1. Introduction
Chlorofluorocarbons (CFCs) are trace gases that are of concern, because their breakdown in the stratosphere releases Cl (chlorine) radicals, which destroy ozone (O
3) molecules through catalytic cycles. Less stratospheric ozone leads to the biosphere being exposed to higher levels of UV radiation. The most prominent manifestation of stratospheric ozone depletion is the Antarctic ozone hole, which was first reported in 1985 [
1] and grew rapidly with increasing abundances of chlorine and bromine containing substances during the 1980s and 1990s [
2]. The Montreal Protocol was first signed in 1987 and, together with its subsequent amendments, regulates the production and consumption of ozone depleting substances (ODSs). CFCs-11, -12 and -113, which together accounted for 62% of tropospheric chlorine in 2008 [
3], are primarily responsible for the production of stratospheric Cl. Under the Montreal Protocol, their tropospheric concentrations peaked in the mid-1990s at 270 ppt and 84 ppt (CFC-11, -113) and the early 2000s at 540 ppt (CFC-12) [
3]. CFC-11 has a lifetime of 52 years [
4], a value of one by definition for the ozone depletion potential (ODP) and a global warming potential (GWP) of 4660 over a time interval of 100 years [
5]. CFC-12 has a lifetime of 102 years [
4], an ODP of 0.82 and a GWP of 10,200 ([
5,
6]). The ODP is a measure of how harmful on a mass emitted basis a substance can be to the ozone layer, relative to an equivalent mass of CFC-11 emitted. The GWP is a measure of the ability of a greenhouse gas to trap heat in the atmosphere relative to that of carbon dioxide (CO
2) on a mass emitted basis and depends on the radiative properties and the residence time in the atmosphere. Most CFCs have comparably high ODPs and GWPs.
Although hydrochlorofluorocarbons (HCFCs) also contain chlorine, they are less destructive for stratospheric O
3 due to the incorporated hydrogen atom, making them prone to removal by hydroxyl radicals (OH) in the troposphere and, therefore, less likely to reach the stratosphere. HCFCs typically exhibit lifetimes of about a decade or less (e.g., 12 years for HCFC-22 and 9.4 years for HCFC-142b [
4]). Their ODPs are therefore smaller (between 0.02 for HCFC-123 and 0.11 for HCFC-141b [
6]), but not negligible. Their GWPs are, on average, also smaller than those of the CFCs, but show a large range from 79 for HCFC-123 to 1980 for HCFC-142b.
Here, we report measurements of three substances, two CFCs and one HCFC, namely CFC-216ca (1,3-dichloro-1,1,2,2,3,3-hexafluoropropane), CFC-216ba (1,2-dichloro-1,1,2,3,3,3-hexafluoropropane) and HCFC-225ca (3,3-dichloro-1,1,1,2, 2-pentafluoropropane). The two CFC isomers were used in the production of refrigerants [
7] and in some niche applications, e.g., as feedstock and intermediates in chemical synthesis [
8–
11]. Due to their ozone depleting properties, they have been controlled under the London Amendment to the Montreal Protocol (1990). Under this amendment, a phase-out from 1993 to 1996 was scheduled for non A5-countries (developed countries) and from 2001 to 2010 for A5-countries (developing countries) [
12]. HCFC-225ca gas was mainly used for coating, as an (aerosol) solvent, in adhesives, and as a replacement for CFC-113 as a cleaning agent [
13,
14]. It has a GWP
100 of 127 [
5] and has been controlled under the Copenhagen and Vienna Amendments (1992 and 1995) [
12]. For the production of HCFC-225ca, the Protocol laid down a freeze at the base level for production by 2004, a 75% reduction by 2010, 90% by 2015, 99.5% by 2020 and a complete phase-out for HCFC-225ca was prescribed by 2030 (2040 for developing countries) [
12]. All CFCs and HCFCs have the potential to impact stratospheric ozone and radiative forcing. It is therefore important to identify the presence, abundance and emission trends of these chemicals in the background atmosphere to help quantify their environmental impact and to ensure that there are no future ‘surprises’ in relation to previously unidentified chemicals in the atmosphere.
2. Methods
2.1. Air Sampling
The main dataset was established using Cape Grim air samples. Since 1978, pristine air samples have been regularly collected in stainless steel flasks at Cape Grim, Australia (41°S, 145°E), located at the tip of northwest Tasmania [
15]. It is 100s of kilometers from major urban CFC and HCFC sources, and samples were collected under “baseline” conditions (with wind direction 190°–290° at
[
16]), which are representative of a large area of the remote Southern Hemisphere. Air samples collected before 1994 were stored in 35-L stainless steel tanks (at around 30 atm) and sub-sampled. Since 1994, air is compressed (with a metal bellows pump) into 3-L Silco-containers (Silcosteel-treated, Restec Corp.) and electrochemically passivated tanks (Rasmussen) [
17]. This archive has been used to reconstruct long-term records of numerous greenhouse gases and ODSs (e.g., [
3,
16–
19]).
