
Detailed Source-Specific Molecular Composition of Ambient Aerosol Organic Matter Using Ultrahigh Resolution Mass Spectrometry and 1H NMR

Abstract
:1. Introduction
2. Experiments
2.1. Aerosol Sample Collection
2.2. Aerosol Mass and Carbon Measurements
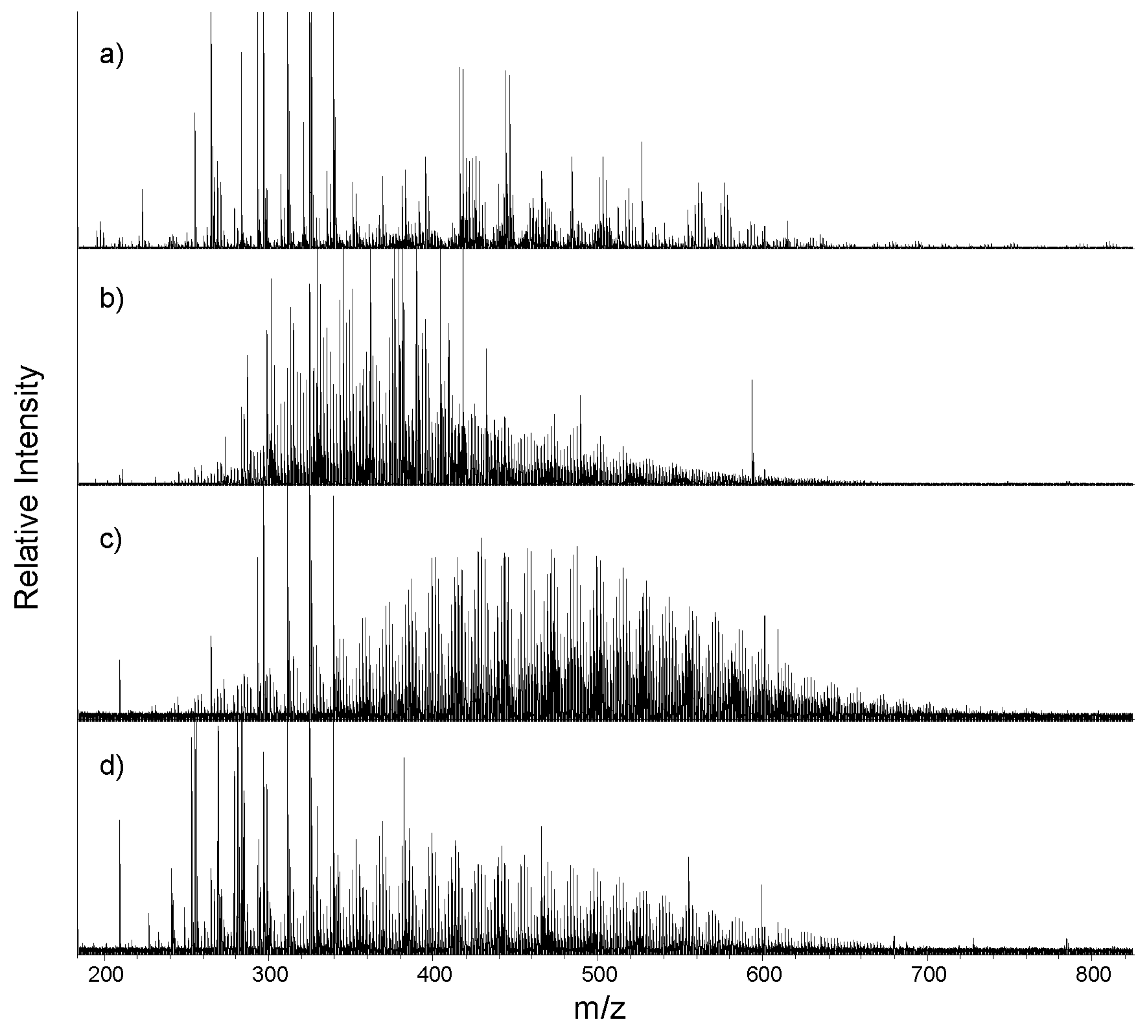
