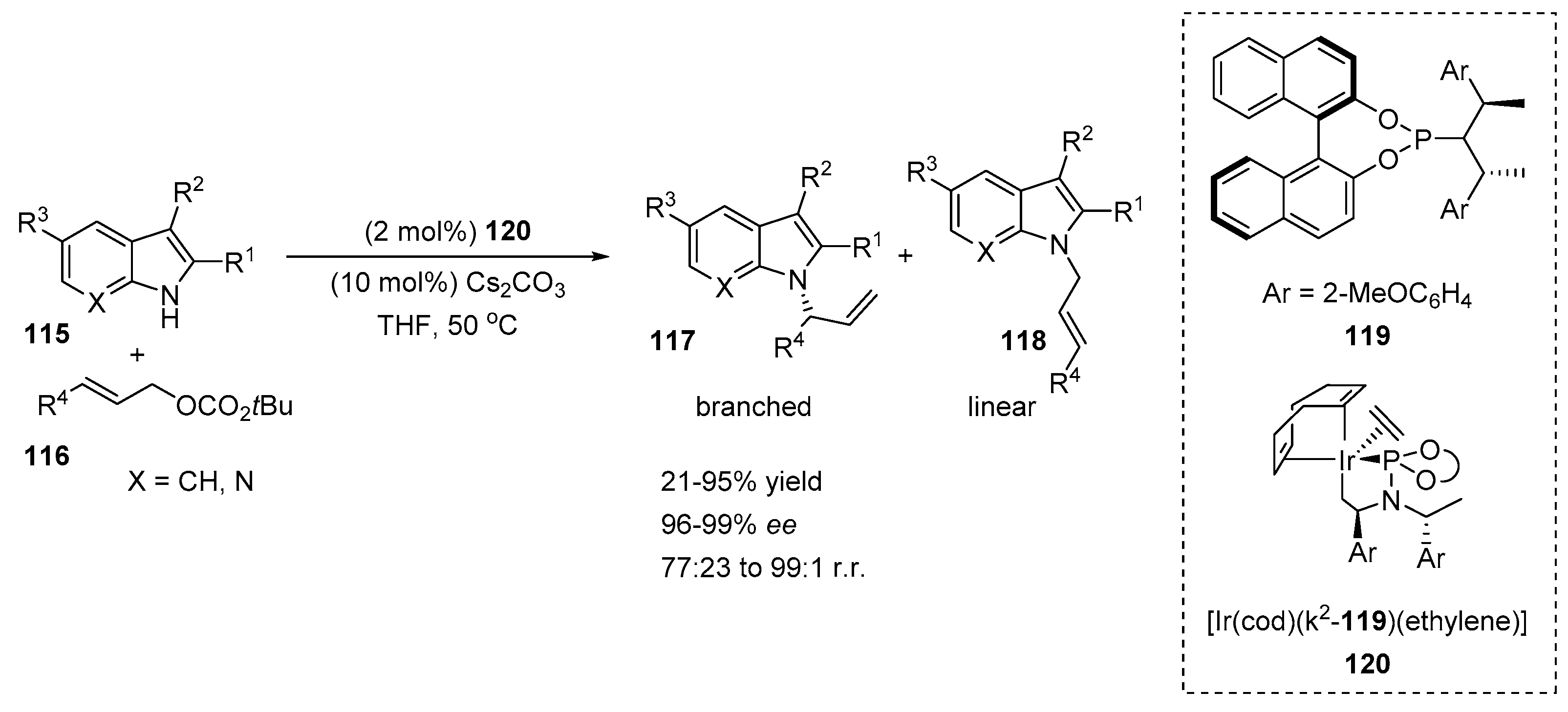
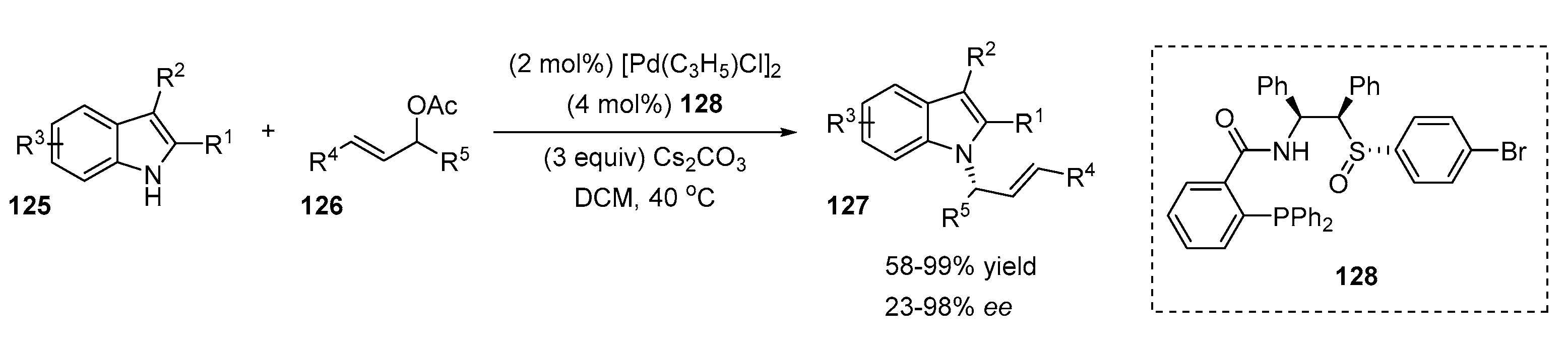
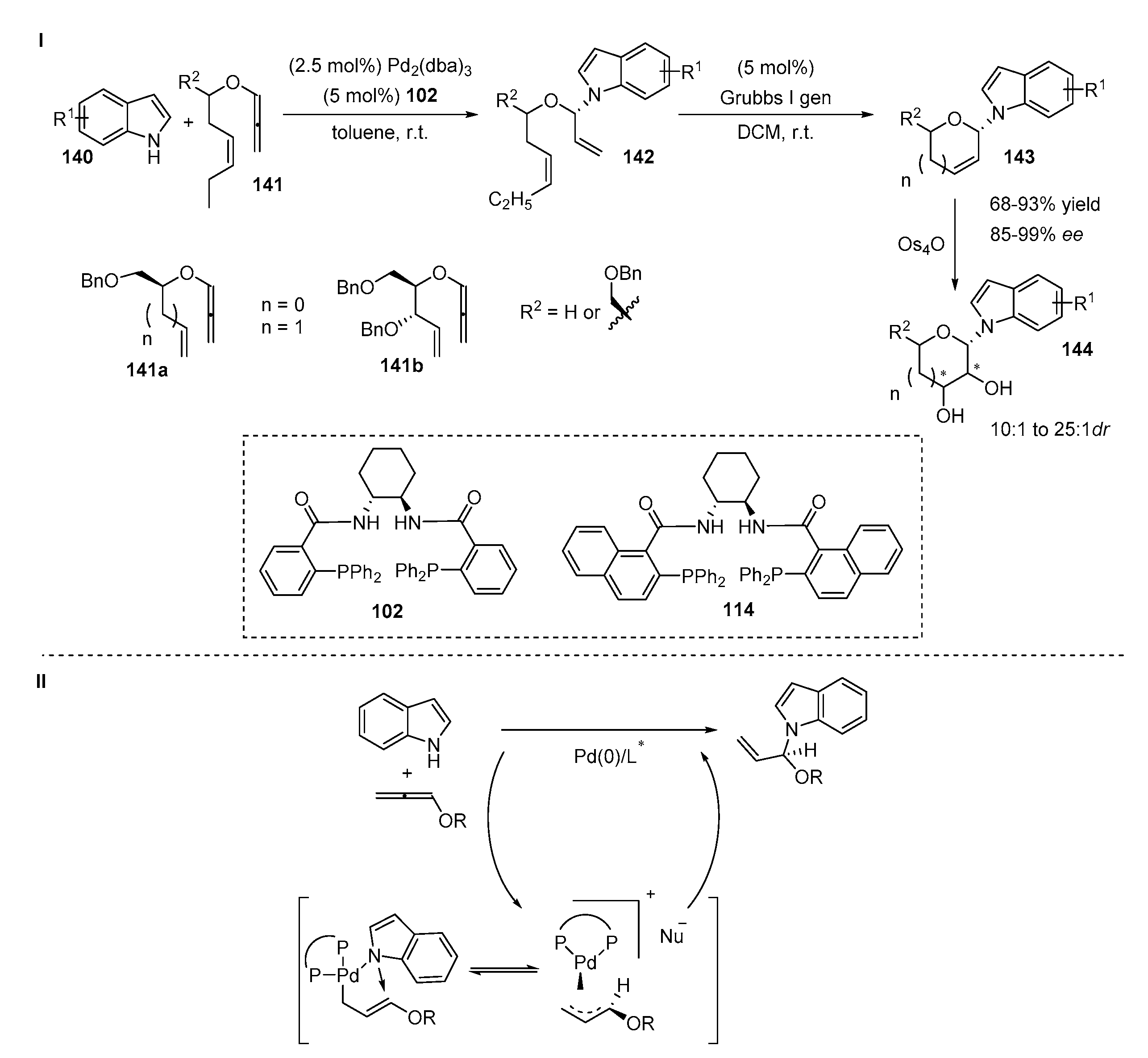
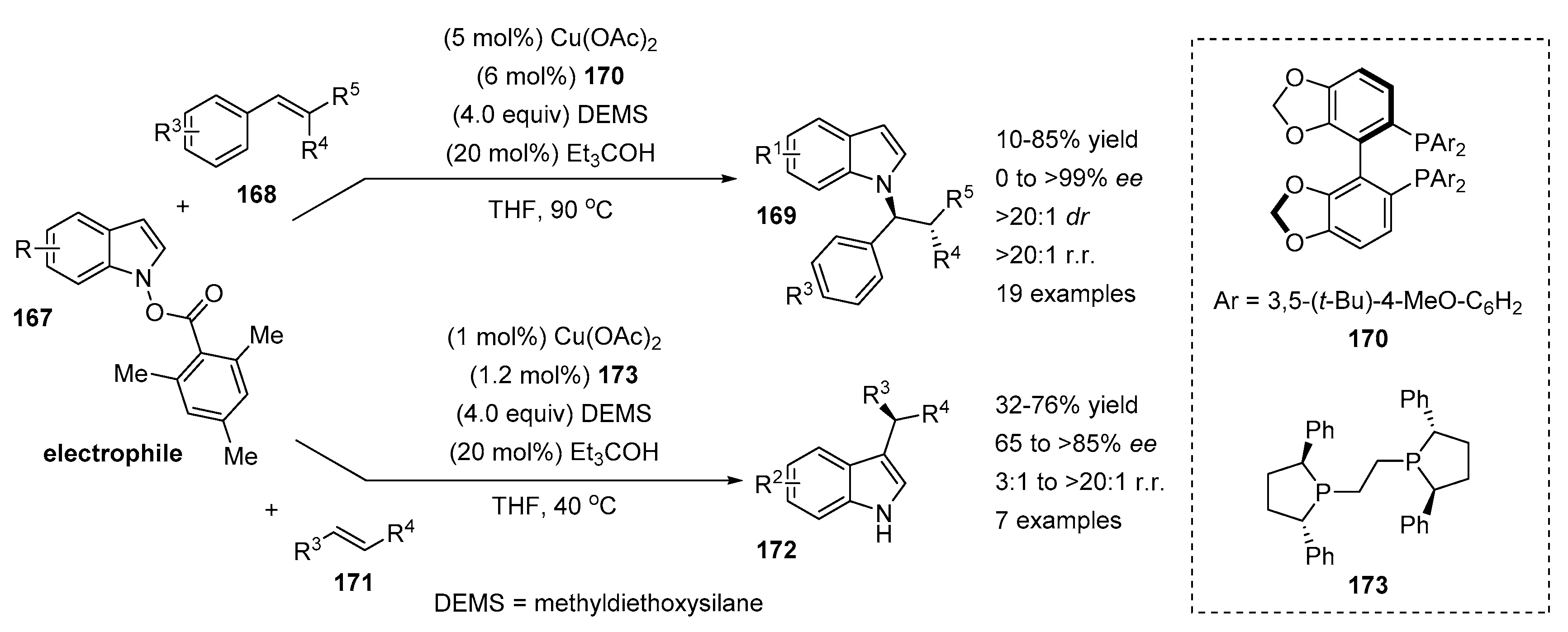
4.2. N-Allylation of Indoles
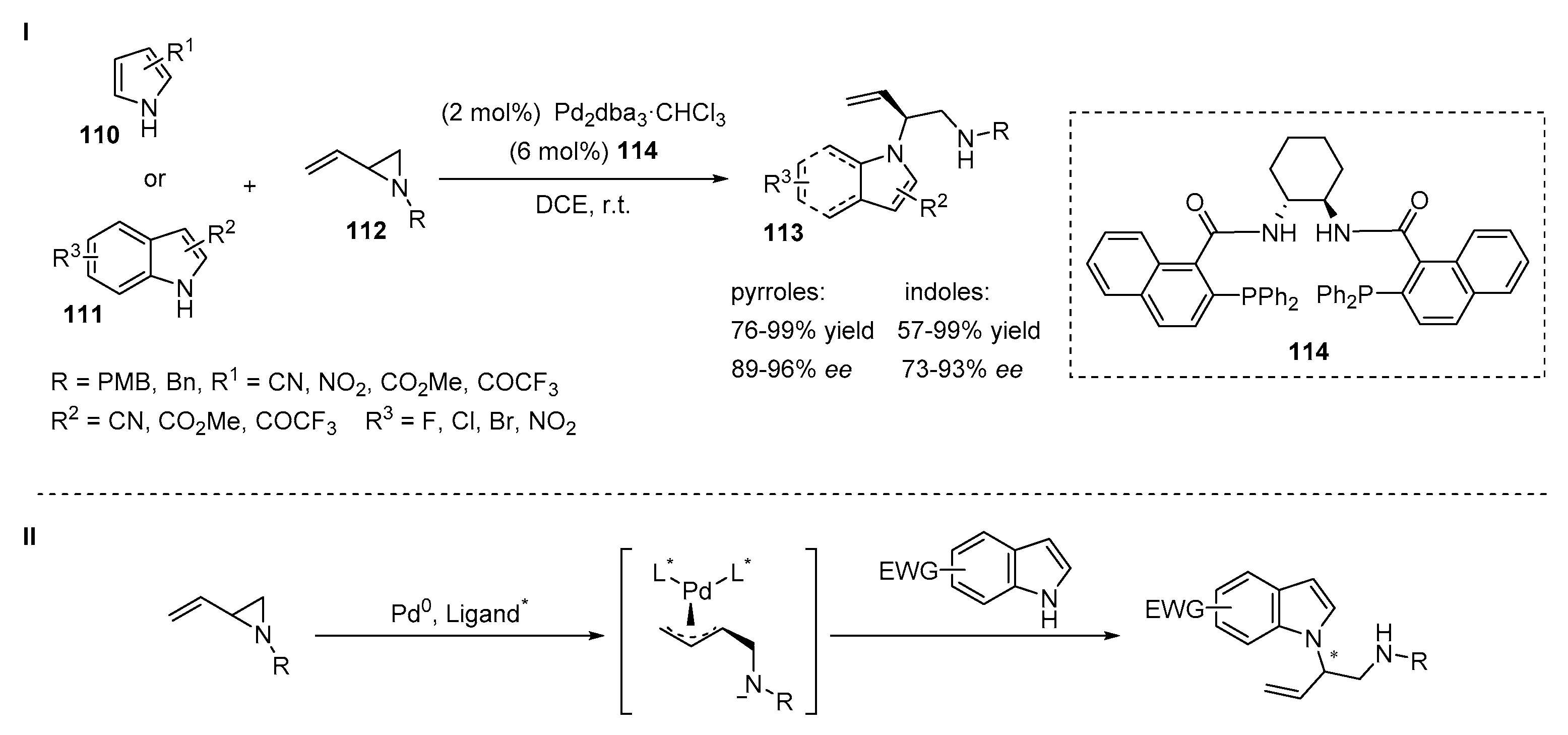
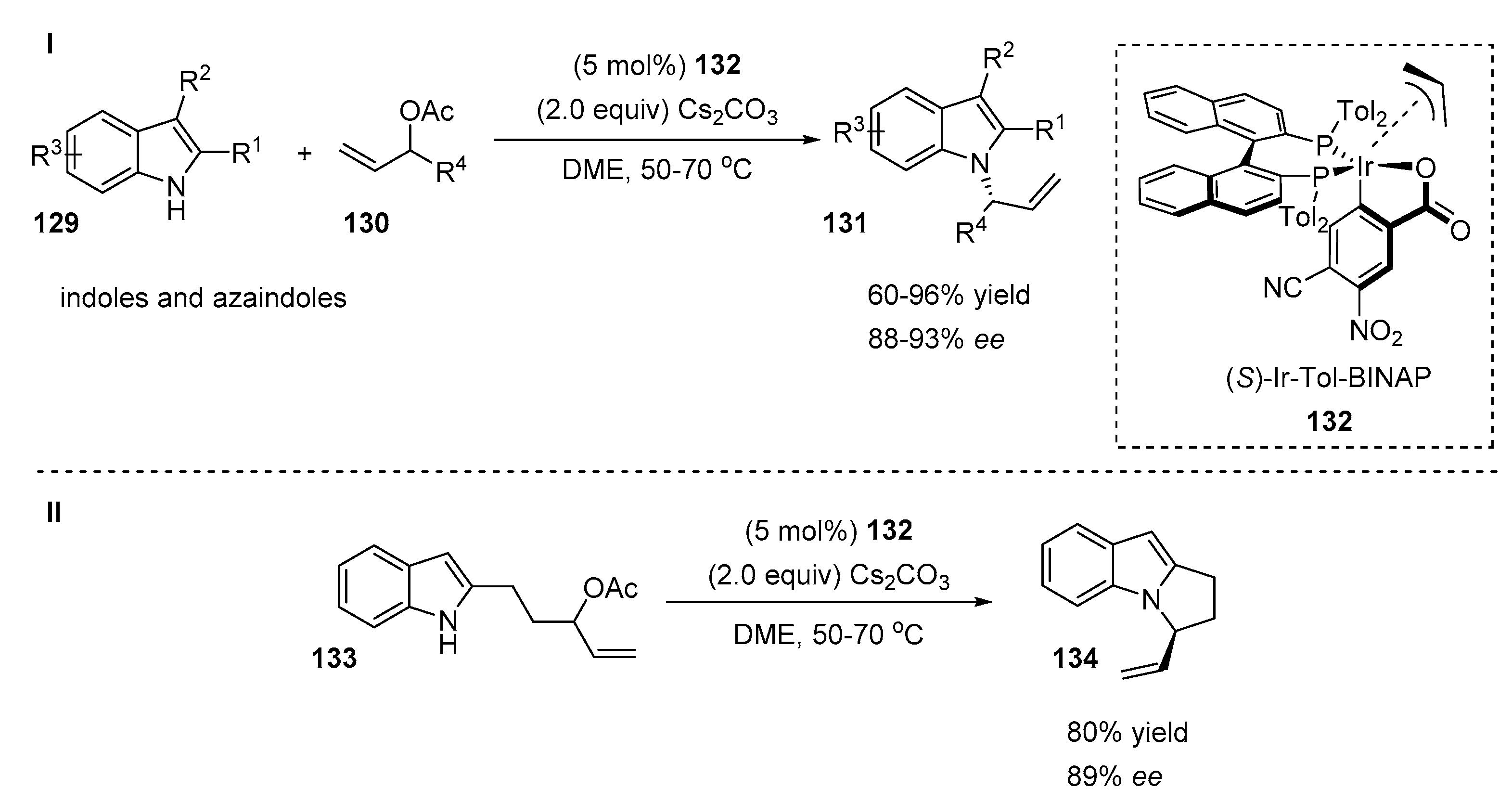
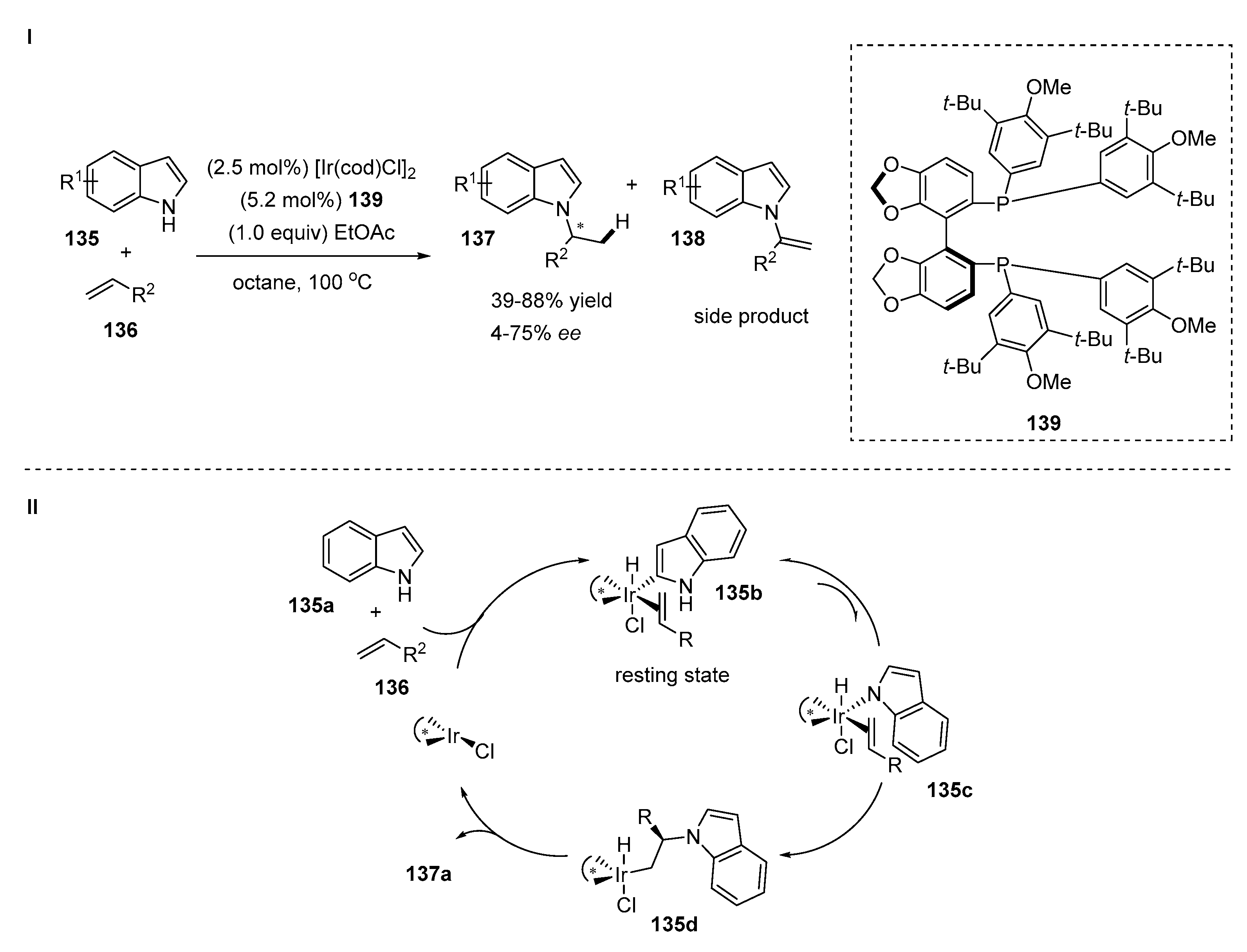
4.3. Aminations of Alkenes with Indoles
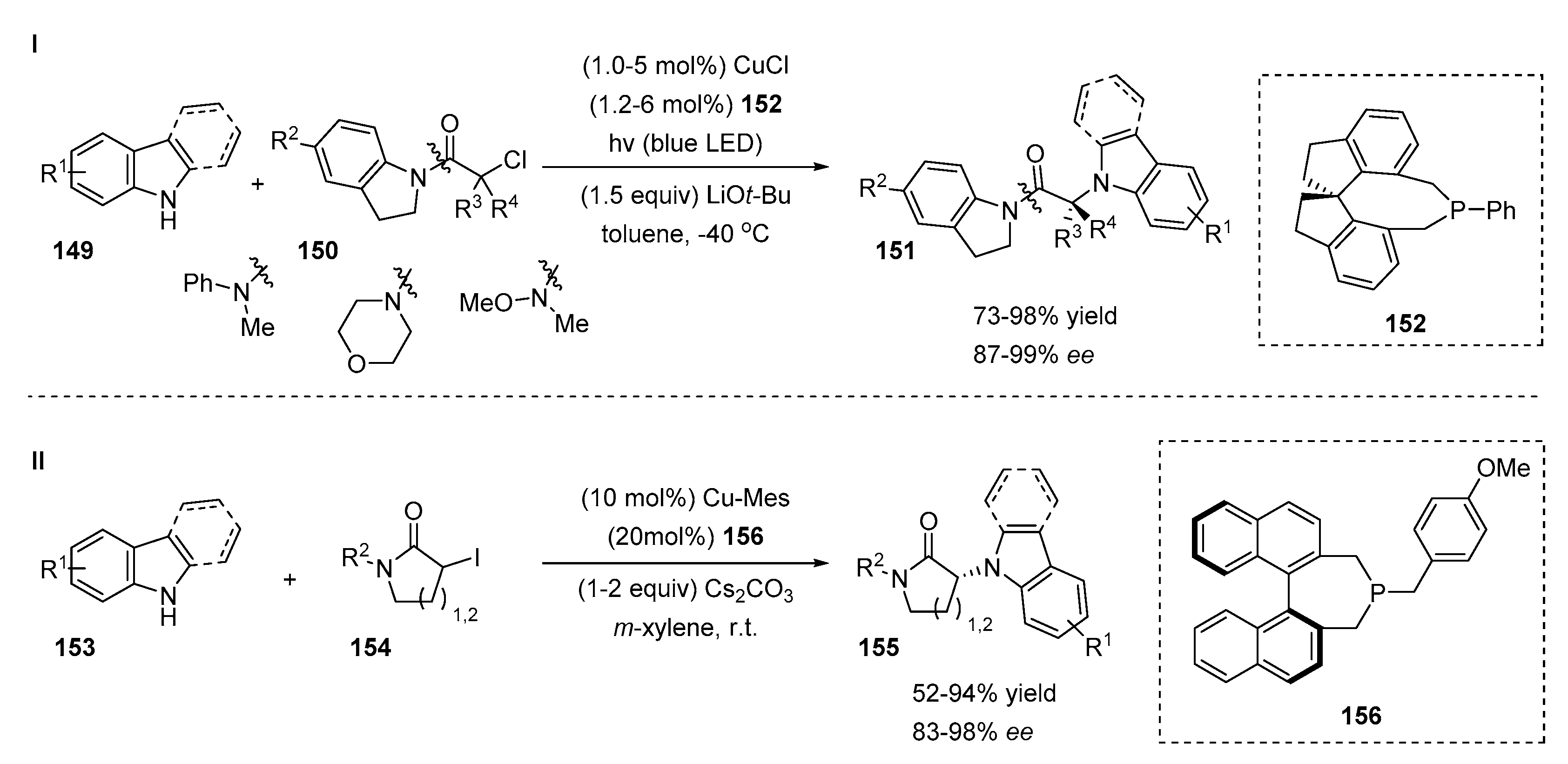
4.4. Copper-Catalyzed N-Selective Additions of Indoles to Alkylhalides
5. Other Direct Methods
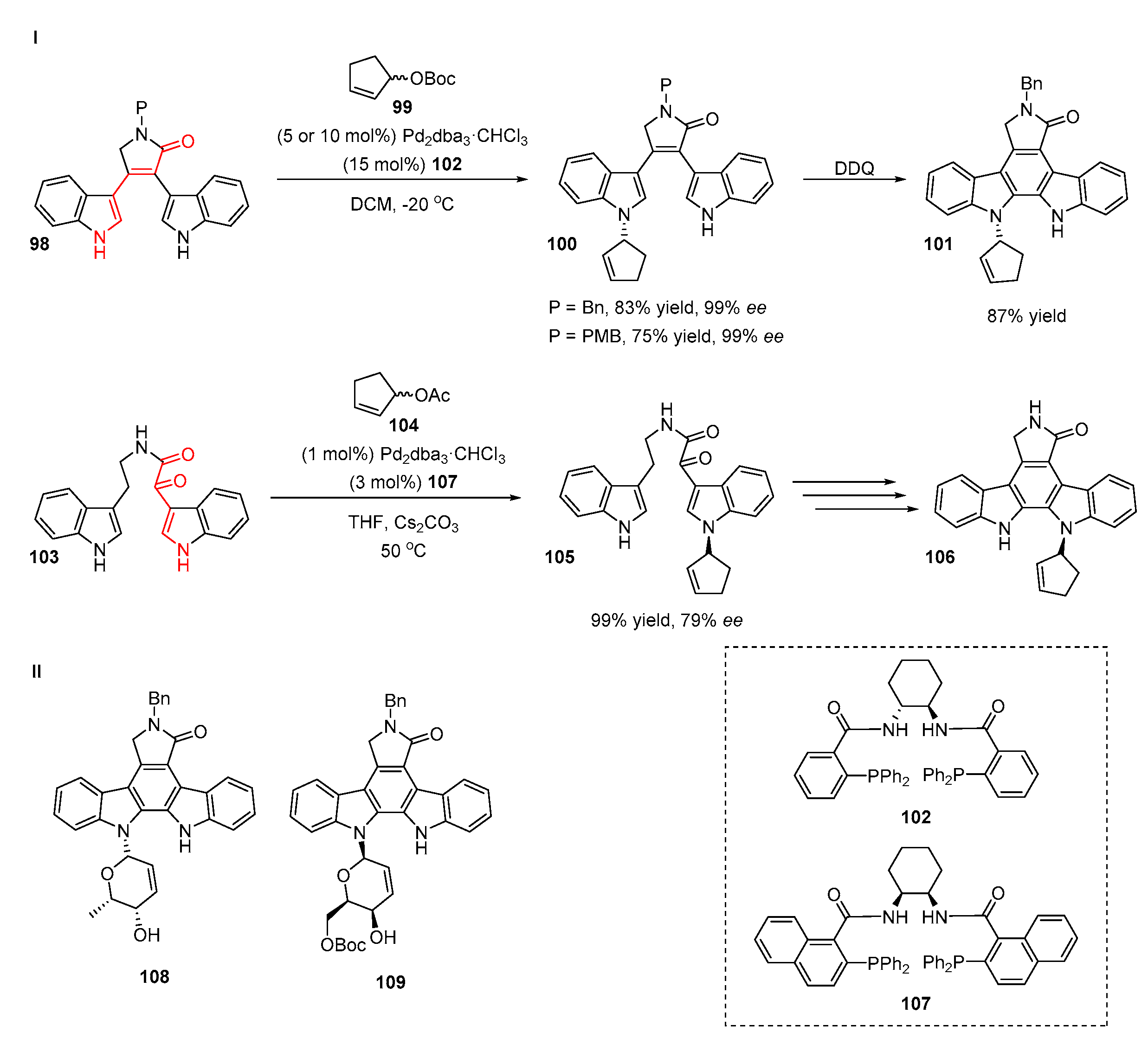
6. Transition-Metal Catalyzed Indirect Methods
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vitaku, E.; Smith, D.T.; Njardsrson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.P.; Singh, O.M. Recent Progress in Biological Activities of Indole and Indole Alkaloids. Mini-Rev. Med. Chem. 2018, 18, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, N.K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.H.; Verma, A.K.; Choi, E.H. Biomedical Importance of Indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, J.; Ding, T.-M.; Yan, Z.-Q.; Hou, S.-H.; Zhu, G.-D.; Zhang, S.-Y. Asymmetric N-Hydroxyalkylation of Indoles with Ethyl Glyoxalates Catalyzed by a Chiral Phosphoric Acid: Highly Enantioselective Synthesis of Chiral N,O-Aminal Indole Derivatives. Org. Lett. 2019, 21, 2795–2799. [Google Scholar] [CrossRef]
- Jang, S.H.; Kim, H.W.; Jeong, W.; Moon, D.; Rhe, Y.H. Palladium-Catalyzed Asymmetric Nitrogen-Selective Addition of Indoles to alkoxyallenes. Org. Lett. 2018, 20, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Schempp, T.T.; Znieg, J.R.; Stivala, C.E.; Krische, M.J. Regio-and Enantioselective Iridium-Catalyzed N-Allylation of Indoles and Related Azoles with Racemic Branched Alkyl-Substituted Allylic Acetates. Angew. Chem. Int. Ed. 2019, 58, 7762–7766. [Google Scholar] [CrossRef]
- Ye, Y.; Kim, S.-T.; Jeong, J.; Baik, M.-H.; Buchwald, S.L. CuH-Catalyzed Enantioselective Alkylation of Indole Derivatives with Ligand-Controlled Regiodivergence. J. Am. Chem. Soc. 2019, 141, 3901–3909. [Google Scholar] [CrossRef] [PubMed]
