Complement Activation in the Treatment of B-Cell Malignancies


{kind=link}
{kind=link}
Abstract
1. Introduction
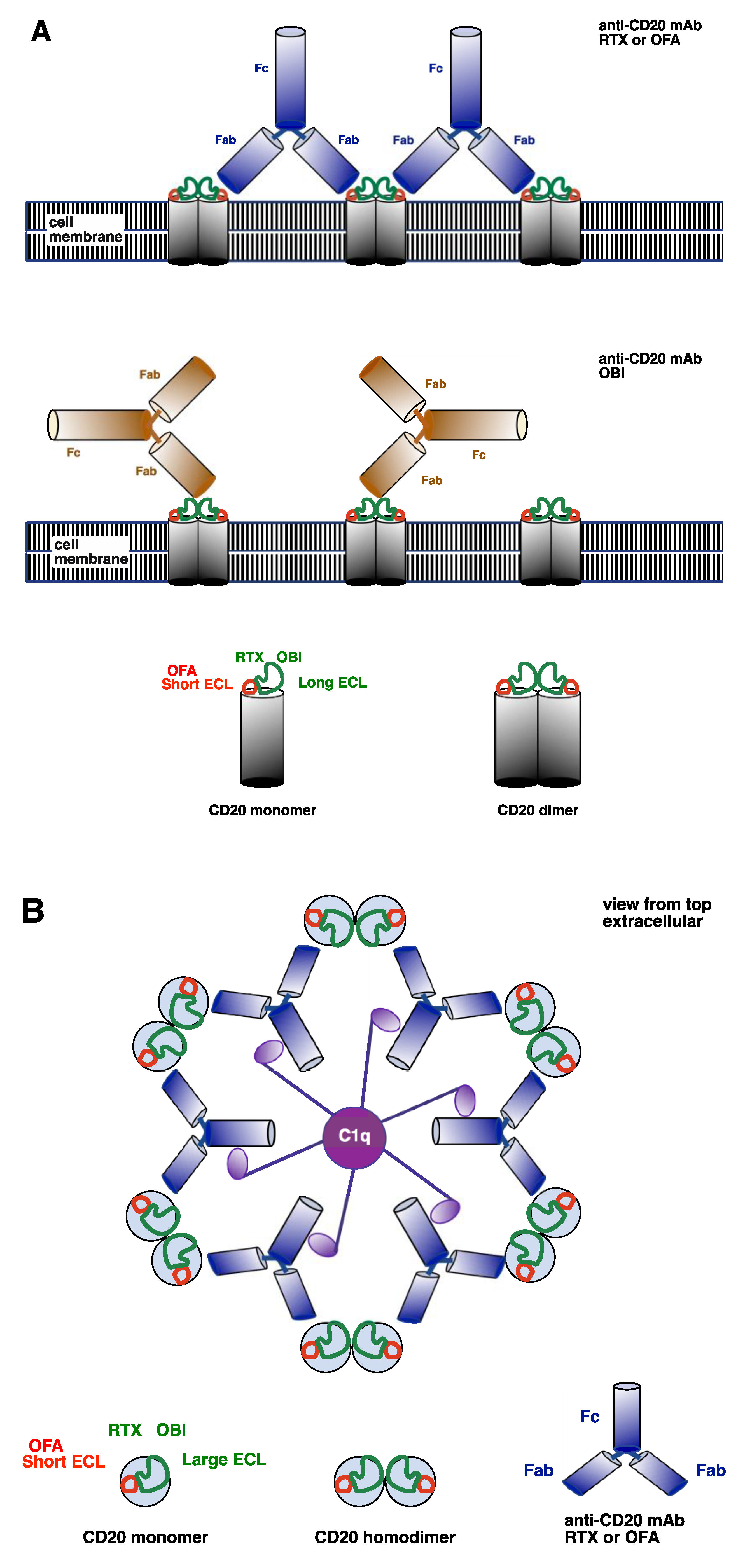
2. Complement-Activating Therapeutic mAb
3. Mechanism of Action of mAb
4. Complement Activation and CDC
4.1. Pharmacokinetics
4.2. Pharmacodynamics
4.3. Complement Levels
4.4. Complement Regulatory Proteins (CRP)
4.5. mAb Antigen
4.6. Intrinsic Cellular Resistance
5. Complement Activation and cADCP
5.1. cADCP Signaling
5.2. Crosstalk between FcγR- and cADCP
5.3. Importance of cADCP in mAb Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Coiffier, B.; Lepage, E.; Brière, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Neste, E.V.D.; Salles, G.; Gaulard, P.; et al. CHOP Chemotherapy plus Rituximab Compared with CHOP Alone in Elderly Patients with Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2002, 346, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Habermann, T.M.; Weller, E.A.; Morrison, V.A.; Gascoyne, R.D.; Cassileth, P.A.; Cohn, J.B.; Dakhil, S.R.; Woda, B.; Fisher, R.I.; Peterson, B.A.; et al. Rituximab-CHOP Versus CHOP Alone or with Maintenance Rituximab in Older Patients with Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2006, 24, 3121–3127. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.; Imrie, K.; Belch, A.; Cunningham, D.; Flores, E.; Catalano, J.; Solal-Celigny, P.; Offner, F.; Walewski, J.; Raposo, J.; et al. CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood 2005, 105, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Schulz, H.; Bohlius, J.F.; Trelle, S.; Skoetz, N.; Reiser, M.; Kober, T.; Schwarzer, G.; Herold, M.; Dreyling, M.; Hallek, M.; et al. Immunochemotherapy with Rituximab and Overall Survival in Patients with Indolent or Mantle Cell Lymphoma: A Systematic Review and Meta-analysis. J. Natl. Cancer Inst. 2007, 99, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Fischer, K.; Fingerle-Rowson, G.; Fink, A.M.; Busch, R.; Mayer, J.; Hensel, M.; Hopfinger, G.; Hess, G.; Von Grünhagen, U.; et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: A randomised, open-label, phase 3 trial. Lancet 2010, 376, 1164–1174. [Google Scholar] [CrossRef]
- Uchida, J.; Hamaguchi, Y.; Oliver, J.A.; Ravetch, J.V.; Poe, J.C.; Haas, K.M.; Tedder, T.F. The Innate Mononuclear Phagocyte Network Depletes B Lymphocytes through Fc Receptor–dependent Mechanisms during Anti-CD20 Antibody Immunotherapy. J. Exp. Med. 2004, 199, 1659–1669. [Google Scholar] [CrossRef]
- Bowles, J.A.; Wang, S.-Y.; Link, B.K.; Allan, B.; Beuerlein, G.; Campbell, M.-A.; Marquis, D.; Ondek, B.; Wooldridge, J.E.; Smith, B.J.; et al. Anti-CD20 monoclonal antibody with enhanced affinity for CD16 activates NK cells at lower concentrations and more effectively than rituximab. Blood 2006, 108, 2648–2654. [Google Scholar] [CrossRef]
- Beum, P.V.; Lindorfer, M.A.; Taylor, R.P. Within Peripheral Blood Mononuclear Cells, Antibody-Dependent Cellular Cytotoxicity of Rituximab-Opsonized Daudi cells Is Promoted by NK Cells and Inhibited by Monocytes due to Shaving. J. Immunol. 2008, 181, 2916–2924. [Google Scholar] [CrossRef]