2.3. FTICR-MS Analysis
2.4. Molecular Formula Assignments
2.5. 1H NMR Spectroscopy
2.6. Principal Component Analysis
2.6.1. FTICR-MS PCA
2.6.2. 1H NMR PCA
3. Results and Discussion
3.1. Aerosol Loadings
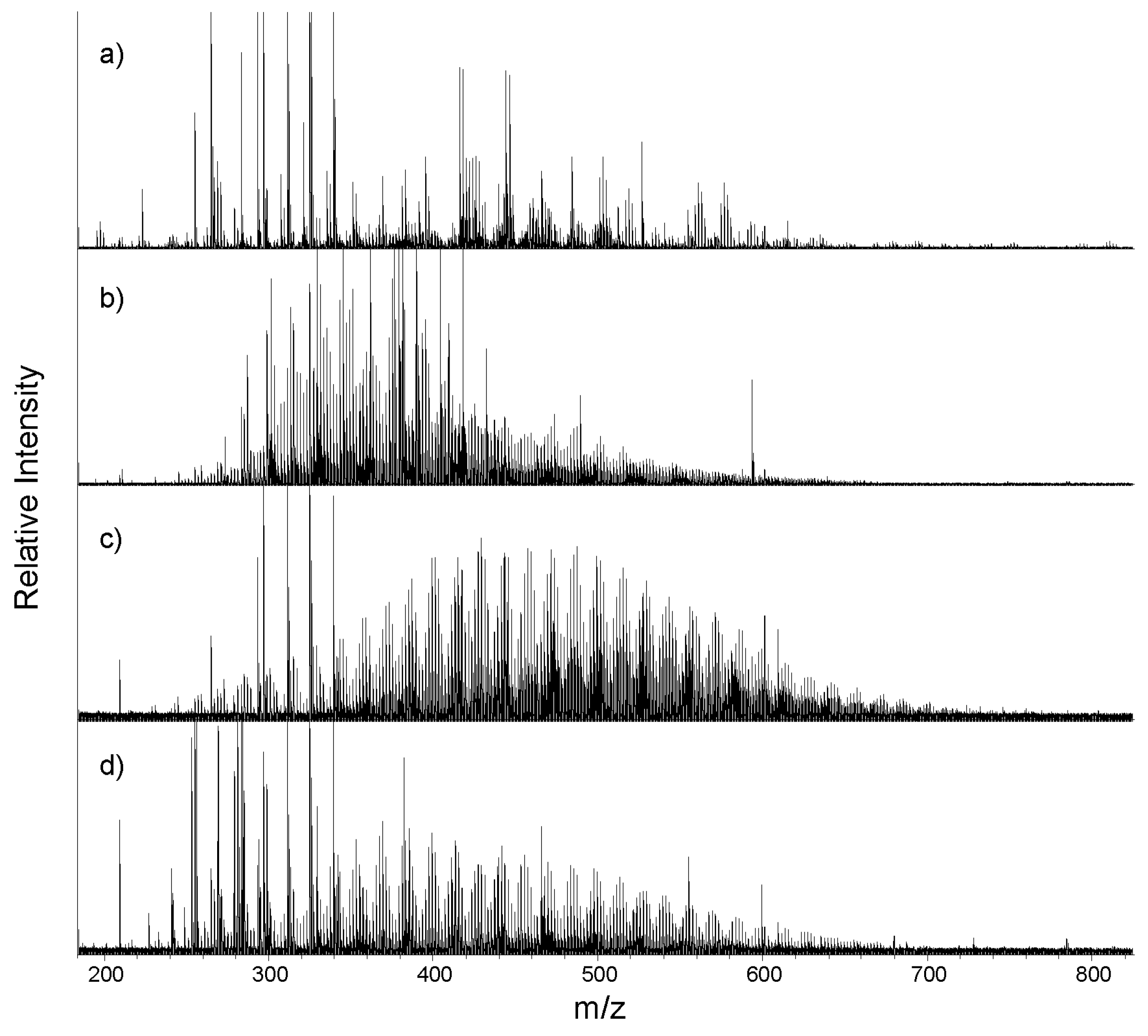
3.2. Mass Spectra and Average Source Molecular Characteristics