- Bandini, M.; Eichholzer, A. Catalytic Functionalization of Indoles in a New Dimension. Angew. Chem. Int. Ed. 2009, 48, 9608–9644. [Google Scholar] [CrossRef]
- Bartoli, G.; Bencivenni, G.; Dalpozzo, R. Organocatalytic strategies for the asymmetric functionalization of indoles. Chem. Soc. Rev. 2010, 39, 4449–4465. [Google Scholar] [CrossRef]
- Dalpozzo, R. Strategies for the asymmetric functionalization of indoles: An update. Chem. Soc. Rev. 2015, 44, 742–778. [Google Scholar] [CrossRef]
- Karchava, A.V.; Melkonyan, F.S.; Yurovskaja, M.A. New Strategies for the synthesis of N-alkylated indoles. Chem. Heterocycl. Compd. 2012, 48, 391–407. [Google Scholar] [CrossRef]
- Lakhdar, S.; Westermaier, M.; Terrier, F.; Goumont, R.; Boubaker, T.; Ofial, A.R.; Mayr, H. Nucleophilic Reactivities of Indoles. J. Org. Chem. 2006, 71, 9088–9095. [Google Scholar] [CrossRef] [PubMed]
- Otero, N.; Mandado, M.; Mosquera, R.A. Nucleophilicity of Indole Derivatives: Activating and Deactivating Effects Based on Proton Affinities and Electron Density Properties. J. Phys. Chem. A 2007, 111, 5557–5562. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, D. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Berkessel, A.; Gröger, H.; MacMillan, D. Asymmetric Organocatalysis; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Torres, R.R. Stereoselective Organocatalysis, 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Dalko, P.I. (Ed.) Comprehensive Enantioselective Organocatalysis; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Cui, H.-L.; Feng, X.; Peng, J.; Lei, J.; Jiang, K.; Chen, Y.-C. Chemoselective Asymmetric N-Allylic Alkylation of Indoles with Morita–Baylis–Hillman Carbonates. Angew. Chem. Int. Ed. 2009, 48, 5737–5740. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wei, Y.; Shi, M. Asymmetric substitutions of O-Boc-protected Morita–Baylis–Hillman adducts with pyrrole and indole derivatives. Org. Biomol. Chem. 2012, 10, 1396–1405. [Google Scholar] [CrossRef]