The Cape Grim dataset is complemented by the analysis of air samples collected in the upper troposphere during recent flights of the CARIBIC observatory (Civil Aircraft for the Regular Investigation of the atmosphere Based on an Instrument Container,
www.caribic-atmospheric.com [
20]), between Frankfurt (Germany) and Bangkok (Thailand). Air samples from four flights were examined for HCFC-225ca, CFC-216ca and CFC-216ba (21 February 2012, 21 March 2012, 8 November 2013, and 4 December 2013). Finally, stratospheric air samples collected on-board the M55 Geophysica high altitude research aircraft during deployments in 2009 (mid-latitudes) and 2010 (high latitudes) have been analyzed for CFC-216ba and CFC-216ca.
2.2. Analytical System
Air samples were analysed for CFC-216ba, CFC-216ca and HCFC-225ca by gas chromatographic separation followed by mass spectrometric detection (GC-MS). The analytical system has been described in detail in [
21]. In brief, samples are dried with magnesium perchlorate (Mg(ClO
4)
2), trace gases pre-concentrated on a cold trap and subsequently injected onto a GC.
The GC column is connected to a high sensitivity trisector mass spectrometer (Micromass/Waters Autospec) tuned to a mass resolution of 1000 and operated in electron impact selected ion recording (EI-SIR) [
21]. On initial analysis using 30-m and 49-m columns, CFC-216ba and CFC-216ca were found to elute close together, forming a close double peak. To enhance separation and ensure the independence of the derived long-term trends of the isomers, a 60-m column was installed for all Cape Grim analyses and measured on
m/
z 185.0 (see, also, the discussion below). The two CFCs were detected at retention times of 20.45 min and 20.54 min. HCFC-225ca eluted at a retention time of 23.60 min. CFC-216ba and CFC-216ca were measured on the fragment ions,
(mass-to-charge ratio
m/
z 84.97),
(
m/
z 86.97),
(
m/
z 185.0) and
(
m/
z 187.0). HCFC-225ca was measured on
,
(
m/
z 82.95 and 84.94) and on
(
m/
z 97.97).
For CFC-216ba and CFC-216ca, the earlier analyses had been performed on m/z 84.97 and 86.97 only. These ions are frequently observed in air sample chromatograms and, thus, prone to interference. In order to validate the dataset, we supplemented it with additional measurements on m/z 185.0 and 187.0, which are much more exclusive. While the two datasets agree very well for CFC-216ba, we observed interferences for CFC-216ca and, therefore, excluded samples measured only on m/z 84.97 and 86.97 (i.e., all stratospheric and a subset of the Cape Grim samples) from further considerations.
No significant blank was observed for any of the compounds discussed here by trapping purified research-grade helium on every measurement day. All air sample measurements were bracketed by working standard measurements with two to three samples in between standards. The linearity of the system was checked by pre-concentrating and measuring different amounts of the working standard (49, 102, 194, 199, 224 and 301 mL). The response behaviour was found to be linear within the average 2
σ standard deviation of the standard for both CFC-216ba and CFC-216ca. HCFC-225ca had not been included in this test, but we trust the linearity of the analytical system, as it has been demonstrated for a great variety of halocarbons previously: [
19] (c-C
4F
8), [
21] (HFC-227ea), [
22] (C
4F
10, C
5F
12, C
6F
14, C
7F
16), [
23] (Halon-1211, Halon-1301, Halon-1202, Halon-2402), [
24] (CFC-12, CFC-11, CFC-113, HCFC-22, HCFC-141b, HCFC-142b, CH
3CCl
3, CCl
4, SF
6; also verified atmospheric trends at Cape Grim, Tasmania, by direct comparison with NOAA scales over several decades) and [
25] (CFC-113a, CFC-112, CFC-112a, HCFC-133a).
The uncertainty calculation of our measurements is based on the repeatability of sample and air standard measurements, summed in quadrature.
2.3. Calibration
Two calibration scales were established for CFC-216ca and HCFC-225ca by statically diluting the pure compounds (obtained from Sigma Aldrich Ltd. and Apollo Scientific Ltd., UK) to parts per trillion (ppt) levels with pure nitrogen gas using two 99.7-L aluminium drums (for details of this system, see [
21]). The dilutions were analyzed with the pre-concentration system GC-MS and used to assign mixing ratios to a high pressure clean air sample used as the working standard. In order to evaluate the accuracy of the calibration procedure, a reference substance (here, CFC-11) with known atmospheric mixing ratios was also added to the dilution drums. Calculated mixing ratios of CFC-11 in the working standard were compared with the internationally recognized NOAA-ESRLcalibration scale and agreed within 4.2%. An analysis of the dilutions was carried out with the MS run in scan mode (scanning m/z 46 to 226), and the results confirmed the purity of both CFC-216ca and HCFC-225ca. The overall uncertainty of the calibration system has been previously estimated to be not more than 7% [
19,
22].
No direct calibration could be made for CFC-216ba, as it was not available for purchase as a pure substance. Therefore, mixing ratios of CFC-216ba were estimated by assuming similar instrument sensitivity as that of its isomer, CFC-216ca. For the quantification of CFC-216ba, we used a fragmentation correction factor, which is calculated from the relative abundance of the ion used to quantify the substance (here,
m/
z 185) divided by the sum of the relative abundances of the 10 major ions. The corrected relative sensitivity is then given by the relative sensitivity divided by the fragmentation factor. The methodology is derived and explained in more detail in [
26]. To test the validity of this approach, we tested the predictability of mixing ratios for two other recently-calibrated CFC isomer pairs: CFC-112/CFC-112a and CFC-113/CFC-113a [
25]. The mixing ratio of the other isomer could be predicted within 23%. We expect the predictability to be better for the CFC-216ca/CFC-216ba isomer pair, as these two gases were measured on the same ion with the exact same dwell time. We have nevertheless added an additional calibration uncertainty of 23% for CFC-216ba.