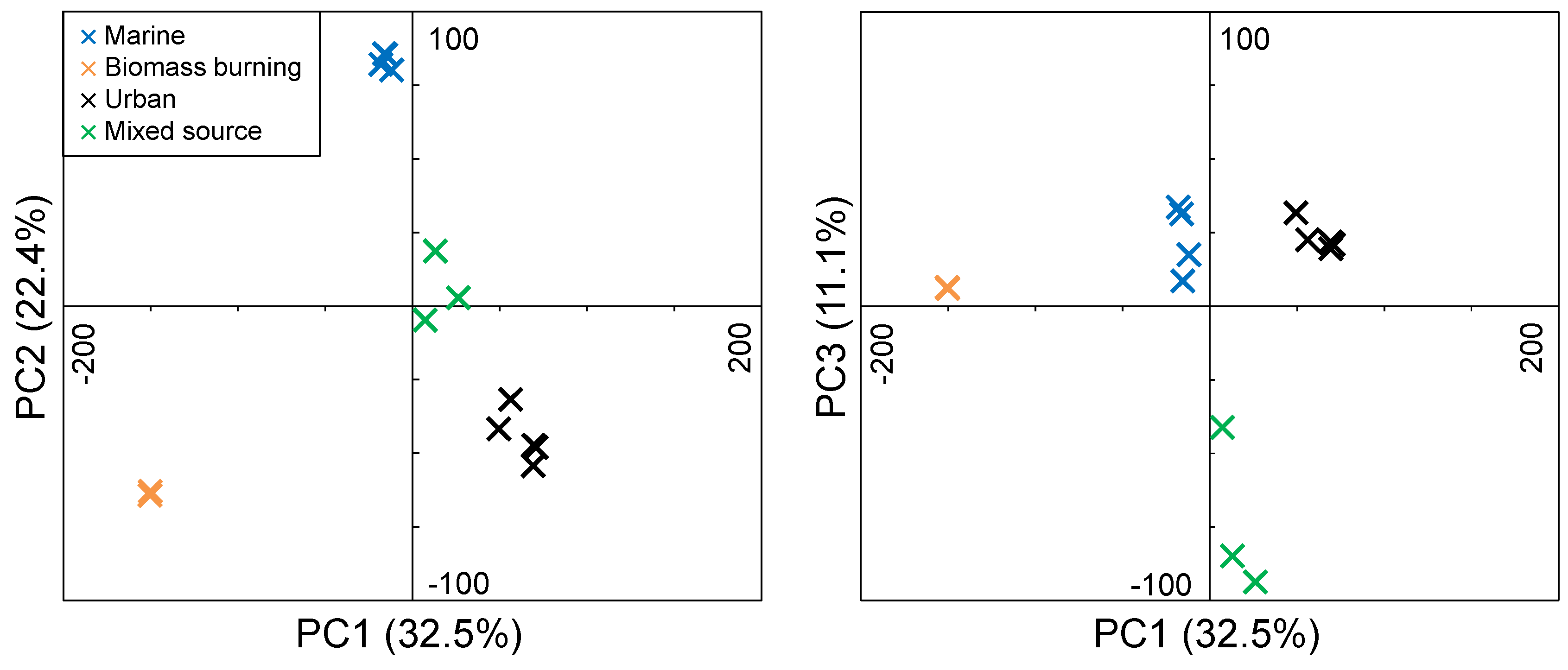
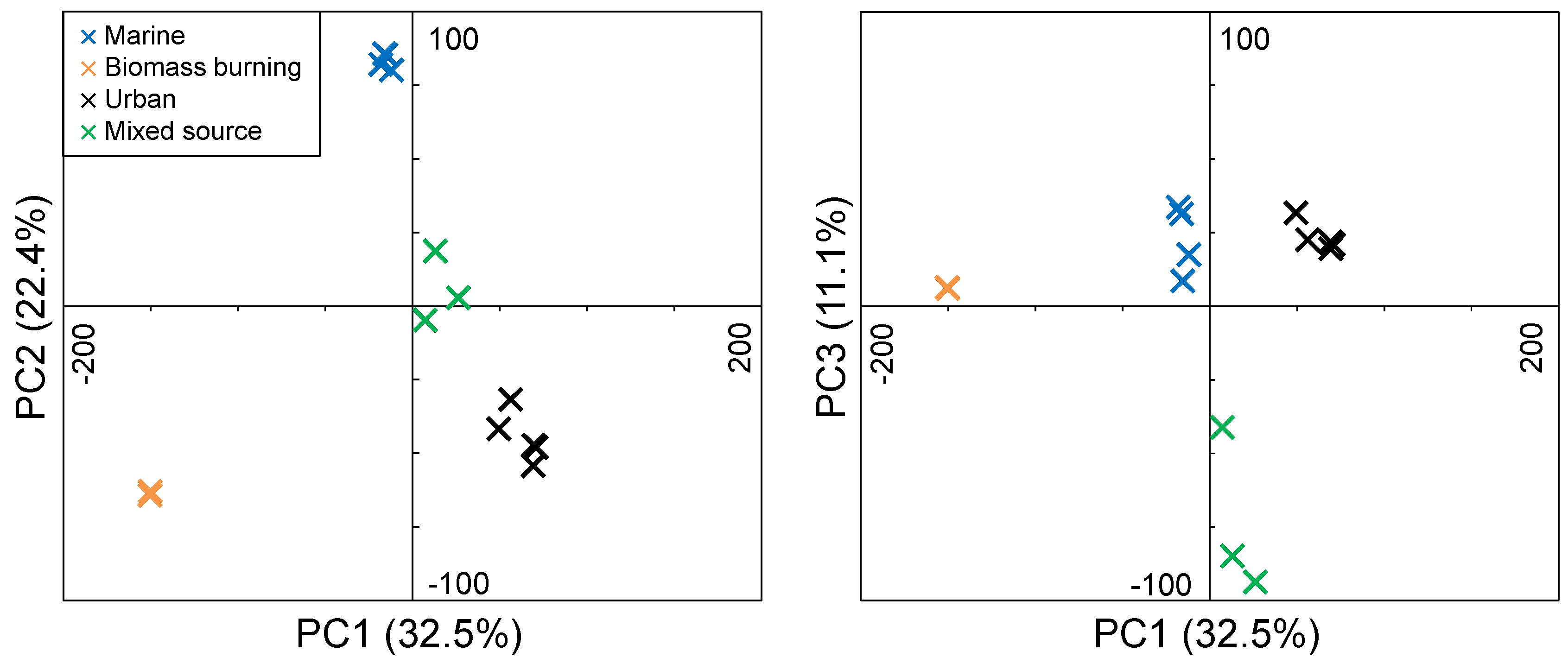
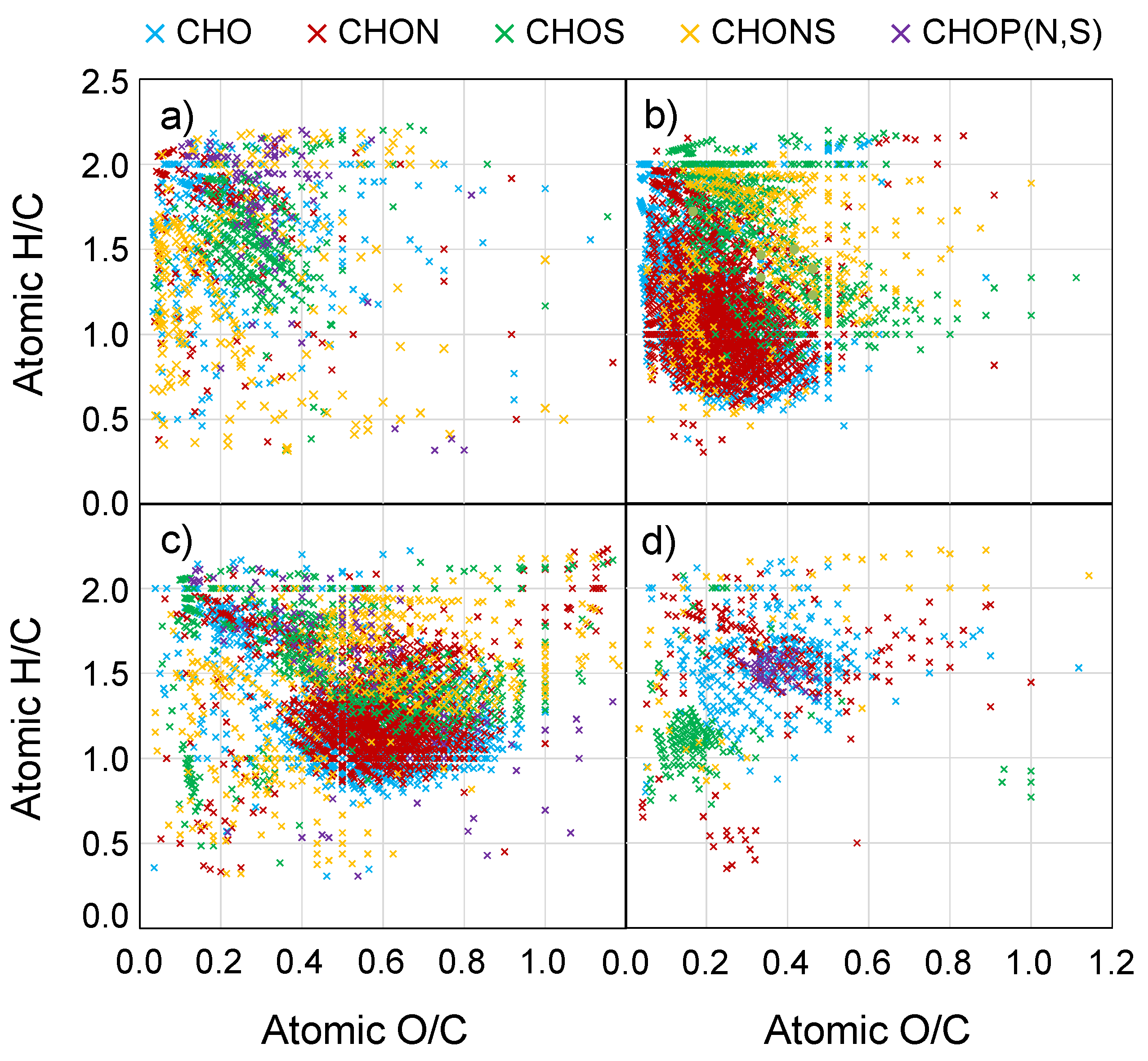
3.3. FTICR-MS PCA
3.3.1. Marine Aerosols
3.3.2. Biomass Burning Aerosols
3.3.3. Urban Aerosols
3.3.4. Mixed Source Aerosols
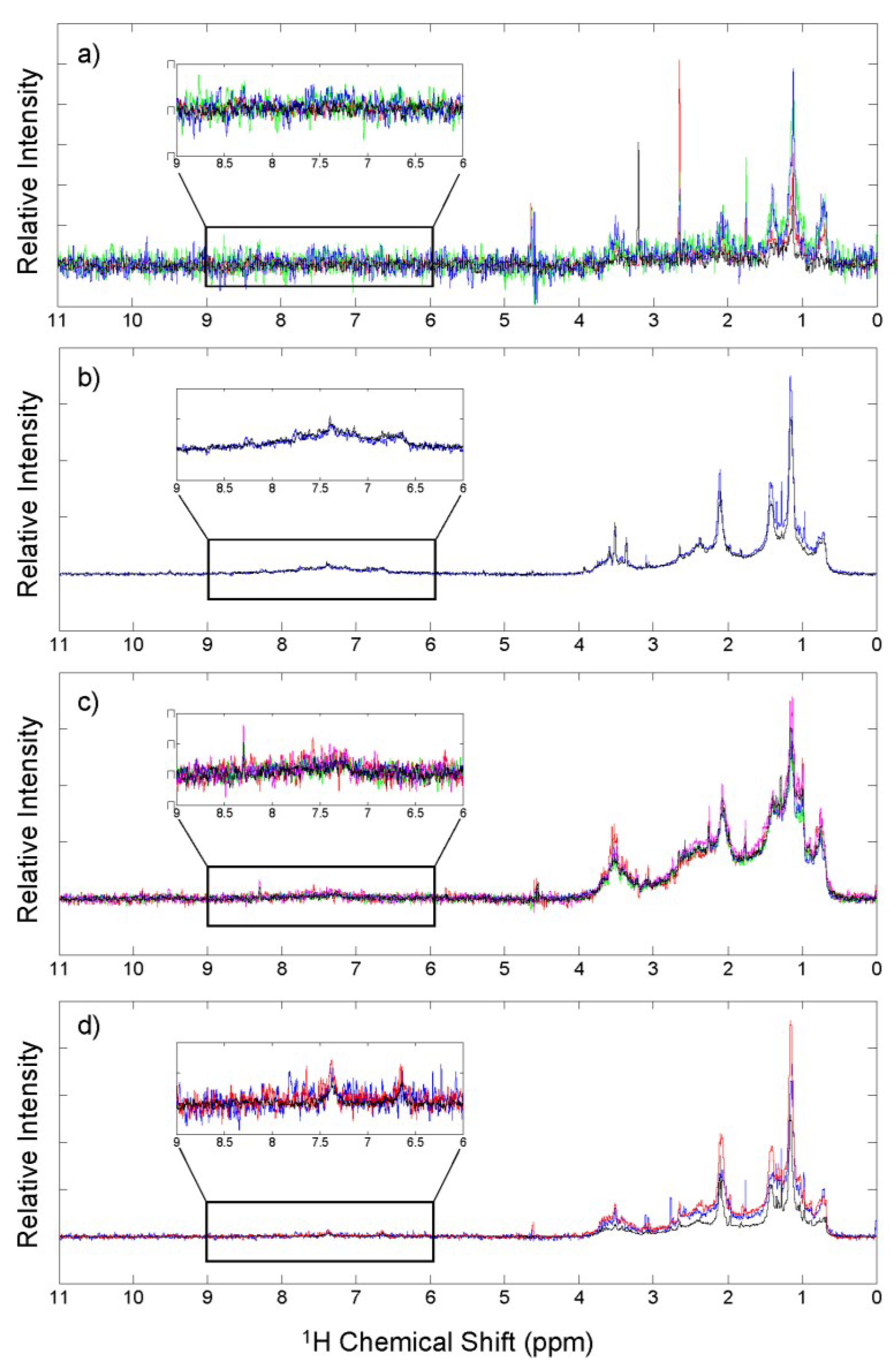