- Zi, Y.; Lange, M.; Schultz, C.; Vilotijević, I. Latent Nucleophiles in Lewis Base Catalyzed Enantioselective N-Allylations of N-Heterocycles. Angew. Chem. Int. Ed. 2019, 58, 10727–10731. [Google Scholar] [CrossRef]
- Zhang, N.; Li, Y.; Chen, Z.; Qin, W. Direct Preparation of Indole Hemiaminals through Organocatalytic Nucleophilic Addition of Indole to Aldehydes. Synthesis 2018, 50, 4063–4070. [Google Scholar]
- Xie, Y.; Zhao, Y.; Qian, B.; Yang, L.; Xia, C.; Huang, H. Enantioselective N–H Functionalization of Indoles with α,β-Unsaturated γ-Lactams Catalyzed by Chiral Brønsted Acids. Angew. Chem. Int. Ed. 2011, 50, 5682–5686. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E. Scaffold Diversity from N-Acyliminium Ions. Chem. Rev. 2017, 117, 7811–7856. [Google Scholar] [CrossRef]
- Shi, Y.-C.; Wang, S.-G.; Yin, Q.; You, S.-L. N-alkylation of indole via ring-closing metathesis/isomerization/Mannich cascade under ruthenium/chiral phosphoric acid sequential catalysis. Org. Chem. Front. 2014, 1, 39–43. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, B.; Chen, Z.; Hu, J.; Zeng, X.; Zhong, G. Chiral phosphoric acid catalyzed enantioselective N-alkylation of indoles with in situ generated cyclic N-acyl ketimines. Chem. Commun. 2018, 54, 9230–9233. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Sun, J. Catalytic Asymmetric N-Alkylation of Indoles and Carbazoles through 1,6-Conjugate Addition of Aza-para-quinone Methides. Angew. Chem. Int. Ed. 2017, 56, 4583–4587. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Gu, Q.; You, S.-L. Chemoselective N–H functionalization of indole derivatives via the Reissert-type reaction catalyzed by a chiral phosphoric acid. Org. Biomol. Chem. 2018, 16, 6146–6154. [Google Scholar] [CrossRef]
- Yagil, G. The Proton Dissocation Constant of Pyrrole, Indole and Related Compounds. Tetrahedron 1967, 23, 2855–2861. [Google Scholar] [CrossRef]