- Montalvao, F.; Garcia, Z.; Celli, S.; Breart, B.; Deguine, J.; Van Rooijen, N.; Bousso, P. The mechanism of anti-CD20–mediated B cell depletion revealed by intravital imaging. J. Clin. Investig. 2013, 123, 5098–5103. [Google Scholar] [CrossRef]
- Gül, N.; Babes, L.; Siegmund, K.; Korthouwer, R.; Bögels, M.; Braster, R.; Vidarsson, G.; Hagen, T.L.T.; Kubes, P.; Van Egmond, M. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J. Clin. Investig. 2014, 124, 812–823. [Google Scholar] [CrossRef]
- Gül, N.; Van Egmond, M. Antibody-Dependent Phagocytosis of Tumor Cells by Macrophages: A Potent Effector Mechanism of Monoclonal Antibody Therapy of Cancer. Cancer Res. 2015, 75, 5008–5013. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, C.L.; Montalvao, F.; Celli, S.; Michonneau, D.; Breart, B.; Garcia, Z.; Perro, M.; Freytag, O.; Gerdes, C.A.; Bousso, P. Intravital imaging reveals improved Kupffer cell-mediated phagocytosis as a mode of action of glycoengineered anti-CD20 antibodies. Sci. Rep. 2016, 6, srep34382. [Google Scholar] [CrossRef] [PubMed]
- Church, A.K.; VanDerMeid, K.R.; Baig, N.A.; Baran, A.M.; Witzig, T.E.; Nowakowski, G.S.; Zent, C.S. Anti-CD20 monoclonal antibody-dependent phagocytosis of chronic lymphocytic leukaemia cells by autologous macrophages. Clin. Exp. Immunol. 2015, 183, 90–101. [Google Scholar] [CrossRef]
- VanDerMeid, K.R.; Elliott, M.R.; Baran, A.M.; Barr, P.M.; Chu, C.C.; Zent, C.S. Cellular Cytotoxicity of Next-Generation CD20 Monoclonal Antibodies. Cancer Immunol. Res. 2018, 6, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.C.; Pinney, J.J.; Whitehead, H.E.; Rivera-Escalera, F.; VanDerMeid, K.R.; Zent, C.S.; Elliott, M.R. High-resolution quantification of discrete phagocytic events by live cell time-lapse high-content microscopy imaging. J. Cell Sci. 2020, 133, jcs237883. [Google Scholar] [CrossRef]
- Pinney, J.J.; Rivera-Escalera, F.; Chu, C.C.; Whitehead, H.E.; VanDerMeid, K.R.; Nelson, A.M.; Barbeau, M.C.; Zent, C.S.; Elliott, M.R. Macrophage hypophagia as a mechanism of innate immune exhaustion in mAb-induced cell clearance. Blood 2020, 136, 2065–2079. [Google Scholar] [CrossRef]
- Pavlasova, G.; Mraz, M. The regulation and function of CD20: An “enigma” of B-cell biology and targeted therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in cancer: Untangling an intricate relationship. Nat. Rev. Immunol. 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Bordron, A.; Bagacean, C.; Tempescul, A.; Berthou, C.; Bettacchioli, E.; Hillion, S.; Renaudineau, Y. Complement System: A Neglected Pathway in Immunotherapy. Clin. Rev. Allergy Immunol. 2020, 58, 155–171. [Google Scholar] [CrossRef]
- Kennedy, A.D.; Beum, P.V.; Solga, M.D.; DiLillo, D.J.; Lindorfer, M.A.; Hess, C.E.; Densmore, J.J.; Williams, M.E.; Taylor, R.P. Rituximab Infusion Promotes Rapid Complement Depletion and Acute CD20 Loss in Chronic Lymphocytic Leukemia. J. Immunol. 2004, 172, 3280–3288. [Google Scholar] [CrossRef]