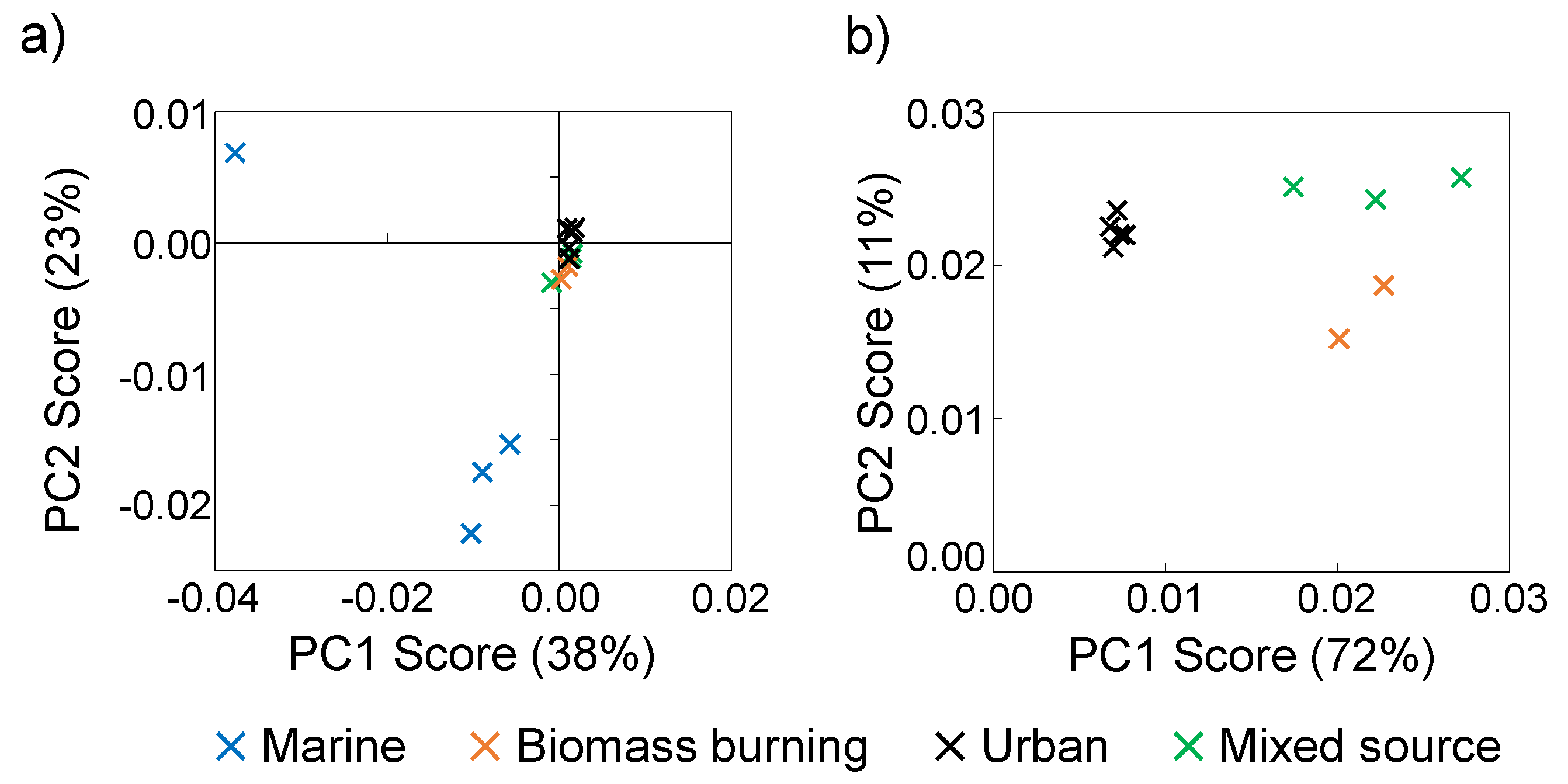
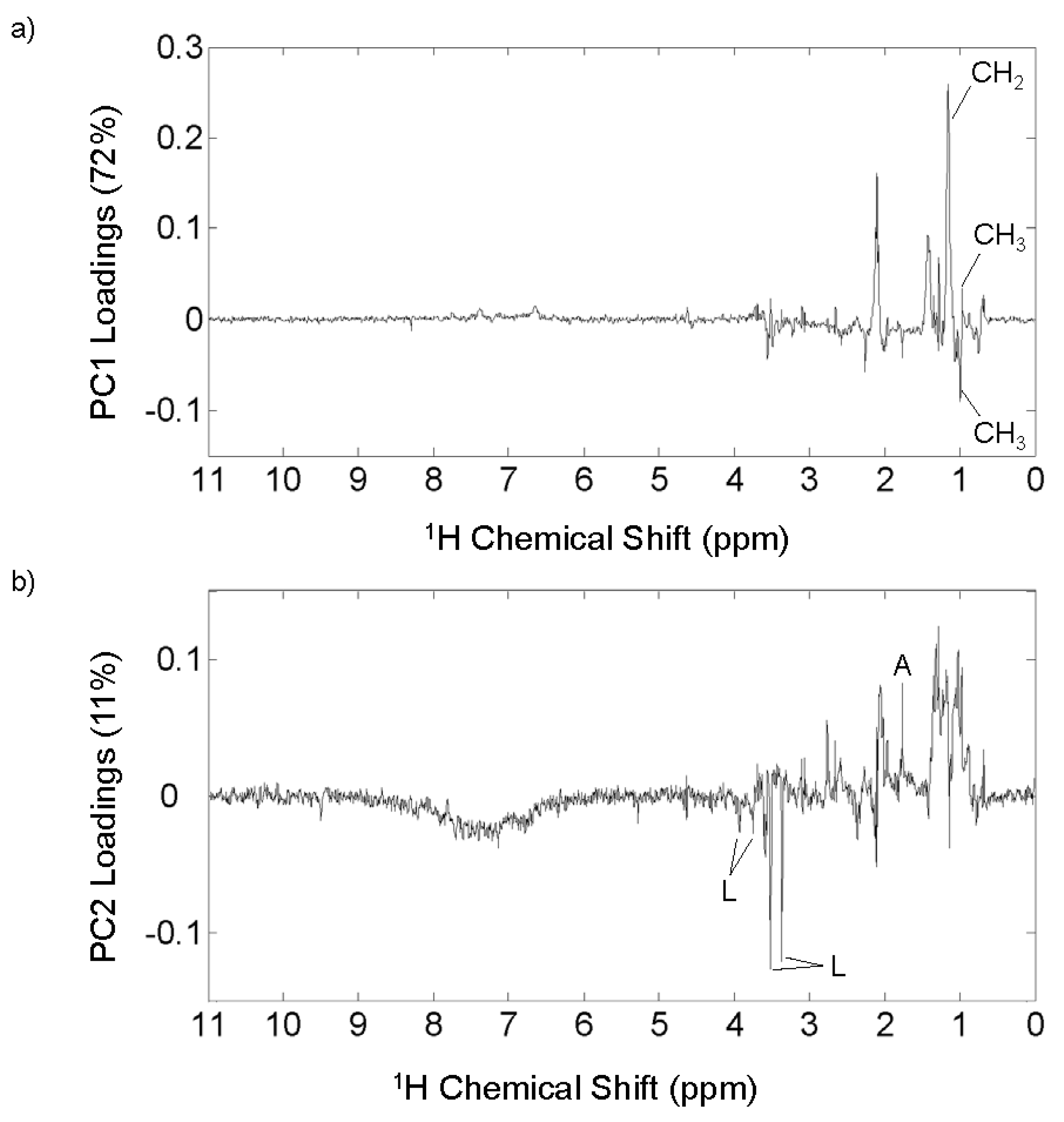
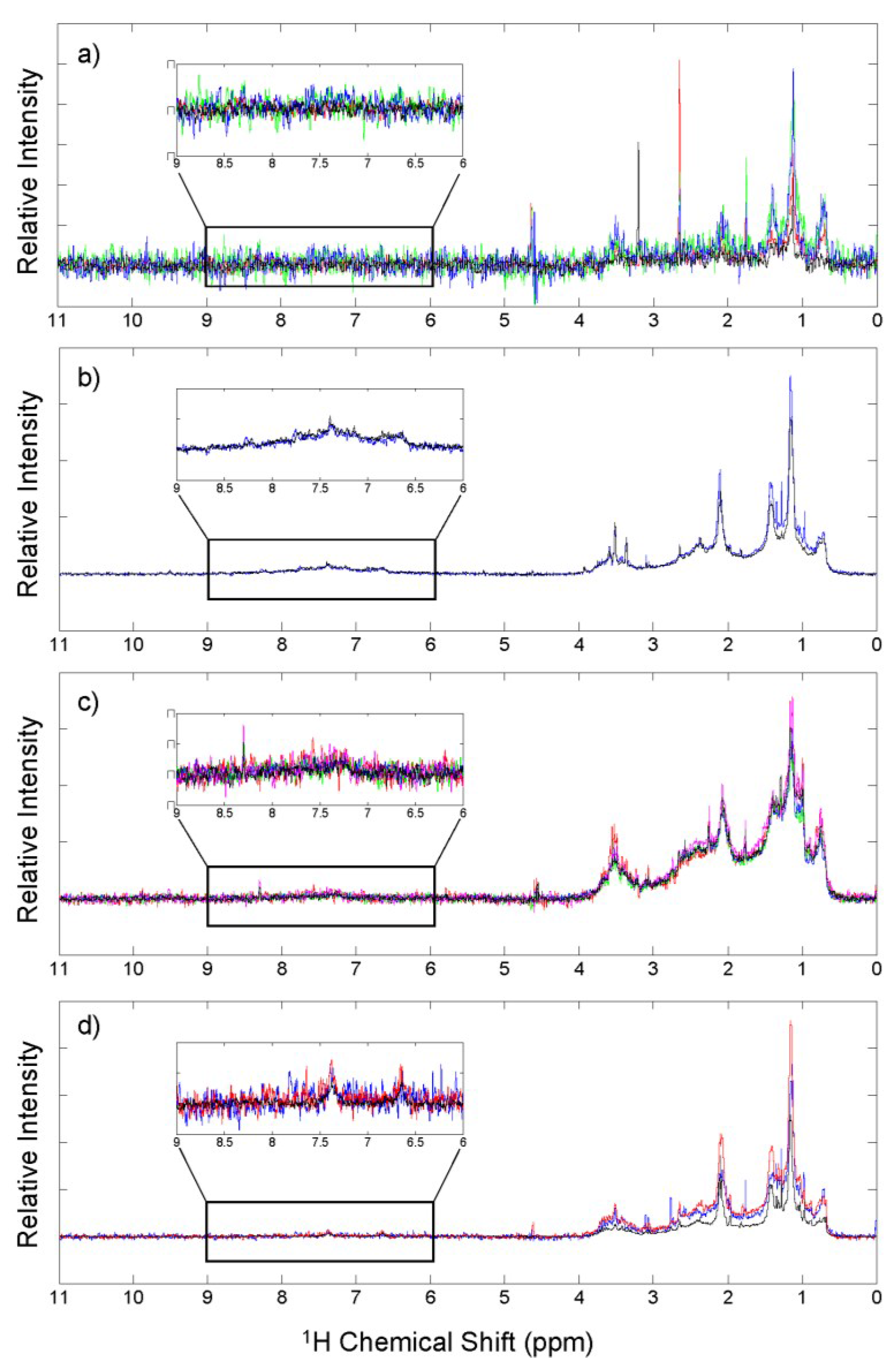
3.4. 1H NMR Analysis
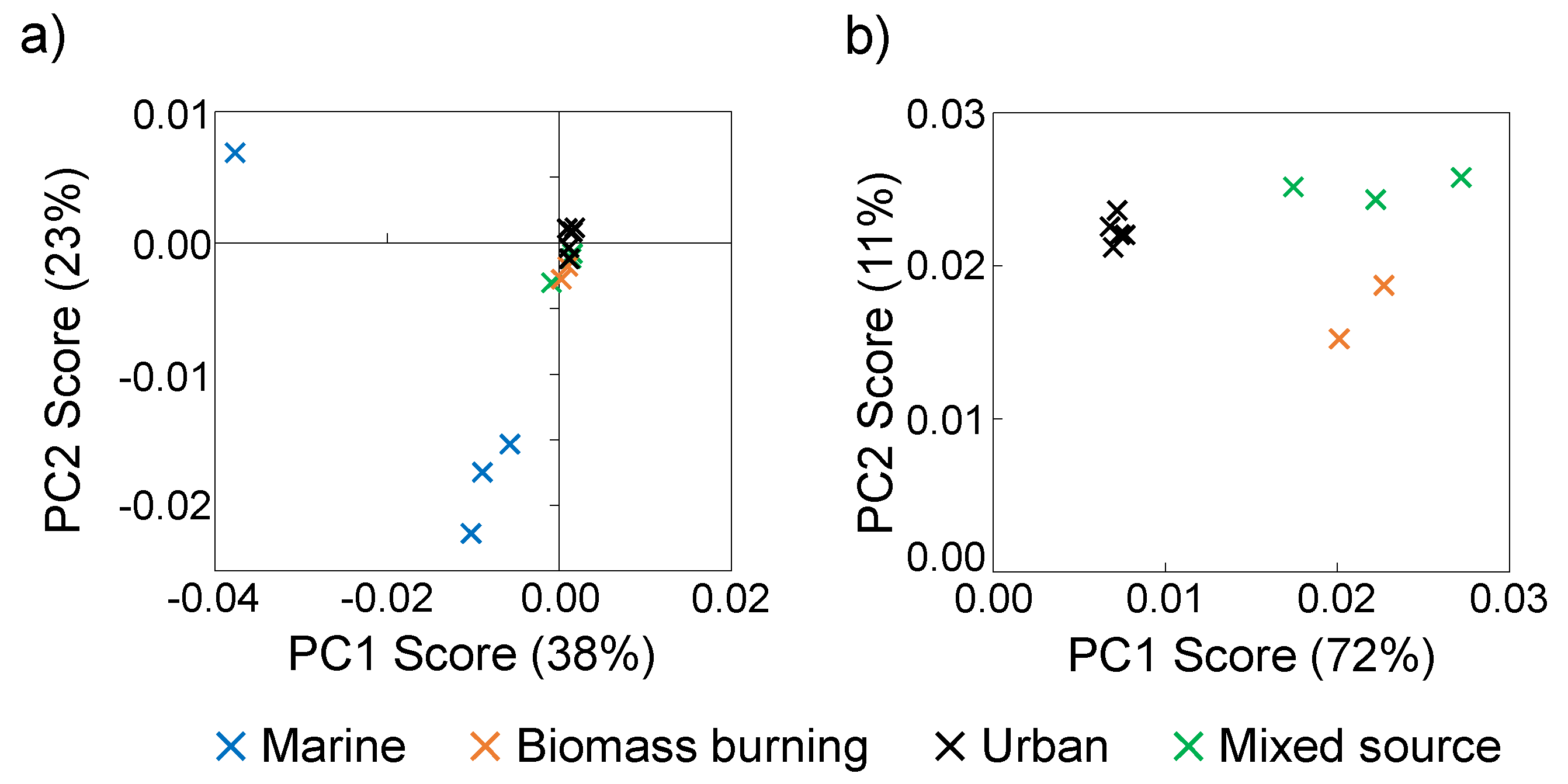
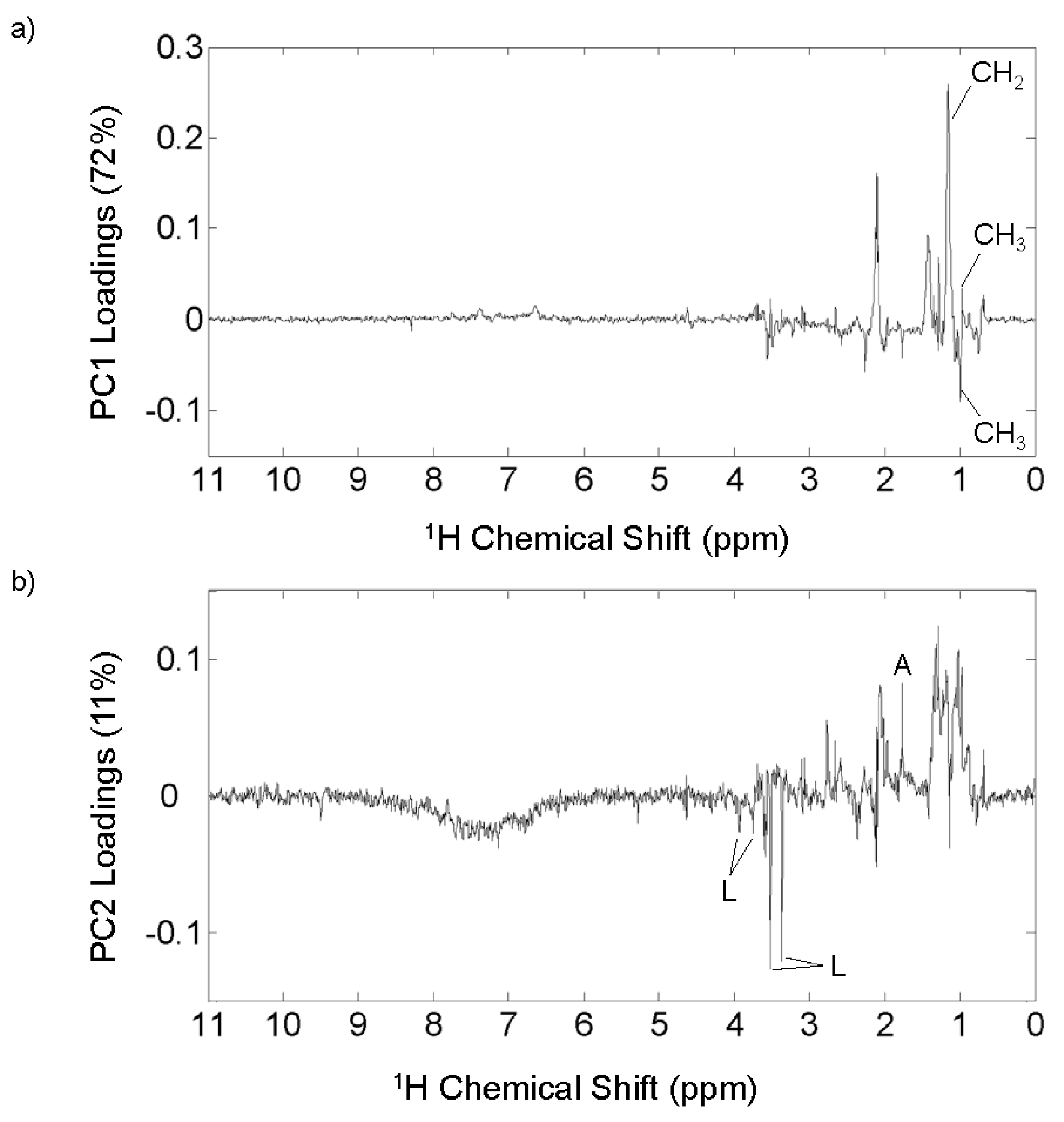
3.5. 1H NMR PCA
4. Conclusions
Supplementary Materials
- Supplementary methods.