- Bandini, M.; Eichholzer, A.; Tragni, M.; Umani-Ronchi, A. Enantioselective Phase-Transfer-Catalyzed Intramolecular Aza-Michael Reaction: Effective Route to Pyrazino-Indole Compounds. Angew. Chem. Int. Ed. 2008, 47, 3238–3241. [Google Scholar] [CrossRef]
- Bandini, M.; Bottoni, A.; Eichholzer, A.; Miscione, G.P.; Stenta, M. Asymmetric Phase-Transfer-Catalyzed Intramolecular N-Alkylation of Indoles and Pyrroles: A Combined Experimental and Theoretical Investigation. Chem. Eur. J. 2010, 16, 12462–12473. [Google Scholar] [CrossRef]
- Trubitsõn, D.; Martõnova, J.; Erkman, K.; Metsala, A.; Saame, J.; Kõster, K.; Järving, I.; Leito, I.; Kanger, T. Enantioselective N-Alkylation of Nitroindoles under Phase-Transfer Catalysis. Synthesis 2020, 52, 1047–1059. [Google Scholar] [CrossRef]
- Wang, C.; Raabe, G.; Enders, D. Enantioselective Synthesis of 3H-Pyrrolo[1,2-a]indole-2-carbaldehydes via an Organocatalytic Domino Aza-Michael/Aldol Condensation Reaction. Synthesis 2009, 24, 4119–4124. [Google Scholar]
- Hong, L.; Sun, W.; Liu, C.; Wang, L.; Wang, R. Asymmetric Organocatalytic N-Alkylation of Indole-2-carbaldehydes with α,β-Unsaturated Aldehydes: One-Pot Synthesis of Chiral Pyrrolo[1,2-α]indole-2-carbaldehydes. Chem. Eur. J. 2010, 16, 440–444. [Google Scholar] [CrossRef]
- Greb, A.; Deckers, K.; Selig, P.; Merkens, C.; Enders, D. Quadruple Domino Organocatalysis: An Asymmetric Aza-Michael/Michael/ Michael/Aldol Reaction Sequence Leading to Tetracyclic Indole Structures with Six Stereocenters. Chem. Eur. J. 2012, 18, 10226–10229. [Google Scholar]
- Cai, Q.; Zheng, C.; You, S.-L. Enantioselective Intramolecular Aza-Michael Additions of Indoles Catalyzed by Chiral Phosphoric Acids. Angew. Chem. Int. Ed. 2010, 49, 8666–8669. [Google Scholar] [CrossRef] [PubMed]
- Ni, Q.J.; Zhang, H.; Grossmann, A.; Loh, C.C.J.; Merkens, C.; Enders, D. Asymmetric Synthesis of Pyrroloindolones by N-heterocyclic Carbene Catalyzed [2+3] Annulation of α-Chloroaldehydes with Nitrovinylindoles. Angew. Chem. Int. Ed. 2013, 52, 13562–13566. [Google Scholar] [CrossRef] [PubMed]