2.4. Emission Modelling
Global annual emissions of CFC-216ba, CFC-216ca and HCFC-225ca were derived using a 2D atmospheric chemical transport model. The model has 24 latitudinal bands and 12 vertical levels between 0 and 24 km altitude (see [
23] for further details). The emissions were implemented by fitting the model output at the relevant latitude band to the Cape Grim measurements. The emission priors were mainly located in mid- to high latitudes (mainly between 36°N and 56°N) of the northern hemisphere prior to 1991. They were subsequently shifted to lower latitudes (mainly between 24°N and 42°N) to reflect a move of production and use to developing (Article 5) countries based on previously assumed changes to the emission distributions of similar anthropogenic compounds, such as halons ([
23,
27]).
The lifetimes of the two CFCs were determined by adjusting the diffusive loss from the top of the model to give appropriate lifetimes, as described above. The lifetime of HCFC-225ca is determined primarily by the loss from the reaction with OH. We use a reaction rate with OH of
[
28] and a mean global OH concentration that gives a lifetime with respect to OH of 6.1 years for methyl chloroform [
3] using a reaction rate coefficient of
[
29]. This gives a lifetime of HCFC-225ca with respect to the loss to OH of 2.0 years, which is similar to the recommendation of [
3]. The stratospheric loss was adjusted to give a total atmospheric lifetime of 1.9 years, as recommended in [
3].
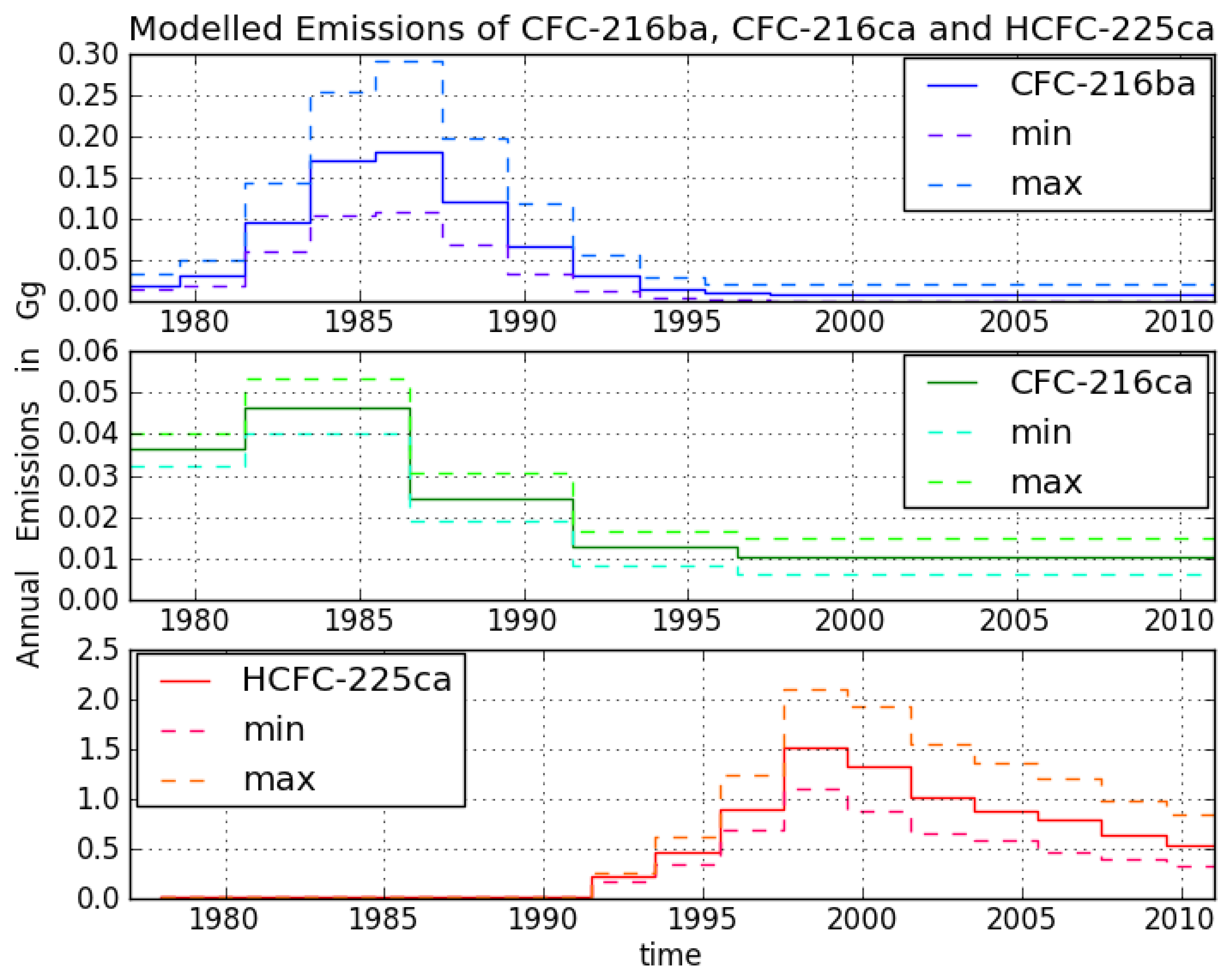
The uncertainty ranges on the emission estimates of CFC-216ba and CFC-216ca were determined by accounting for a combined measurement uncertainty of 3% for the mean measurement uncertainty and 7% for the calibration uncertainty (additionally, a further 23% was applied to the calibration uncertainty of CFC-216ba).