- Table S1: Percent area contributions from the major proton regions and calculated H/C ratios in 1H NMR spectra for aerosol PSOM.
- Table S2: Total formulas and average elemental properties for aerosol WSOMPPL and PSOM from each emission source determined using FTICR mass spectra. The distribution of molecular formulas based on atomic content and AImod structure type are listed as number of formulas with the percentage of total formulas in parentheses directly below.
- Table S3: Total formulas and average elemental properties for aerosol WSOM−, WSOM+, and PSOM− from each emission source determined using FTICR-MS. Atomic content and structure type values are expressed as the number of formulas. The values in parentheses are the percentage of total molecular formulas in each sample, an average for each source.
- Table S4: Total formulas and average elemental properties for aerosol WSOM−, WSOM+, and PSOM from each emission source identified by PCA. Distributions of formulas based on atomic content and AImod structure type are listed as percentage of total formulas.
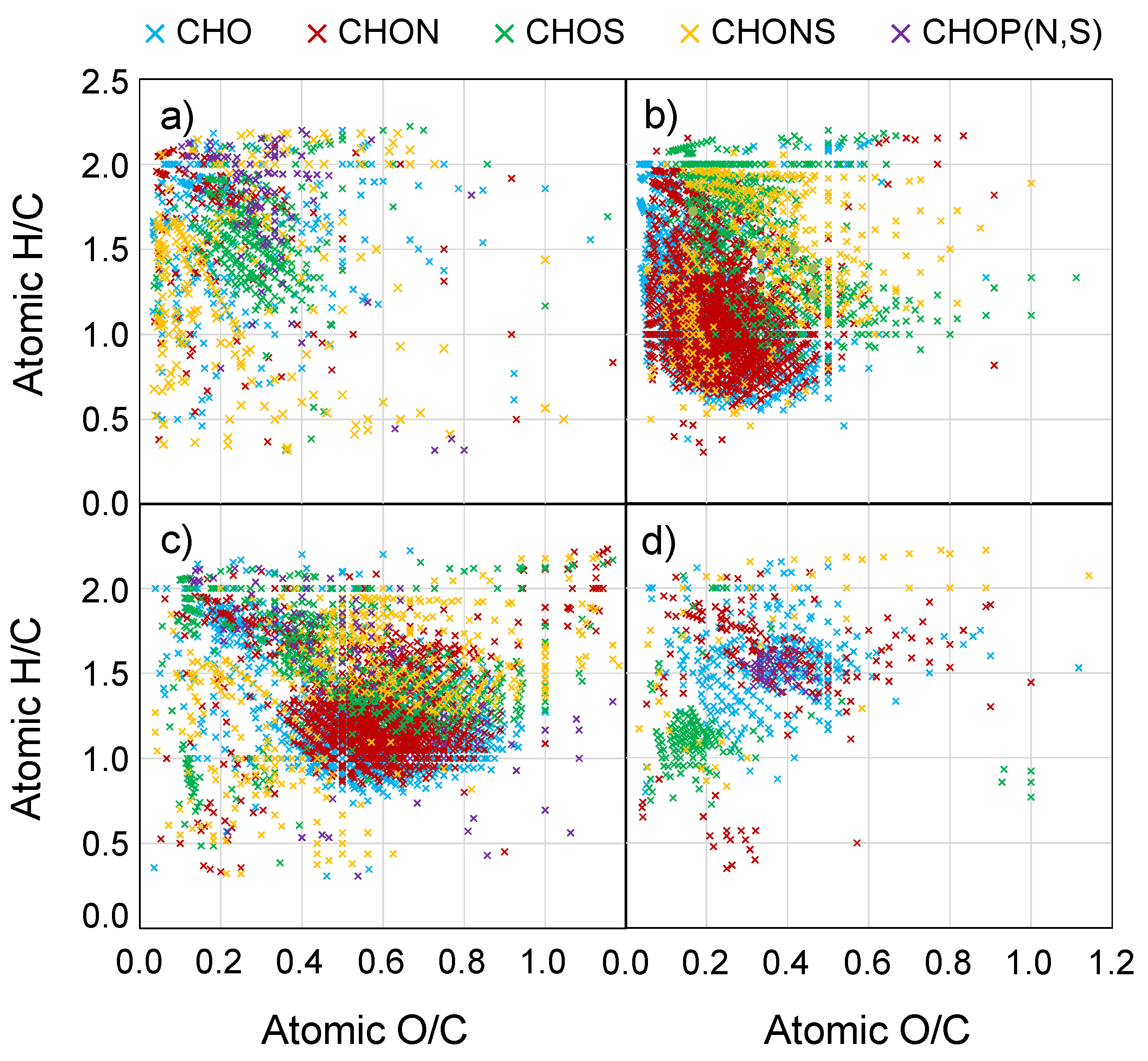
- Figure S1: Van Krevelen diagrams for molecular formulas identified in the FTICR mass spectra for the marine, biomass burning, urban, and mixed source aerosols. Each row represents a different source, and each column represents only those formulas with a specific elemental makeup (CHO, CHON, or CHOS). Each “×” represents one or more molecular formulas.
- Figure S2: The loadings for (a) PC1 and PC2 and (b) PC1 and PC3 from the PCA analysis of the FTICR-MS molecular formulas.
- Figure S3: Venn diagrams showing the relative distribution of PCA molecular formulas present in any of the three solvent/ionization methods (WSOM−, WSOM+, and PSOM) for each aerosol source. Areas of overlap represent percentages of molecular formulas that appear in two or more of those samples. Areas with no overlap represent the percentage of molecular formulas unique to that individual solvent/ionization method.