- Zent, C.S.; Secreto, C.R.; LaPlant, B.R.; Bone, N.D.; Call, T.G.; Shanafelt, T.D.; Jelinek, D.F.; Tschumper, R.C.; Kay, N.E. Direct and complement dependent cytotoxicity in CLL cells from patients with high-risk early–intermediate stage chronic lymphocytic leukemia (CLL) treated with alemtuzumab and rituximab. Leuk. Res. 2008, 32, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Zent, C.S.; Elliott, M.R. Maxed out macs: Physiologic cell clearance as a function of macrophage phagocytic capacity. FEBS J. 2017, 284, 1021–1039. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Romain, G.; Yan, W.; Watanabe, M.; Charab, W.; Todorova, B.; Lee, J.; Triplett, K.; Donkor, M.; Lungu, O.I.; et al. IgG Fc domains that bind C1q but not effector Fcgamma receptors delineate the importance of complement-mediated effector functions. Nat. Immunol. 2017, 18, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Lukácsi, S.; Nagy-Baló, Z.; Erdei, A.; Sándor, N.; Bajtay, Z. The role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in complement-mediated phagocytosis and podosome formation by human phagocytes. Immunol. Lett. 2017, 189, 64–72. [Google Scholar] [CrossRef]
- Hamblin, T.J.; Abdul-Ahad, A.K.; Gordon, J.; Stevenson, F.K.; Stevenson, G.T. Preliminary experience in treating lymphocytic leukaemia with antibody to immunoglobulin idiotypes on the cell surfaces. Br. J. Cancer 1980, 42, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Maloney, D.G.; Grillo-Lopez, A.J.; White, C.A.; Bodkin, D.; Schilder, R.J.; Neidhart, J.A.; Janakiraman, N.; Foon, K.A.; Liles, T.-M.; Dallaire, B.K.; et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood 1997, 90, 2188–2195. [Google Scholar] [CrossRef]
- Coiffier, B.; Haioun, C.; Ketterer, N.; Engert, A.; Tilly, H.; Ma, D.; Johnson, P.; Lister, A.; Feuring-Buske, M.; Radford, J.A.; et al. Rituximab (anti-CD20 monoclonal antibody) for the treatment of patients with relapsing or refractory aggressive lymphoma: A multicenter phase II study. Blood 1998, 92, 27. [Google Scholar]
- Teeling, J.L.; French, R.R.; Cragg, M.S.; Brakel, J.V.D.; Pluyter, M.; Huang, H.; Chan, C.; Parren, P.W.H.I.; Hack, C.E.; DeChant, M.; et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood 2004, 104, 1793–1800. [Google Scholar] [CrossRef]
- Teeling, J.L.; Mackus, W.J.M.; Wiegman, L.J.J.M.; Brakel, J.H.N.V.D.; Beers, S.A.; French, R.R.; Van Meerten, T.; Ebeling, S.; Vink, T.; Slootstra, J.W.; et al. The Biological Activity of Human CD20 Monoclonal Antibodies Is Linked to Unique Epitopes on CD20. J. Immunol. 2006, 177, 362–371. [Google Scholar] [CrossRef]
- Mössner, E.; Brünker, P.; Moser, S.; Püntener, U.; Schmidt, C.; Herter, S.; Grau, R.; Gerdes, C.; Nopora, A.; Van Puijenbroek, E.; et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell–mediated B-cell cytotoxicity. Blood 2010, 115, 4393–4402. [Google Scholar] [CrossRef]
- Hale, G.; Clark, M.R.; Marcus, R.; Winter, G.; Dyer, M.J.; Phillips, J.; Riechmann, L.; Waldmann, H. Remission induction in non-hodgkin lymphoma with reshaped human monoclonal antibody campath-1h. Lancet 1988, 332, 1394–1399. [Google Scholar] [CrossRef]