The modelling uncertainty was estimated to be 5% based on previous work with the model, which has shown it to recreate measurements of long-lived gases at Cape Grim with mainly northern hemisphere emissions and well-established emission histories (e.g., CFC-11, CFC-12) to within 5% [
27]. Furthermore, the uncertainty in the model fit to the measurements was considered based on the square root of the mean of the sum of the squares of the deviation of the actual mixing ratios from the model fits for each compound (4.7% for CFC-216ba, 1.9% for CFC-216ca). The measurement, modelling and model fit uncertainties were then combined using the square root of the sum of the squares, and the resulting uncertainty factor was applied to the ‘best fit’ model output to give an envelope of upper and lower uncertainty bounds.
The model output was then fitted to the upper bound of this envelope using the lower estimate of the lifetime to give the maximum emissions and fitted to the lower bound of the envelope using the upper estimate of the lifetime to give the minimum emissions.
The range on the emission estimates for HCFC-225ca is determined in a similar fashion by creating an envelope based on a combined uncertainty of 3% mean measurement uncertainty, 7% calibration uncertainty, 5% modelling uncertainty and 2.3% uncertainty in the model fit to the measurements. The model output was then fitted to the upper and lower bound of this envelope using the upper and lower estimates of the reaction rate constant with OH (a factor of 3 [