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AImod | modified aromaticity index |
| BC | black carbon |
| CTO-375 | chemothermal oxidation at 375 °C |
| ESI | electrospray ionization |
| FTICR-MS | Fourier transform ion cyclotron resonance mass spectrometry |
| H/C | hydrogen-to-carbon atomic ratio |
| 1H NMR | proton nuclear magnetic resonance spectroscopy |
| NMR | nuclear magnetic resonance spectroscopy |
| O/C | oxygen-to-carbon atomic ratio |
| OA | organic aerosols |
| OM | organic matter |
| PC | principal component |
| PCA | principal component analysis |
| PPL | solid phase extraction medium |
| PSOC | pyridine-soluble organic carbon |
| PSOM | pyridine-soluble organic matter |
| SOA | secondary organic aerosol |
| TC | total carbon |
| TSP | total suspended particulates |
| UHR-MS | ultrahigh resolution mass spectrometry |
| WSOC | water-soluble organic carbon |
| WSOM | water-soluble organic matter |
| WSOMPPL | PPL-extracted water-soluble organic matter |
References
- Jimenez, J.L.; Canagaratna, M.R.; Donahue, N.M.; Prevot, A.S.H.; Zhang, Q.; Kroll, J.H.; DeCarlo, P.F.; Allan, J.D.; Coe, H.; Ng, N.L.; et al. Evolution of organic aerosols in the atmosphere. Science 2009, 326, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Kanakidou, M.; Seinfeld, J.; Pandis, S.; Barnes, I.; Dentener, F.; Facchini, M.; Van Dingenen, R.; Ervens, B.; Nenes, A.; Nielsen, C.; et al. Organic aerosol and global climate modelling: A review. Atmos. Chem. Phys. 2005, 5, 1053–1123. [Google Scholar] [CrossRef]
- Chan, M.N.; Choi, M.Y.; Ng, N.L.; Chan, C.K. Hygroscopicity of water-soluble organic compounds in atmospheric aerosols: Amino acids and biomass burning derived organic species. Environ. Sci. Technol. 2005, 39, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Cerully, K.; Bougiatioti, A.; Hite, J., Jr.; Guo, H.; Xu, L.; Ng, N.; Weber, R.; Nenes, A. On the link between hygroscopicity, volatility, and oxidation state of ambient and water-soluble aerosols in the southeastern united states. Atmos. Chem. Phys. 2015, 15, 8679–8694. [Google Scholar] [CrossRef]
- Andreae, M.O.; Gelencsér, A. Black carbon or brown carbon? The nature of light-absorbing carbonaceous aerosols. Atmos. Chem. Phys. 2006, 6, 3131–3148. [Google Scholar] [CrossRef]
- Wozniak, A.S.; Shelley, R.U.; Sleighter, R.L.; Abdulla, H.A.; Morton, P.L.; Landing, W.M.; Hatcher, P.G. Relationships among aerosol water soluble organic matter, iron and aluminum in European, North African, and marine air masses from the 2010 US GEOTRACES cruise. Mar. Chem. 2013, 54, 557–898. [Google Scholar] [CrossRef]
- Wozniak, A.S.; Shelley, R.U.; McElhenie, S.D.; Landing, W.M.; Hatcher, P.G. Aerosol water soluble organic matter characteristics over the North Atlantic Ocean: Implications for iron-binding ligands and iron solubility. Mar. Chem. 2015, 173, 162–172. [Google Scholar] [CrossRef]
- Samburova, V.; Szidat, S.; Hueglin, C.; Fisseha, R.; Baltensperger, U.; Zenobi, R.; Kalberer, M. Seasonal variation of high-molecular-weight compounds in the water-soluble fraction of organic urban aerosols. J. Geophys. Res. Atmos. 2005, 110. [Google Scholar] [CrossRef]
- Takegawa, N.; Miyakawa, T.; Kondo, Y.; Jimenez, J.L.; Zhang, Q.; Worsnop, D.R.; Fukuda, M. Seasonal and diurnal variations of submicron organic aerosol in Tokyo observed using the Aerodyne aerosol mass spectrometer. J. Geophys. Res. Atmos. 2006, 111. [Google Scholar] [CrossRef]
- Baduel, C.; Voisin, D.; Jaffrezo, J. Seasonal variations of concentrations and optical properties of water soluble HULIS collected in urban environments. Atmos. Chem. Phys. 2010, 10, 4085–4095. [Google Scholar] [CrossRef]
- Alves, C.; Vicente, A.; Pio, C.; Kiss, G.; Hoffer, A.; Decesari, S.; Prevôt, A.S.H.; Cruz Minguillón, M.; Querol, X.; Hillamo, R. Organic compounds in aerosols from selected European sites—Biogenic versus anthropogenic sources. Atmos. Environ. 2012, 59, 243–255. [Google Scholar] [CrossRef]
- Kieber, R.J.; Adams, M.B.; Willey, J.D.; Whitehead, R.F.; Avery, G.B.; Mullaugh, K.M.; Mead, R.N. Short term temporal variability in the photochemically mediated alteration of chromophoric dissolved organic matter (CDOM) in rainwater. Atmos. Environ. 2012, 50, 112–119. [Google Scholar] [CrossRef]
- Chalbot, M.G.; Kavouras, I.G. Nuclear magnetic resonance spectroscopy for determining the functional content of organic aerosols: A review. Environ. Pollut. 2014, 191, 232–249. [Google Scholar] [CrossRef] [PubMed]
- Duarte, R.M.; Freire, S.M.; Duarte, A.C. Investigating the water-soluble organic functionality of urban aerosols using two-dimensional correlation of solid-state 13C NMR and FTIR spectral data. Atmos. Environ. 2015, 116, 245–252. [Google Scholar] [CrossRef]
- Decesari, S.; Facchini, M.; Matta, E.; Lettini, F.; Mircea, M.; Fuzzi, S.; Tagliavini, E.; Putaud, J. Chemical features and seasonal variation of fine aerosol water-soluble organic compounds in the Po Valley, Italy. Atmos. Environ. 2001, 35, 3691–3699. [Google Scholar] [CrossRef]
- Moretti, F.; Tagliavini, E.; Decesari, S.; Facchini, M.; Rinaldi, M.; Fuzzi, S. NMR determination of total carbonyls and carboxyls: A tool for tracing the evolution of atmospheric oxidized organic aerosols. Environ. Sci. Technol. 2008, 42, 4844–4849. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Kopplin, P.; Gelencser, A.; Dabek-Zlotorzynska, E.; Kiss, G.; Hertkorn, N.; Harir, M.; Hong, Y.; Gebefugi, I. Analysis of the unresolved organic fraction in atmospheric aerosols with ultrahigh-resolution mass spectrometry and nuclear magnetic resonance spectroscopy: Organosulfates as photochemical smog constituents. Anal. Chem. 2010, 82, 8017–8026. [Google Scholar] [CrossRef] [PubMed]
- Shakya, K.M.; Place, P.F.; Griffin, R.J.; Talbot, R.W. Carbonaceous content and water-soluble organic functionality of atmospheric aerosols at a semi-rural New England location. J. Geophys. Res. Atmos. 2012, 117, D03301. [Google Scholar] [CrossRef]
- Altieri, K.E.; Seitzinger, S.P.; Carlton, A.G.; Turpin, B.J.; Klein, G.C.; Marshall, A.G. Oligomers formed through in-cloud methylglyoxal reactions: Chemical composition, properties, and mechanisms investigated by ultra-high resolution FT-ICR mass spectrometry. Atmos. Environ. 2008, 42, 1476–1490. [Google Scholar] [CrossRef]
- Wozniak, A.; Bauer, J.; Sleighter, R.; Dickhut, R.; Hatcher, P. Technical note: Molecular characterization of aerosol-derived water soluble organic carbon using ultrahigh resolution electrospray ionization fourier transform ion cyclotron resonance mass spectrometry. Atmos. Chem. Phys. 2008, 8, 5099–5111. [Google Scholar] [CrossRef]
- Heaton, K.J.; Sleighter, R.L.; Hatcher, P.G.; Hall, W.A., IV; Johnston, M.V. Composition domains in monoterpene secondary organic aerosol. Environ. Sci. Technol. 2009, 43, 6950–6955. [Google Scholar]
- Laskin, J.; Laskin, A.; Roach, P.; Slysz, G.; Anderson, G.; Nizkorodov, S.; Bones, D.; Nguyen, L. High-resolution desorption electrospray ionization mass spectrometry for chemical characterization of organic aerosols. Anal. Chem. 2010, 82, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Mazzoleni, L.; Ehrmann, B.; Shen, X.; Marshall, A.; Collett, J., Jr. Water-soluble atmospheric organic matter in fog: Exact masses and chemical formula identification by ultrahigh-resolution Fourier transform ion cyclotron resonance mass spectrometry. Environ. Sci. Technol. 2010, 44, 3690–3697. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Bateman, A.; Bones, D.; Nizkorodov, S.; Laskin, J.; Laskin, A. High-resolution mass spectrometry analysis of secondary organic aerosol generated by ozonolysis of isoprene. Atmos. Environ. 2010, 44, 1032–1042. [Google Scholar] [CrossRef]
- LeClair, J.P.; Collett, J.L.; Mazzoleni, L.R. Fragmentation analysis of water-soluble atmospheric organic matter using ultrahigh-resolution FT-ICR mass spectrometry. Environ. Sci. Technol. 2012, 46, 4312–4322. [Google Scholar] [CrossRef] [PubMed]
- Mazzoleni, L.R.; Saranjampour, P.; Dalbec, M.M.; Samburova, V.; Hallar, A.G.; Zielinska, B.; Lowenthal, D.H.; Kohl, S. Identification of water-soluble organic carbon in non-urban aerosols using ultrahigh-resolution FT-ICR mass spectrometry: Organic anions. Environ. Chem. 2012, 9, 285–297. [Google Scholar] [CrossRef]
- Mead, R.; Mullaugh, K.; Brooks Avery, G.; Kieber, R.; Willey, J.; Podgorski, D. Insights into dissolved organic matter complexity in rainwater from continental and coastal storms by ultrahigh resolution Fourier transform ion cyclotron resonance mass spectrometry. Atmos. Chem. Phys. 2013, 13, 4829–4838. [Google Scholar] [CrossRef]
- Mitra, S.; Wozniak, A.S.; Miller, R.; Hatcher, P.G.; Buonassissi, C.; Brown, M. Multiproxy probing of rainwater dissolved organic matter (DOM) composition in coastal storms as a function of trajectory. Mar. Chem. 2013, 154, 67–76. [Google Scholar] [CrossRef]
- Kourtchev, I.; Fuller, S.J.; Giorio, C.; Healy, R.M.; Wilson, E.; O’Connor, I.; Wenger, J.C.; McLeod, M.; Aalto, J.; Ruuskanen, T.M. Molecular composition of biogenic secondary organic aerosols using ultrahigh-resolution mass spectrometry: Comparing laboratory and field studies. Atmos. Chem. Phys. 2014, 14, 2155–2167. [Google Scholar] [CrossRef]
- Willoughby, A.S.; Wozniak, A.S.; Hatcher, P.G. A molecular-level approach for characterizing water-insoluble components of ambient organic aerosol particulates using ultrahigh-resolution mass spectrometry. Atmos. Chem. Phys. 2014, 14, 10299–10314. [Google Scholar] [CrossRef]
- Wozniak, A.S.; Willoughby, A.S.; Gurganus, S.C.; Hatcher, P.G. Distinguishing molecular characteristics of aerosol water soluble organic matter from the 2011 Trans-North Atlantic US GEOTRACES cruise. Atmos. Chem. Phys. 2014, 14, 8419–8434. [Google Scholar] [CrossRef]
- Kourtchev, I.; Doussin, J.-F.; Giorio, C.; Mahon, B.; Wilson, E.; Maurin, N.; Pangui, E.; Venables, D.; Wenger, J.; Kalberer, M. Molecular composition of fresh and aged secondary organic aerosol from a mixture of biogenic volatile compounds: A high-resolution mass spectrometry study. Atmos. Chem. Phys. 2015, 15, 5683–5695. [Google Scholar] [CrossRef]
- Reinhardt, A.; Emmenegger, C.; Gerrits, B.; Panse, C.; Dommen, J.; Baltensperger, U.; Zenobi, R.; Kalberer, M. Ultrahigh mass resolution and accurate mass measurements as a tool to characterize oligomers in secondary organic aerosols. Anal. Chem. 2007, 79, 4074–4082. [Google Scholar] [CrossRef] [PubMed]
- Geotraces: An international study of the marine biogeochemical cycles of trace elements and their isotopes. Available online: http://www.geotraces.org/ (accessed on 1 June 2016).