- Bera, S.; Daniliuc, C.G.; Studer, A. Oxidative N-heterocyclic Carbene Catalyzed Dearomatization of Indoles to Spirocyclic Indolenines with a Quaternary Carbon Stereocenter. Angew. Chem., Int. Ed. 2017, 56, 7402–7406. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.; Yang, S.; Xu, W.; Liu, J.; Perveen, S.; Kong, X.; Zehra, S.T.; Fang, X. Carbene-catalyzed asymmetric Friedel–Crafts alkylation-annulation sequence and rapid synthesis of indole-fused polycyclic alkaloids. Commun. Chem. 2019, 2, 85. [Google Scholar] [CrossRef]
- Zhu, S.-Y.; Zhang, Y.; Chen, X.-F.; Huang, J.; Shi, S.-H.; Hui, X.-P. Highly enantioselective synthesis of functionalized azepino[1,2α]indoles via NHC-catalyzed [3+4] annulation. Chem. Commun. 2019, 55, 4363–4366. [Google Scholar] [CrossRef]
- Sun, S.; Lang, M.; Wang, J. N-Heterocyclic Carbene-Catalyzed β-Indolylation of α-Bromoenals with Indoles. Adv. Synth. Catal. 2019, 361, 5704–5708. [Google Scholar] [CrossRef]
- Mukherjee, S.; Shee, S.; Poisson, T.; Besset, T.; Biju, A.T. Enantioselective N-Heterocyclic Carbene-Catalyzed Cascade Reaction for the Synthesis of Pyrroloquinolines via N-H functionalization of indoles. Org. Lett. 2018, 20, 6998–7002. [Google Scholar] [CrossRef]
- Yang, X.; Luo, G.; Zhou, L.; Liu, B.; Zhang, X.; Gao, H.; Jin, Z.; Chi, Y.R. Enantioselective Indole N−H Functionalization Enabled by Addition of Carbene Catalyst to Indole Aldehyde at Remote Site. ACS Catal. 2019, 9, 10971–10976. [Google Scholar] [CrossRef]
- Zhou, B.; Ghosh, A.K. Bifunctional cinchona alkaloid-squaramide-catalyzed highly enantioselective aza-Michael addition of indolines to α,β-unsaturated ketones. Tetrahedron Lett. 2013, 54, 3500–3502. [Google Scholar]