28], to give the uncertainty range of the emissions).
3. Results and Discussion
As the knowledge of the atmospheric lifetime is a prerequisite for the determination of the emissions of a trace gas, we first discuss the stratospheric dataset followed by trends and emissions derived from the Cape Grim archive.
3.1. Lifetime and ODP Estimation
Stratospheric lifetimes of CFCs are equivalent to their total atmospheric lifetimes, as these compounds do not have significant tropospheric sinks [
4]. We here use a dataset of 97 stratospheric samples that were collected with a whole air sampler [
21,
30] on-board the high-altitude aircraft, Geophysica, during two sampling campaigns from Germany and Sweden, respectively, in 2009 and 2010 [
31]. Samples collected between 10 and 21 km were analyzed for their CFC-216ba content to establish the first estimate of its lifetime. As explained in Section 2.2, the results for CFC-216ca could not be used. Mixing ratios of long-lived trace gases form compact correlations with each other in the stratosphere. In [
24], the slope of these correlations at the tropopause to calculate stratospheric lifetimes is used for 10 major ozone-relevant gases, namely: CFC-11, CFC-12, CFC-113, HCFC-22, HCFC-141b, HCFC-142b, H-1211, H1301, CCl
4 and CH
3CCl
3. The technique works well for compounds that break down sufficiently fast in the stratosphere and, in return, exhibit a significant decomposition in the vicinity of the tropopause. This is not observed for CFC-216ba. Additionally, its very low abundances limit the precision of its measurement.
We therefore use a correlation between the stratospheric lifetimes and fractional release factors (FRFs,
i.e., the fraction of a compound that has decomposed) established by [
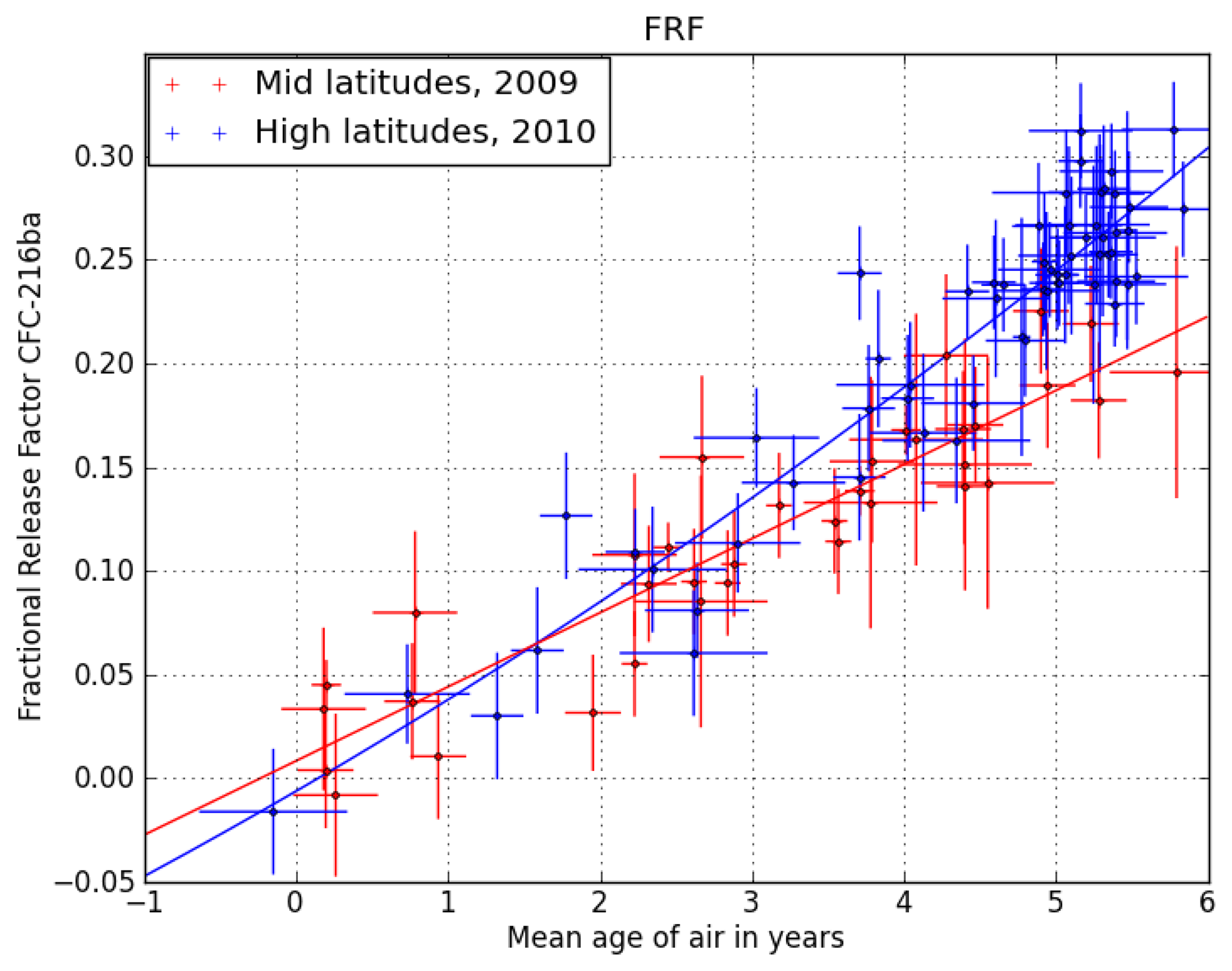
24] to estimate the stratospheric lifetime of CFC-216ba.
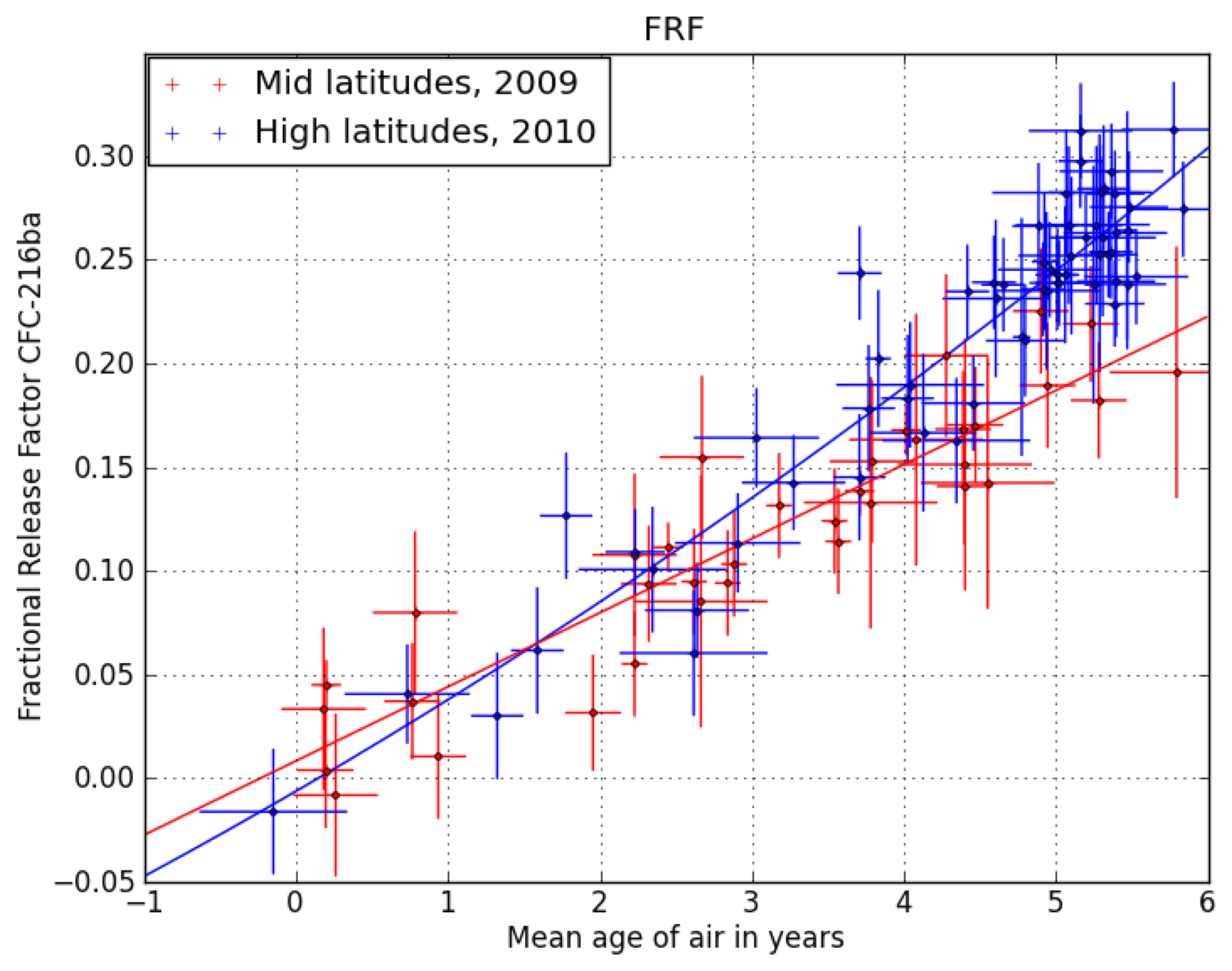
Figure 1 shows the correlation of CFC-216ba FRFs with mean transit times (or mean ages, derived from measurements of the passive tracer, SF
6) of the sampled stratospheric air at mid- and high latitudes. Vertical transport times in the stratosphere are on the order of years; so normally, a correction for intermittent changes in tropospheric abundances is required. However, as the tropospheric abundances of CFC-216ba have not changed significantly over the past two decades (see Section 3.2), no such correction is needed here. Using the relation in
Figure 1, we calculate the FRF of CFC-216ba at two key points,