- National Oceanic and Atmospheric Administration: Pacific Marine Environmental Laboratory. WACS2 (2014) Western Atlantic Climate Study. Available online: http://saga.pmel.noaa.gov/field_WACS2 (accessed on 1 June 2016).
- Gustafsson, Ö.; Haghseta, F.; Chan, C.; MacFarlane, J.; Gschwend, P. Quantification of the dilute sedimentary soot phase: Implications for pah speciation and bioavailability. Environ. Sci. Technol. 1997, 31, 203–209. [Google Scholar] [CrossRef]
- Dittmar, T.; Koch, B.; Hertkorn, N.; Kattner, G. A simple and efficient method for the solid-phase extraction of dissolved organic matter (SPE-DOM) from seawater. Limnol. Oceanogr. Methods 2008, 6, 230–235. [Google Scholar] [CrossRef]
- Sleighter, R.; McKee, G.; Liu, Z.; Hatcher, P. Naturally present fatty acids as internal calibrants for Fourier transform mass spectra of dissolved organic matter. Limnol. Oceanogr. Methods 2008, 6, 246–253. [Google Scholar] [CrossRef]
- Stubbins, A.; Spencer, R.G.M.; Chen, H.; Hatcher, P.G.; Mopper, K.; Hernes, P.J.; Mwamba, V.L.; Mangangu, A.M.; Wabakanghanzi, J.N.; Six, J. Illuminated darkness: Molecular signatures of Congo River dissolved organic matter and its photochemical alteration as revealed by ultrahigh precision mass spectrometry. Limnol. Oceanogr. 2010, 55, 1467–1477. [Google Scholar] [CrossRef]
- Sleighter, R.; Liu, Z.; Xue, J.; Hatcher, P. Multivariate statistical approaches for the characterization of dissolved organic matter analyzed by ultrahigh resolution mass spectrometry. Environ. Sci. Technol. 2010, 44, 7576–7582. [Google Scholar] [CrossRef] [PubMed]
- Gurganus, S.C.; Wozniak, A.S.; Hatcher, P.G. Molecular characteristics of the water soluble organic matter in size-fractionated aerosols collected over the North Atlantic Ocean. Mar. Chem. 2015, 170, 37–48. [Google Scholar] [CrossRef]
- Hammes, K.; Schmidt, M.W.; Smernik, R.J.; Currie, L.A.; Ball, W.P.; Nguyen, T.H.; Louchouarn, P.; Houel, S.; Gustafsson, Ö.; Elmquist, M. Comparison of quantification methods to measure fire-derived (black/elemental) carbon in soils and sediments using reference materials from soil, water, sediment and the atmosphere. Glob. Biogeochem. Cycles 2007, 21. [Google Scholar] [CrossRef]
- Heald, C.; Kroll, J.; Jimenez, J.; Docherty, K.; DeCarlo, P.; Aiken, A.; Chen, Q.; Martin, S.; Farmer, D.; Artaxo, P. A simplified description of the evolution of organic aerosol composition in the atmosphere. Geophys. Res. Lett. 2010, 37. [Google Scholar] [CrossRef]
- Kroll, J.H.; Donahue, N.M.; Jimenez, J.L.; Kessler, S.H.; Canagaratna, M.R.; Wilson, K.R.; Altieri, K.E.; Mazzoleni, L.R.; Wozniak, A.S.; Bluhm, H. Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol. Nat. Chem. 2011, 3, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Alfarra, M.R.; Worsnop, D.R.; Allan, J.D.; Coe, H.; Canagaratna, M.R.; Jimenez, J.L. Deconvolution and quantification of hydrocarbon-like and oxygenated organic aerosols based on aerosol mass spectrometry. Environ. Sci. Technol. 2005, 39, 4938–4952. [Google Scholar] [CrossRef] [PubMed]
- Aiken, A.C.; Decarlo, P.F.; Kroll, J.H.; Worsnop, D.R.; Huffman, J.A.; Docherty, K.S.; Ulbrich, I.M.; Mohr, C.; Kimmel, J.R.; Sueper, D. O/C and OM/OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry. Environ. Sci. Technol. 2008, 42, 4478–4485. [Google Scholar] [CrossRef] [PubMed]
- Koch, B.; Dittmar, T. From mass to structure: An aromaticity index for high-resolution mass data of natural organic matter. Rapid Commun. Mass Spectrom. 2006, 20, 926–932. [Google Scholar] [CrossRef]
- Claeys, M.; Wang, W.; Vermeylen, R.; Kourtchev, I.; Chi, X.; Farhat, Y.; Surratt, J.D.; Gómez-González, Y.; Sciare, J.; Maenhaut, W. Chemical characterisation of marine aerosol at Amsterdam Island during the austral summer of 2006–2007. J. Aerosol Sci. 2010, 41, 13–22. [Google Scholar] [CrossRef]
- Hawkins, L.; Russell, L.; Covert, D.; Quinn, P.; Bates, T. Carboxylic acids, sulfates, and organosulfates in processed continental organic aerosol over the southeast Pacific Ocean during VOCALS-REX 2008. J. Geophys. Res. 2010, 115. [Google Scholar] [CrossRef]
- Iinuma, Y.; Müller, C.; Berndt, T.; Böge, O.; Claeys, M.; Herrmann, H. Evidence for the existence of organosulfates from β-pinene ozonolysis in ambient secondary organic aerosol. Environ. Sci. Technol. 2007, 41, 6678–6683. [Google Scholar] [CrossRef] [PubMed]
- Minerath, E.C.; Casale, M.T.; Elrod, M.J. Kinetics feasibility study of alcohol sulfate esterification reactions in tropospheric aerosols. Environ. Sci. Technol. 2008, 42, 4410–4415. [Google Scholar] [CrossRef] [PubMed]
- Minerath, E.C.; Elrod, M.J. Assessing the potential for diol and hydroxy sulfate ester formation from the reaction of epoxides in tropospheric aerosols. Environ. Sci. Technol. 2009, 43, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, M.; Lee, C.; Aller, J. Characterization of the proteinaceous matter in marine aerosols. Mar. Chem. 2005, 96, 359–377. [Google Scholar] [CrossRef]
- Hawkins, L.N.; Russell, L.M. Polysaccharides, proteins, and phytoplankton fragments: Four chemically distinct types of marine primary organic aerosol classified by single particle spectromicroscopy. Adv. Meteorol. 2010, 2010, 1–14. [Google Scholar] [CrossRef]
- Russell, P.; Bergstrom, R.; Shinozuka, Y.; Clarke, A.; DeCarlo, P.; Jimenez, J.; Livingston, J.; Redemann, J.; Dubovik, O.; Strawa, A. Absorption angstrom exponent in AERONET and related data as an indicator of aerosol composition. Atmos. Chem. Phys. 2010, 10, 1155–1169. [Google Scholar] [CrossRef]
- Surratt, J.D.; Kroll, J.H.; Kleindienst, T.E.; Edney, E.O.; Claeys, M.; Sorooshian, A.; Ng, N.L.; Offenberg, J.H.; Lewandowski, M.; Jaoui, M. Evidence for organosulfates in secondary organic aerosol. Environ. Sci. Technol. 2007, 41, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Noziere, B.; Ekström, S.; Alsberg, T.; Holmström, S. Radical-initiated formation of organosulfates and surfactants in atmospheric aerosols. Geophys. Res. Lett. 2010, 37. [Google Scholar] [CrossRef]
- McNeill, V.F.; Woo, J.L.; Kim, D.D.; Schwier, A.N.; Wannell, N.J.; Sumner, A.J.; Barakat, J.M. Aqueous-phase secondary organic aerosol and organosulfate formation in atmospheric aerosols: A modeling study. Environ. Sci. Technol. 2012, 46, 8075–8081. [Google Scholar] [CrossRef] [PubMed]
- Cullis, C.; Hirschler, M. Atmospheric sulphur: Natural and man-made sources. Atmos. Environ. 1980, 14, 1263–1278. [Google Scholar] [CrossRef]
- Möller, D. Estimation of the global man-made sulphur emission. Atmos. Environ. 1984, 18, 19–27. [Google Scholar] [CrossRef]
- Andreae, M.O. Dimethylsulfoxide in marine and freshwaters. Limnol. Oceanogr. 1980, 25, 1054–1063. [Google Scholar] [CrossRef]
- Masiello, C. New directions in black carbon organic geochemistry. Mar. Chem. 2004, 92, 201–213. [Google Scholar] [CrossRef]
- Decesari, S.; Facchini, M.; Matta, E.; Mircea, M.; Fuzzi, S.; Chughtai, A.; Smith, D. Water soluble organic compounds formed by oxidation of soot. Atmos. Environ. 2002, 36, 1827–1832. [Google Scholar] [CrossRef]
- Kamegawa, K.; Nishikubo, K.; Kodama, M.; Adachi, Y.; Yoshida, H. Oxidative degradation of carbon blacks with nitric acid II. Formation of water-soluble polynuclear aromatic compounds. Carbon 2002, 40, 1447–1455. [Google Scholar] [CrossRef]
- Wagner, S.; Dittmar, T.; Jaffé, R. Molecular characterization of dissolved black nitrogen via electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Org. Geochem. 2015, 79, 21–30. [Google Scholar] [CrossRef]
- Goldberg, E. Black Carbon in the Environment: Properties and Distribution; John Wiley and Sons: New York, NY, USA, 1985. [Google Scholar]