- Xia, M.Q.; Hale, G.; Waldmann, H.; Meng-Qi, X. Efficient complement-mediated lysis of cells containing the CAMPATH-1 (CDw52) antigen. Mol. Immunol. 1993, 30, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Österborg, A.; Fassas, A.S.; Anagnostopoulos, A.; Dyer, M.J.S.; Catovsky, D.; Mellstedt, H. Humanized CD52 monoclonal antibody campath-1H as first-line treatment in chronic lymphocytic leukaemia. Br. J. Haematol. 1996, 93, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Keating, M.J.; Flinn, I.; Jain, V.; Binet, J.-L.; Hillmen, P.; Byrd, J.; Albitar, M.; Brettman, L.; Santabarbara, P.; Wacker, B.; et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: Results of a large international study. Blood 2002, 99, 3554–3561. [Google Scholar] [CrossRef] [PubMed]
- Bowen, A.L.; Zomas, A.; Emmett, E.; Matutes, E.; Dyer, M.J.; Catovsky, D. Subcutaneous CAMPATH-1H in fludarabine-resistant/relapsed chronic lymphocytic and B-prolymphocytic leukaemia. Br. J. Haematol. 1997, 96, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Hillmen, P.; Skotnicki, A.B.; Robak, T.; Jaksic, B.; Dmoszynska, A.; Wu, J.; Sirard, C.; Mayer, J. Alemtuzumab Compared with Chlorambucil As First-Line Therapy for Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2007, 25, 5616–5623. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.; Dearden, C.E. B and T cell prolymphocytic leukaemia. Best Pr. Res. Clin. Haematol. 2019, 32, 217–228. [Google Scholar] [CrossRef]
- Van De Donk, N.W.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef] [PubMed]
- De Weers, M.; Tai, Y.-T.; Van Der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Moreno, L.; Perez, C.; Zabaleta, A.; Manrique, I.; Alignani, D.; Ajona, D.; Blanco, L.; Lasa, M.; Maiso, P.; Rodriguez, I.; et al. The Mechanism of Action of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef]
- Golay, J.; Zaffaroni, L.; Vaccari, T.; Lazzari, M.; Borleri, G.M.; Bernasconi, S.; Tedesco, F.; Rambaldi, A.; Introna, M. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood 2000, 95, 3900–3908. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Solga, M.D.; Schuman, T.A.; Chi, A.W.; Lindorfer, M.A.; Sutherland, W.M.; Foley, P.L.; Taylor, R.P. An anti-C3b(i) mAb enhances complement activation, C3b(i) deposition, and killing of CD20+ cells by rituximab. Blood 2003, 101, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Lui, G.; Chaperot, L.; Gressin, R.; Molens, J.-P.; Jacob, M.-C.; Sotto, J.-J.; Leroux, D.; Bensa, J.-C.; Plumas, J. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood 2003, 101, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Zent, C.S.; Chen, J.B.; Kurten, R.C.; Kaushal, G.P.; Lacy, H.M.; Schichman, S.A. Alemtuzumab (CAMPATH 1H) does not kill chronic lymphocytic leukemia cells in serum free medium. Leuk. Res. 2004, 28, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Manganini, M.; Rambaldi, A.; Introna, M. Effect of alemtuzumab on neoplastic B cells. Haematologica 2004, 89, 1476–1483. [Google Scholar]
- Reff, M.E.; Carner, K.; Chambers, K.S.; Chinn, P.C.; Leonard, J.E.; Raab, R.; Newman, R.A.; Hanna, N.; Anderson, D.R. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 1994, 83, 435–445. [Google Scholar] [CrossRef]