i.e., a mean age of three years at mid- and of 5.5 years at high latitudes. The above-mentioned data set of [
24] only allows for the derivation of stratospheric lifetimes relative to a reference compound. We here scale the available lifetime dataset to the most recent estimate of the lifetime of CFC-11, which is 52 years [
4].
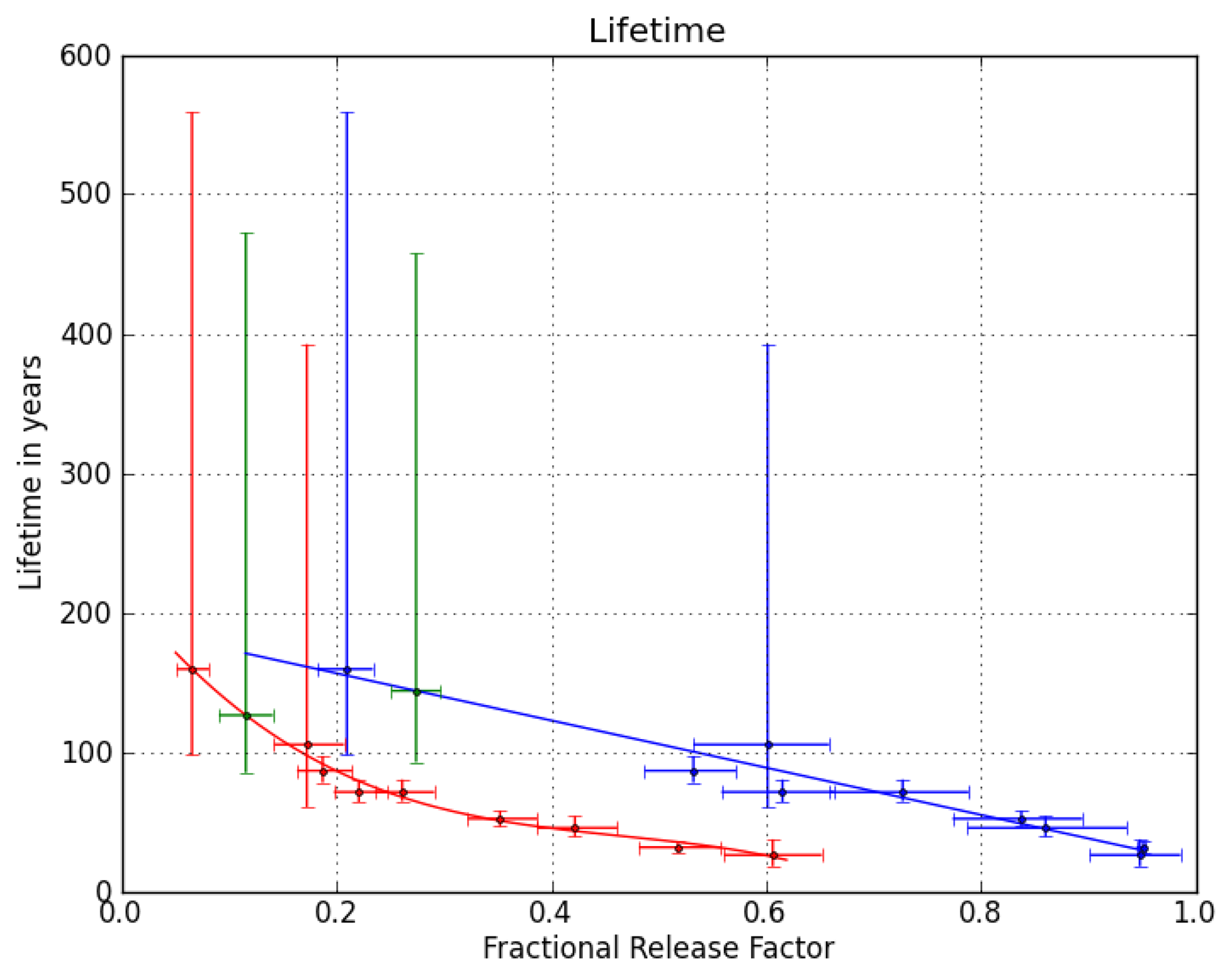
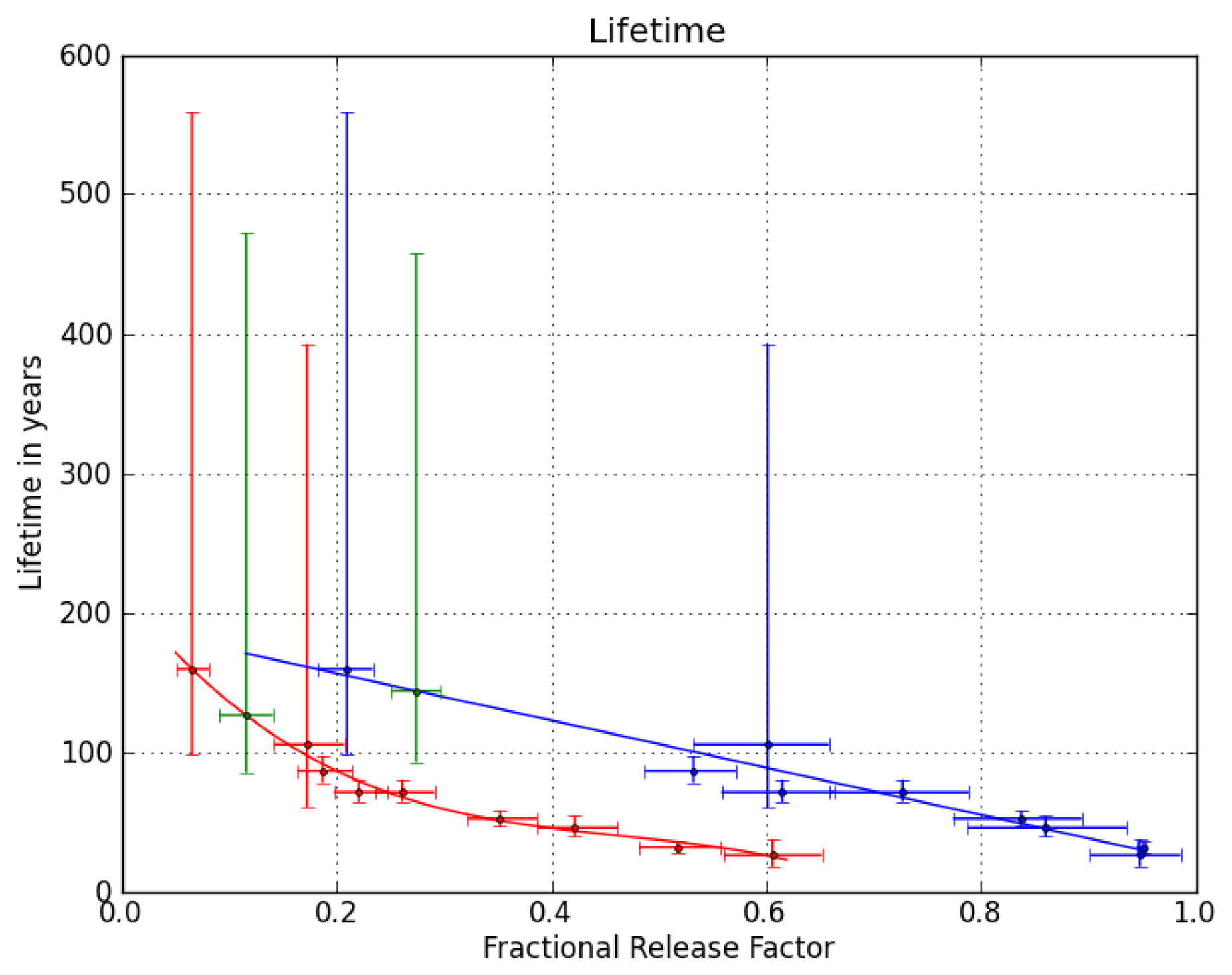
Figure 2 shows the relation between stratospheric lifetimes and FRFs at the two key points for nine of the compounds presented in [
24],
i.e., all but the longest-lived (HCFC-142b). Using the derived polynomial fit functions and the FRFs of CFC-216ba at the two key points yields two very similar stratospheric lifetimes of 126 years (1
σ uncertainty range of 85–472 years) at a mean age of three years in mid-latitudes and 144 years (93–458 years) at a mean age of 5.5 years in high latitudes. We combine these two numbers to a best estimate of 135 (85–472) years for the stratospheric lifetime of CFC-216ba. Given the chemical similarity of the two CFC-216 isomers and the lack of reliable stratospheric data for CFC-216ca, we assume the same stratospheric lifetime and uncertainty range for both compounds. It should be noted, though, that the lifetimes of other CFC isomer pairs have been found to differ by 14% (CFC-112/CFC-112a) and 18% (CFC-113/CFC-113a), respectively ([
24,
25]).