- Bateman, A.P.; Nizkorodov, S.A.; Laskin, J.; Laskin, A. High-resolution electrospray ionization mass spectrometry analysis of water-soluble organic aerosols collected with a particle into liquid sampler. Anal. Chem. 2010, 82, 8010–8016. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Aiona, P.K.; Laskin, A.; Laskin, J.; Nizkorodov, S.A. Effect of solar radiation on the optical properties and molecular composition of laboratory proxies of atmospheric brown carbon. Environ. Sci. Technol. 2014, 48, 10217–10226. [Google Scholar] [CrossRef] [PubMed]
- Washenfelder, R.A.; Attwood, A.R.; Brock, C.A.; Guo, H.; Xu, L.; Weber, R.J.; Ng, N.L.; Allen, H.M.; Ayres, B.R.; Baumann, K. Biomass burning dominates brown carbon absorption in the rural southeastern United States. Geophys. Res. Lett. 2015, 42, 1–12. [Google Scholar] [CrossRef]
- Knicker, H.; Almendros, G.; González-Vila, F.J.; Martín, F.; Lüdemann, H.-D. 13C- and 15N-NMR spectroscopic examination of the transformation of organic nitrogen in plant biomass during thermal treatment. Soil Biol. Biochem. 1996, 28, 1053–1060. [Google Scholar] [CrossRef]
- Laskin, A.; Smith, J.; Laskin, J. Molecular characterization of nitrogen-containing organic compounds in biomass burning aerosols using high-resolution mass spectrometry. Environ. Sci. Technol. 2009, 43, 3764–3771. [Google Scholar] [CrossRef] [PubMed]
- Hertkorn, N.; Benner, R.; Frommberger, M.; Schmitt-Kopplin, P.; Witt, M.; Kaiser, K.; Kettrup, A.; Hedges, J.I. Characterization of a major refractory component of marine dissolved organic matter. Geochim. Cosmochim. Acta 2006, 70, 2990–3010. [Google Scholar] [CrossRef]
- Ohno, T.; He, Z.; Sleighter, R.L.; Honeycutt, C.W.; Hatcher, P.G. Ultrahigh resolution mass spectrometry and indicator species analysis to identify marker components of soil-and plant biomass-derived organic matter fractions. Environ. Sci. Technol. 2010, 44, 8594–8600. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.S.; Bauer, J.E.; Dickhut, R.M. Characteristics of water-soluble organic carbon associated with aerosol particles in the eastern United States. Atmos. Environ. 2012, 46, 181–188. [Google Scholar] [CrossRef]
- Altieri, K.E.; Turpin, B.J.; Seitzinger, S.P. Oligomers, organosulfates, and nitrooxy organosulfates in rainwater identified by ultra-high resolution electrospray ionization FT-ICR mass spectrometry. Atmos. Chem. Phys. 2009, 9, 2533–2542. [Google Scholar] [CrossRef]
- Surratt, J.D.; Gómez-González, Y.; Chan, A.W.H.; Vermeylen, R.; Shahgholi, M.; Kleindienst, T.E.; Edney, E.O.; Offenberg, J.H.; Lewandowski, M.; Jaoui, M. Organosulfate formation in biogenic secondary organic aerosol. J. Phys. Chem. A 2008, 112, 8345–8378. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, M. Aromatic sulfur compounds other than condensed thiophenes in fossil fuels: Enrichment and identification. Energy Fuels 1988, 2, 214–219. [Google Scholar] [CrossRef]
- Panda, S.K.; Andersson, J.T.; Schrader, W. Mass-spectrometric analysis of complex volatile and nonvolatile crude oil components: A challenge. Anal. Bioanal. Chem. 2007, 389, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.; Faul, K.; Paytan, A. Phosphorus associations in aerosols: What can they tell us about P bioavailability? Mar. Chem. 2010, 120, 44–56. [Google Scholar] [CrossRef]
- Chen, H.; Abdulla, H.A.; Sanders, R.L.; Myneni, S.C.; Mopper, K.; Hatcher, P.G. Production of black carbon-like and aliphatic molecules from terrestrial dissolved organic matter in the presence of sunlight and iron. Environ. Sci. Technol. Lett. 2014, 1, 399–404. [Google Scholar] [CrossRef]
- Waggoner, D.C.; Chen, H.; Willoughby, A.S.; Hatcher, P.G. Formation of black carbon-like and alicyclic aliphatic compounds by hydroxyl radical initiated degradation of lignin. Org. Geochem. 2015, 82, 69–76. [Google Scholar] [CrossRef]
- Decesari, S.; Finessi, E.; Rinaldi, M.; Paglione, M.; Fuzzi, S.; Stephanou, E.; Tziaras, T.; Spyros, A.; Ceburnis, D.; O’Dowd, C. Primary and secondary marine organic aerosols over the North Atlantic Ocean during the map experiment. J. Geophys. Res. Atmos. 2011, 116. [Google Scholar] [CrossRef]
- Simoneit, B.R.T.; Schauer, J.J.; Nolte, C.; Oros, D.R.; Elias, V.O.; Fraser, M.; Rogge, W.; Cass, G.R. Levoglucosan, a tracer for cellulose in biomass burning and atmospheric particles. Atmos. Environ. 1999, 33, 173–182. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aerosol Source | n | TSP (μg·m−3) | TC (μg·m−3) | %BC | %WSOC | %PSOC |
|---|---|---|---|---|---|---|
| * Marine | 4 | - | 0.5 ± 0.7 | - | 39.6 ± 25.1 | - |
| Biomass burning | 2 | 73.2 ± 5.3 | 24.8 ± 4.4 | 6.5 ± 0.7 | 33.6 ± 2.6 | 66 ± 20 |
| Urban | 5 | 47.1 ± 11.0 | 6.3 ± 1.3 | 3.4 ± 3.2 | 40.8 ± 5.5 | 44 ± 6 |
| Mixed source | 3 | 24.1 ± 2.9 | 5.7 ± 0.7 | 1.9 ± 2.5 | 50.5 ± 10.6 | 45 ± 13 |
| Marine Aerosols n = 4 | Biomass Burning Aerosols n = 2 | Urban Aerosols n = 5 | Mixed Source Aerosols n = 3 | |
|---|---|---|---|---|
| Average formulas | 2569 ± 736 | 6579 ± 173 | 6527 ± 173 | 4104 ± 467 |
| Total formulas | 4570 | 7891 | 10,701 | 6134 |
| Average O/C | 0.32 ± 0.19 | 0.32 ± 0.19 | 0.45 ± 0.23 | 0.44 ± 0.23 |
| Average H/C | 1.56 ± 0.39 | 1.35 ± 0.39 | 1.44 ± 0.37 | 1.46 ± 0.35 |
| Average AImod | 0.18 ± 0.26 | 0.29 ± 0.27 | 0.18 ± 0.24 | 0.18 ± 0.23 |
| Marine Aerosols | Biomass Burning Aerosols | Urban Aerosols | Mixed Source Aerosols | |
|---|---|---|---|---|
| Total formulas | 1078 | 4174 | 3484 | 693 |
| Average O/C | 0.27 ± 0.17 | 0.26 ± 0.13 | 0.55 ± 0.21 | 0.33 ± 0.18 |
| Average H/C | 1.55 ± 0.41 | 1.24 ± 0.37 | 1.36 ± 0.34 | 1.48 ± 0.34 |
| Average AImod | 0.20 ± 0.25 | 0.37 ± 0.23 | 0.18 ± 0.22 | 0.20 ± 0.21 |
| Atomic Content (%) | ||||
| CHO | 31.6 | 34.7 | 33.4 | 45.0 |
| CHON | 13.4 | 45.4 | 38.0 | 24.4 |
| CHOS | 27.4 | 12.0 | 13.3 | 18.3 |
| CHONS | 19.0 | 7.8 | 12.5 | 6.6 |
| CHOP(N,S) | 8.6 | 0.1 | 2.8 | 5.6 |
| Structure Type (%) | ||||
| Aliphatic | 31.4 | 12.0 | 34.1 | 24.0 |
| Olefinic/alicyclic | 56.6 | 55.6 | 60.6 | 67.8 |
| Aromatic | 5.5 | 26.8 | 3.1 | 4.9 |
| Condensed aromatic | 6.6 | 5.6 | 2.3 | 3.3 |
| Aerosol Source | H-C (0.6–1.8 ppm) | H-C-C= (1.8–3.2 ppm) | H-C-O-R (3.2–4.4 ppm) | Ar-H (6.0–9.0 ppm) |
|---|---|---|---|---|
| Marine | 50.3 ± 5.5 | 34.2 ± 3.9 | 14.5 ± 3.6 | 1.0 ± 0.3 |
| Biomass burning | 50.9 ± 3.4 | 31.1 ± 0.6 | 9.4 ± 0.9 | 9.2 ± 1.9 |
| Urban | 53.2 ± 1.5 | 35.0 ± 2.0 | 9.4 ± 1.1 | 2.4 ± 0.6 |
| Mixed source | 57.0 ± 1.3 | 32.9 ± 0.4 | 8.4 ± 0.8 | 1.7 ± 0.3 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willoughby, A.S.; Wozniak, A.S.; Hatcher, P.G. Detailed Source-Specific Molecular Composition of Ambient Aerosol Organic Matter Using Ultrahigh Resolution Mass Spectrometry and 1H NMR. Atmosphere 2016, 7, 79. https://doi.org/10.3390/atmos7060079
Willoughby AS, Wozniak AS, Hatcher PG. Detailed Source-Specific Molecular Composition of Ambient Aerosol Organic Matter Using Ultrahigh Resolution Mass Spectrometry and 1H NMR. Atmosphere. 2016; 7(6):79. https://doi.org/10.3390/atmos7060079
Chicago/Turabian StyleWilloughby, Amanda S., Andrew S. Wozniak, and Patrick G. Hatcher. 2016. "Detailed Source-Specific Molecular Composition of Ambient Aerosol Organic Matter Using Ultrahigh Resolution Mass Spectrometry and 1H NMR" Atmosphere 7, no. 6: 79. https://doi.org/10.3390/atmos7060079
APA StyleWilloughby, A. S., Wozniak, A. S., & Hatcher, P. G. (2016). Detailed Source-Specific Molecular Composition of Ambient Aerosol Organic Matter Using Ultrahigh Resolution Mass Spectrometry and 1H NMR. Atmosphere, 7(6), 79. https://doi.org/10.3390/atmos7060079