- Williams, M.E.; Densmore, J.J.; Pawluczkowycz, A.W.; Beum, P.V.; Kennedy, A.D.; Lindorfer, M.A.; Hamil, S.H.; Eggleton, J.C.; Taylor, R.P. Thrice-Weekly Low-Dose Rituximab Decreases CD20 Loss via Shaving and Promotes Enhanced Targeting in Chronic Lymphocytic Leukemia. J. Immunol. 2006, 177, 7435–7443. [Google Scholar] [CrossRef]
- Aue, G.; Lindorfer, M.A.; Beum, P.V.; Pawluczkowycz, A.W.; Vire, B.; Hughes, T.; Taylor, R.P.; Wiestner, A. Fractionated subcutaneous rituximab is well-tolerated and preserves CD20 expression on tumor cells in patients with chronic lymphocytic leukemia. Haematologica 2009, 95, 329–332. [Google Scholar] [CrossRef]
- Baig, N.A.; Taylor, R.P.; Lindorfer, M.A.; Church, A.K.; LaPlant, B.R.; Pettinger, A.M.; Shanafelt, T.D.; Nowakowski, G.S.; Zent, C.S. Induced Resistance to Ofatumumab-Mediated Cell Clearance Mechanisms, Including Complement-Dependent Cytotoxicity, in Chronic Lymphocytic Leukemia. J. Immunol. 2014, 192, 1620–1629. [Google Scholar] [CrossRef]
- Zent, C.S.; Taylor, R.P.; Lindorfer, M.A.; Beum, P.V.; LaPlant, B.; Wu, W.; Call, T.G.; Bowen, D.A.; Conte, M.J.; Frederick, L.A.; et al. Chemoimmunotherapy for relapsed/refractory and progressive 17p13-deleted chronic lymphocytic leukemia (CLL) combining pentostatin, alemtuzumab, and low-dose rituximab is effective and tolerable and limits loss of CD20 expression by circulating CLL cells. Am. J. Hematol. 2014, 89, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Cartron, G.; Blasco, H.; Paintaud, G.; Watier, H.; Le Guellec, C. Pharmacokinetics of rituximab and its clinical use: Thought for the best use? Crit. Rev. Oncol. 2007, 62, 43–52. [Google Scholar] [CrossRef]
- Berinstein, N.L.; Grillo-Lopez, A.J.; White, C.A.; Bence-Bruckler, I.; Maloney, D.; Czuczman, M.; Green, D.; Rosenberg, J.; McLaughlin, P.; Shen, D. Association of serum Rituximab (IDEC–C2B8) concentration and anti-tumor response in the treatment of recurrent low-grade or follicular non-Hodgkin’s lymphoma. Ann. Oncol. 1998, 9, 995–1001. [Google Scholar] [CrossRef]
- Pawluczkowycz, A.W.; Beurskens, F.J.; Beum, P.V.; Lindorfer, M.A.; Van De Winkel, J.G.J.; Parren, P.W.H.I.; Taylor, R.P. Binding of Submaximal C1q Promotes Complement-Dependent Cytotoxicity (CDC) of B Cells Opsonized with Anti-CD20 mAbs Ofatumumab (OFA) or Rituximab (RTX): Considerably Higher Levels of CDC Are Induced by OFA than by RTX. J. Immunol. 2009, 183, 749–758. [Google Scholar] [CrossRef]
- Bondza, S.; Broeke, T.T.; Nestor, M.; Leusen, J.H.; Buijs, J. Bivalent binding on cells varies between anti-CD20 antibodies and is dose-dependent. mAbs 2020, 12, 1792673. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Planchais, C.; Fronzes, R.; Mouquet, H.; Reyes, N. Binding mechanisms of therapeutic antibodies to human CD20. Science 2020, 369, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Rougé, L.; Chiang, N.; Steffek, M.; Kugel, C.; Croll, T.I.; Tam, C.; Estevez, A.; Arthur, C.P.; Koth, C.M.; Ciferri, C.; et al. Structure of CD20 in complex with the therapeutic monoclonal antibody rituximab. Science 2020, 367, 1224–1230. [Google Scholar] [CrossRef]
- Diebolder, C.A.; Beurskens, F.J.; De Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.R.; et al. Complement Is Activated by IgG Hexamers Assembled at the Cell Surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef]