- Dou, X.; Yao, W.; Jiang, C.; Lu, Y. Enantioselective N-alkylation of isatins and synthesis of chiral N-alkylated indoles. Chem. Commun. 2014, 50, 11354–11357. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Zhang, W.; You, S.-L. One-Pot Synthesis of Pyrrolo[1,2-a]indoles by Chiral N-Triflyl Phosphoramide Catalyzed Friedel-Crafts Alkylation of 4,7-Dihydroindole with β,γ-Unsaturated α-Keto Esters. Chin. J. Chem. 2012, 30, 2615–2623. [Google Scholar]
- Pellissier, H. Recent Developments in Enantioselective Metal-Catalyzed Domino Reactions. Adv. Synth. Catal. 2019, 361, 1733–1755. [Google Scholar] [CrossRef]
- Cheng, H.-G.; Lu, L.-Q.; Wang, T.; Yang, Q.-Q.; Liu, X.-P.; Li, Y.; Deng, Q.-H.; Chen, J.-R.; Xiao, W.-J. Highly Enantioselective Friedel–Crafts Alkylation/N-Hemiacetalization Cascade Reaction with Indoles. Angew. Chem. Int. Ed. 2013, 52, 3250–3254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, X.; Zhao, X.; Zhang, J.; Zhou, L.; Lin, L.; Feng, X. Enantioselective Friedel–Crafts alkylation for synthesis of 2-substituted indole derivatives. Chem. Commun. 2013, 49, 11311–11313. [Google Scholar] [CrossRef]
- Li, N.-K.; Zhang, J.-Q.; Sun, B.-B.; Li, H.-Y.; Wang, X.-W. Chiral Diphosphine−Palladium-Catalyzed Sequential Asymmetric Double-Friedel−Crafts Alkylation and N-Hemiketalization for Spiropolycyclic Indole Derivatives. Org. Lett. 2017, 19, 1954–1957. [Google Scholar] [CrossRef]
- Krische, M.; Berl, V.; Grenzer, E.M.; Trost, B.M. Chemo-, Regio-, and Enantioselective Pd-Catalyzed Allylic Alkylation of Indolocarbazole Pro-aglycons. Org. Lett. 2002, 4, 2005–2008. [Google Scholar]
- Osipov, M.; Dong, G.; Trost, B.M. Palladium-Catalyzed Dynamic Kinetic Asymmetric Transformations of Vinyl Aziridines with Nitrogen Heterocycles: Rapid Access to Biologically Active Pyrroles and Indoles. J. Am. Chem. Soc. 2010, 132, 15800–15807. [Google Scholar]
- Levi, M.; Hartwing, J.F. Iridium-Catalyzed Regio- and Enantioselective N-Allylation of Indoles. Angew. Chem. Int. Ed. 2009, 48, 7841–7844. [Google Scholar]