- Cragg, M.S.; Glennie, M.J. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood 2004, 103, 2738–2743. [Google Scholar] [CrossRef]
- Herter, S.; Herting, F.; Mundigl, O.; Waldhauer, I.; Weinzierl, T.; Fauti, T.; Muth, G.; Ziegler-Landesberger, D.; Van Puijenbroek, E.; Lang, S.; et al. Preclinical Activity of the Type II CD20 Antibody GA101 (Obinutuzumab) Compared with Rituximab and Ofatumumab In Vitro and in Xenograft Models. Mol. Cancer Ther. 2013, 12, 2031–2042. [Google Scholar] [CrossRef]
- Schlesinger, M.; Broman, I.; Lugassy, G. The complement system is defective in chronic lymphatic leukemia patients and in their healthy relatives. Leukemia 1996, 10, 1509–1513. [Google Scholar] [PubMed]
- Middleton, O.; Cosimo, E.; Dobbin, E.; McCaig, A.M.; Clarke, C.L.; Brant, A.M.; Leach, M.; Michie, A.M.; Wheadon, H. Complement deficiencies limit CD20 monoclonal antibody treatment efficacy in CLL. Leukemia 2015, 29, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Klepfish, A.; Rachmilewitz, E.; Kotsianidis, I.; Patchenko, P.; Schattner, A. Adding fresh frozen plasma to rituximab for the treatment of patients with refractory advanced CLL. QJM Int. J. Med. 2008, 101, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Lazzari, M.; Facchinetti, V.; Bernasconi, S.; Borleri, G.; Barbui, T.; Rambaldi, A.; Introna, M. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: Further regulation by CD55 and CD59. Blood 2001, 98, 3383–3389. [Google Scholar] [CrossRef]
- Hu, W.; Ge, X.; You, T.; Xu, T.; Zhang, J.; Wu, G.; Peng, Z.; Chorev, M.; Aktas, B.H.; Halperin, J.A.; et al. Human CD59 Inhibitor Sensitizes Rituximab-Resistant Lymphoma Cells to Complement-Mediated Cytolysis. Cancer Res. 2011, 71, 2298–2307. [Google Scholar] [CrossRef]
- Beum, P.V.; Kennedy, A.D.; Williams, M.E.; Lindorfer, M.A.; Taylor, R.P. The Shaving Reaction: Rituximab/CD20 Complexes Are Removed from Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia Cells by THP-1 Monocytes. J. Immunol. 2006, 176, 2600–2609. [Google Scholar] [CrossRef]
- Taylor, R.P.; Lindorfer, M.A. Fcgamma-receptor-mediated trogocytosis impacts mAb-based therapies: Historical precedence and recent developments. Blood 2015, 125, 762–766. [Google Scholar] [CrossRef]
- Valgardsdottir, R.; Cattaneo, I.; Klein, C.; Introna, M.; Figliuzzi, M.; Golay, J. Human neutrophils mediate trogocytosis rather than phagocytosis of CLL B cells opsonized with anti-CD20 antibodies. Blood 2017, 129, 2636–2644. [Google Scholar] [CrossRef]
- Beum, P.V.; Peek, E.M.; Lindorfer, M.A.; Beurskens, F.J.; Engelberts, P.J.; Parren, P.W.H.I.; Van De Winkel, J.G.J.; Taylor, R.P.; Labrijn, A.F.; Meesters, J.; et al. Loss of CD20 and Bound CD20 Antibody from Opsonized B Cells Occurs More Rapidly Because of Trogocytosis Mediated by Fc Receptor-Expressing Effector Cells Than Direct Internalization by the B Cells. J. Immunol. 2011, 187, 3438–3447. [Google Scholar] [CrossRef]
- Beers, S.A.; French, R.R.; Chan, H.T.C.; Lim, S.H.; Jarrett, T.C.; Vidal, R.M.; Wijayaweera, S.S.; Dixon, S.V.; Kim, H.; Cox, K.L.; et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: Implications for antibody selection. Blood 2010, 115, 5191–5201. [Google Scholar] [CrossRef]
- Baig, N.A.; Taylor, R.P.; Lindorfer, M.A.; Church, A.K.; Laplant, B.R.; Pavey, E.S.; Nowakowski, G.S.; Zent, C.S. Complement dependent cytotoxicity (CDC) in chronic lymphocytic leukemia (CLL): Ofatumumab enhances alemtuzumab CDC and reveals cells resistant to activated complement. Leuk. Lymphoma 2012, 53, 2218–2227. [Google Scholar] [CrossRef] [PubMed]
- Lindorfer, M.A.; Cook, E.M.; Tupitza, J.C.; Zent, C.S.; Burack, R.; De Jong, R.N.; Beurskens, F.J.; Schuurman, J.; Parren, P.W.; Taylor, R.P. Real-time analysis of the detailed sequence of cellular events in mAb-mediated complement-dependent cytotoxicity of B-cell lines and of chronic lymphocytic leukemia B-cells. Mol. Immunol. 2016, 70, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Jaumouillé, V.; Grinstein, S. The Cell Biology of Phagocytosis. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 61–98. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.D.; Reed, W.; Dalzell, J.G.; Becker, S.E.; Hogg, N. Macrophage cytoskeleton association with CR3 and CR4 regulates receptor mobility and phagocytosis of iC3b-opsonized erythrocytes. J. Leukoc. Biol. 1992, 51, 109–117. [Google Scholar] [CrossRef]
- Wiedemann, A.; Patel, J.C.; Lim, J.; Tsun, A.; van Kooyk, Y.; Caron, E. Two distinct cytoplasmic regions of the beta2 integrin chain regulate RhoA function during phagocytosis. J Cell Biol. 2006, 172, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Olazabal, I.M.; Caron, E.; May, R.C.; Schilling, K.; Knecht, D.A.; Machesky, L.M. Rho-Kinase and Myosin-II Control Phagocytic Cup Formation during CR, but Not FcγR, Phagocytosis. Curr. Biol. 2002, 12, 1413–1418. [Google Scholar] [CrossRef]
- Colucci-Guyon, E.; Niedergang, F.; Wallar, B.J.; Peng, J.; Alberts, A.S.; Chavrier, P. A Role for Mammalian Diaphanous-Related Formins in Complement Receptor (CR3)-Mediated Phagocytosis in Macrophages. Curr. Biol. 2005, 15, 2007–2012. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, F.; Brumell, J.; Al-Alawi, N.; Latour, S.; Cheng, A.; Veillette, A.; Grinstein, S.; Pawson, T. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol. Cell Biol. 1998, 18, 4209–4220. [Google Scholar] [CrossRef] [PubMed]
- Lindorfer, M.A.; Kohl, J.; Taylor, R.P. Interactions between the complement system and Fcg receptors. In Antibody Fc: Linking Adaptive and Innate Immunity; Ackerman, M.E., Nimmerhahn, F., Eds.; Elsevier Press: Philadelphia, PA, USA, 2014; pp. 49–74. [Google Scholar]
- Schreiber, A.D.; Frank, M.M. Role of antibody and complement in the immune clearance and destruction of erythrocytes. I. In vivo effects of IgG and IgM complement-fixing sites. J. Clin. Investig. 1972, 51, 575–582. [Google Scholar] [CrossRef]
- Schreiber, A.D.; Frank, M.M. Role of Antibody and Complement in the Immune Clearance and Destruction of Erythrocytes II. Molecular nature of igg and igm complement-fixing sites and effects of their interaction with serum. J. Clin. Investig. 1972, 51, 583–589. [Google Scholar] [CrossRef]