- Ye, K.-Y.; Cheng, Q.; Zhou, C.-X.; Dai, L.-X.; You, S.-L. An Iridium(I) N-Heterocyclic Carbene Complex Catalyzes Asymmetric Intramolecular Allylic Amination Reactions. Angew. Chem. Int. Ed. 2016, 55, 8113–8116. [Google Scholar] [CrossRef]
- Chen, L.-Y.; Yu, X.-Y.; Chen, J.-R.; Feng, B.; Zhang, H.; Qi, Y.-H.; Xiao, W.-J. Enantioselective Direct Functionalization of Indoles by Pd/SulfoxidePhosphine-Catalyzed N-Allylic Alkylation. Org. Lett. 2015, 17, 1381–1384. [Google Scholar] [CrossRef] [PubMed]
- Sevov, C.S.; Zhou, J.; Hartwig, J.F. Iridium-Catalyzed, Intermolecular Hydroamination of Unactivated Alkenes with Indoles. J. Am. Chem. Soc. 2014, 136, 3200–3207. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.R.; Bahamonde, A.; Farukawa, Y.; Sigman, M.S. Enantioselective N-Alkylation of Indoles via an Intermolecular AzaWacker-Type Reaction. J. Am. Chem. Soc. 2019, 141, 8670–8674. [Google Scholar] [CrossRef]
- Abreu, D.; Belmont, M.; Brachet, E. Synergistic Photoredox/Transition-Metal Catalysis for Carbon–Carbon Bond Formation Reactions. Eur. J. Org. Chem. 2020, 2020, 1327–1378. [Google Scholar] [CrossRef]
- Jiang, C.; Chen, W.; Zheng, W.-H.; Lu, H. Advances in asymmetric visible-light photocatalysis, 2015–2019. Org. Biomol. Chem. 2019, 17, 8673–8689. [Google Scholar] [CrossRef] [PubMed]
- Kainz, Q.M.; Matier, C.D.; Bartoszewicz, A.; Zultanski, S.L.; Peters, J.C.; Fu, G.C. Asymmetric copper-catalyzed C-N cross-couplings induced by visible light. Science 2016, 351, 681–684. [Google Scholar] [CrossRef]
- Bartoszewicz, A.; Matier, C.D.; Fu, G.C. Enantioconvergent Alkylations of Amines by Alkyl Electrophiles: Copper-Catalyzed Nucleophilic Substitutions of Racemic α-Halolactams by Indoles. J. Am. Chem. Soc. 2019, 141, 14864–14869. [Google Scholar] [CrossRef]
- Gnanamani, E.; Hung, C.-I.; Trost, B.M. Controlling Regioselectivity in the Enantioselective N-Alkylation of Indole Analogues Catalyzed by Dinuclear Zinc-ProPhenol. Angew. Chem. Int. Ed. 2017, 56, 10451–10456. [Google Scholar]
- Roy, R.; Saha, S. Scope and advances in the catalytic propargylic substitution reaction. RSC Adv. 2018, 8, 31129–31193. [Google Scholar] [CrossRef]