- Brown, E.J.; Bohnsack, J.F.; Gresham, H.D. Mechanism of inhibition of immunoglobulin G-mediated phagocytosis by monoclonal antibodies that recognize the Mac-1 antigen. J. Clin. Investig. 1988, 81, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Jongstra-Bilen, J.; Harrison, R.; Grinstein, S. Fcgamma-receptors induce Mac-1 (CD11b/CD18) mobilization and accumulation in the phagocytic cup for optimal phagocytosis. J. Biol. Chem. 2003, 278, 45720–45729. [Google Scholar] [CrossRef] [PubMed]
- Shushakova, N.; Skokowa, J.; Schulman, J.; Baumann, U.; Zwirner, J.; Schmidt, R.E.; Gessner, J.E. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcgammaRs in immune complex-induced lung disease. J. Clin. Investig. 2002, 110, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-Y.; Hunter, S.; Chien, P.; Kim, M.-K.; Han-Kim, T.-H.; Indik, Z.K.; Schreiber, A.D. Interaction of Two Phagocytic Host Defense Systems. J. Biol. Chem. 2011, 286, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tohyama, Y.; Kadono, T.; He, J.; Miah, S.M.S.; Hazama, R.; Tanaka, C.; Tohyama, K.; Yamamura, H. Protein-tyrosine kinase Syk is required for pathogen engulfment in complement-mediated phagocytosis. Blood 2006, 107, 4554–4562. [Google Scholar] [CrossRef] [PubMed]
- Di Gaetano, N.; Cittera, E.; Nota, R.; Vecchi, A.; Grieco, V.; Scanziani, E.; Botto, M.; Introna, M.; Golay, J. Complement Activation Determines the Therapeutic Activity of Rituximab In Vivo. J. Immunol. 2003, 171, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Van Spriel, A.B.; Van Ojik, H.H.; Bakker, A.; Jansen, M.J.H.; Van De Winkel, J.G.J. Mac-1 (CD11b/CD18) is crucial for effective Fc receptor–mediated immunity to melanoma. Blood 2003, 101, 253–258. [Google Scholar] [CrossRef]
- Taylor, R.P.; Lindorfer, M.A.; Cook, E.M.; Beurskens, F.J.; Schuurman, J.; Parren, P.W.; Zent, C.S.; VanDerMeid, K.R.; Burack, R.; Mizuno, M.; et al. Hexamerization-enhanced CD20 antibody mediates complement-dependent cytotoxicity in serum genetically deficient in C9. Clin. Immunol. 2017, 181, 24–28. [Google Scholar] [CrossRef]
- Macor, P.; Secco, E.; Mezzaroba, N.; Zorzet, S.; Durigutto, P.; Gaiotto, T.; De Maso, L.; Biffi, S.; Garrovo, C.; Capolla, S.; et al. Bispecific antibodies targeting tumor-associated antigens and neutralizing complement regulators increase the efficacy of antibody-based immunotherapy in mice. Leukemia 2015, 29, 406–414. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zent, C.S.; Pinney, J.J.; Chu, C.C.; Elliott, M.R. Complement Activation in the Treatment of B-Cell Malignancies. Antibodies 2020, 9, 68. https://doi.org/10.3390/antib9040068
Zent CS, Pinney JJ, Chu CC, Elliott MR. Complement Activation in the Treatment of B-Cell Malignancies. Antibodies. 2020; 9(4):68. https://doi.org/10.3390/antib9040068
Chicago/Turabian StyleZent, Clive S., Jonathan J. Pinney, Charles C. Chu, and Michael R. Elliott. 2020. "Complement Activation in the Treatment of B-Cell Malignancies" Antibodies 9, no. 4: 68. https://doi.org/10.3390/antib9040068
APA StyleZent, C. S., Pinney, J. J., Chu, C. C., & Elliott, M. R. (2020). Complement Activation in the Treatment of B-Cell Malignancies. Antibodies, 9(4), 68. https://doi.org/10.3390/antib9040068