- Lauder, K.; Toscani, A.; Scalacci, N.; Castagnolo, D. Synthesis and Reactivity of Propargylamines in Organic Chemistry. Chem. Rev. 2017, 117, 14091–14200. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, S.; Shan, W.; Shao, Z. Direct asymmetric N-propargylation of indoles and carbazoles catalyzed by lithium SPINOL phosphate. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-B.; Zhang, X.; Dai, L.-X.; You, S.-L. Asymmetric N-Allylation of Indoles Through the Iridium-Catalyzed Allylic Alkylation/Oxidation of Indolines. Angew. Chem. Int. Ed. 2012, 51, 5183–5187. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhuo, C.-X.; You, S.-L. Enantioselective synthesis of N-allylindoles via palladium-catalyzed allylic amination/oxidation of indolines. RSC Adv. 2014, 4, 10875–10878. [Google Scholar] [CrossRef]
- Zhu, F.; Hu, X. Enantioselective N-propargylation of indoles via Cu-catalyzed propargylic alkylation/dehydrogenation of indolines. Chin. J. Catal. 2015, 36, 86–92. [Google Scholar] [CrossRef]
- Chen, Q.-A.; Chen, Z.; Dong, V.M. Rhodium-Catalyzed Enantioselective Hydroamination of Alkynes with Indolines. J. Am. Chem. Soc. 2015, 137, 8392–8395. [Google Scholar] [CrossRef]





































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trubitsõn, D.; Kanger, T. Enantioselective Catalytic Synthesis of N-alkylated Indoles. Symmetry 2020, 12, 1184. https://doi.org/10.3390/sym12071184
Trubitsõn D, Kanger T. Enantioselective Catalytic Synthesis of N-alkylated Indoles. Symmetry. 2020; 12(7):1184. https://doi.org/10.3390/sym12071184
Chicago/Turabian StyleTrubitsõn, Dmitri, and Tõnis Kanger. 2020. "Enantioselective Catalytic Synthesis of N-alkylated Indoles" Symmetry 12, no. 7: 1184. https://doi.org/10.3390/sym12071184
APA StyleTrubitsõn, D., & Kanger, T. (2020). Enantioselective Catalytic Synthesis of N-alkylated Indoles. Symmetry, 12(7), 1184. https://doi.org/10.3390/sym12071184