Differences are, however, expected to be smaller for CFC-216ba and CFC-216ca, due to the greater similarity of functional groups in these two molecules. Using the lifetime estimate, the FRF at a mean age of three years and the methodology described in [
24], we calculate an ODP of 0.35 (0.16–1.6) for CFC-216ba and CFC-216ca.
3.2. Cape Grim Measurements and Modelled Emissions
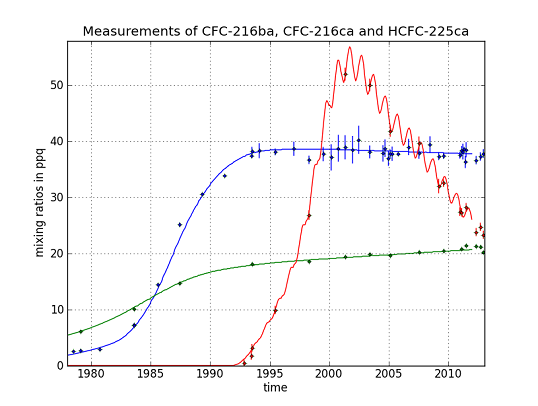
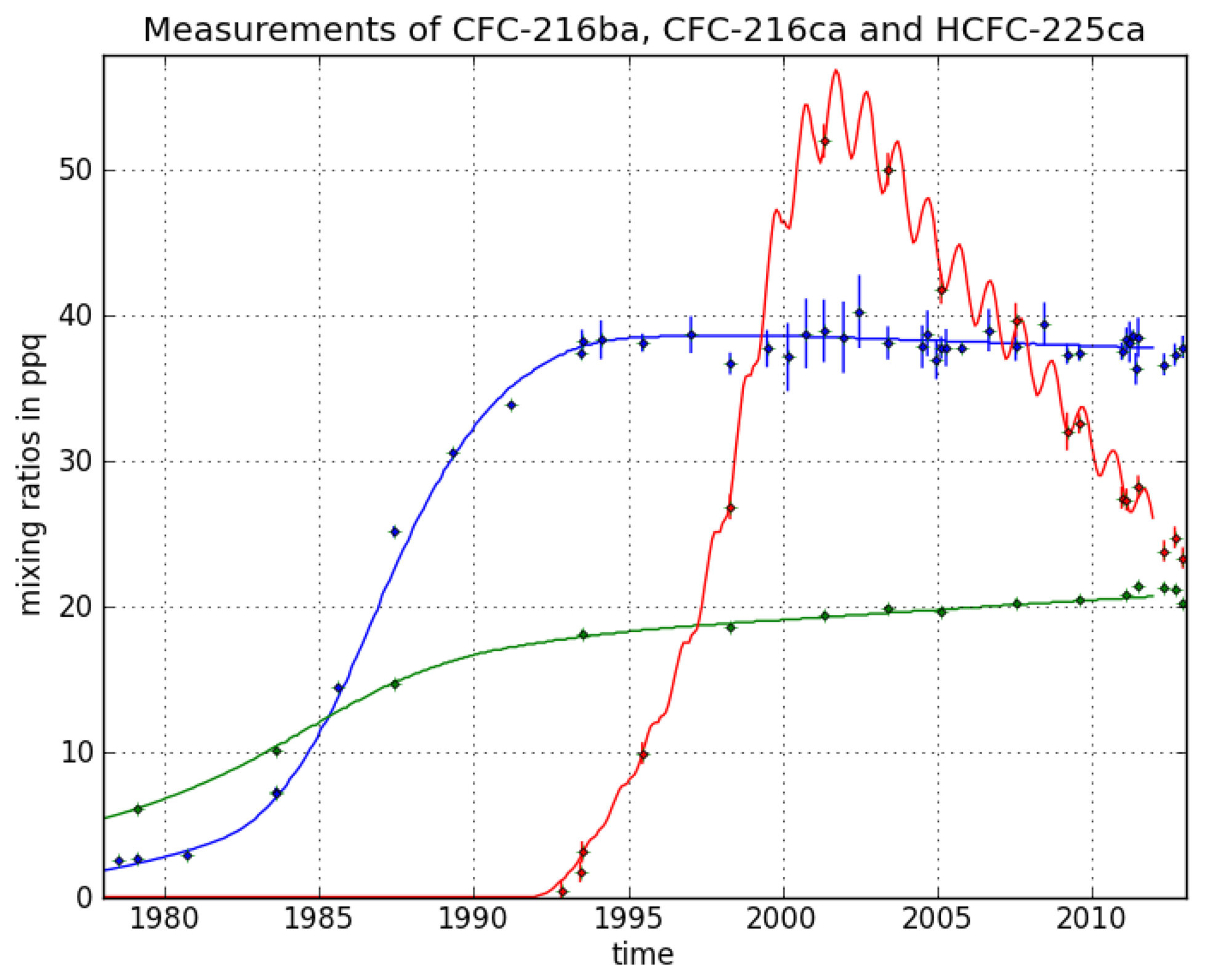
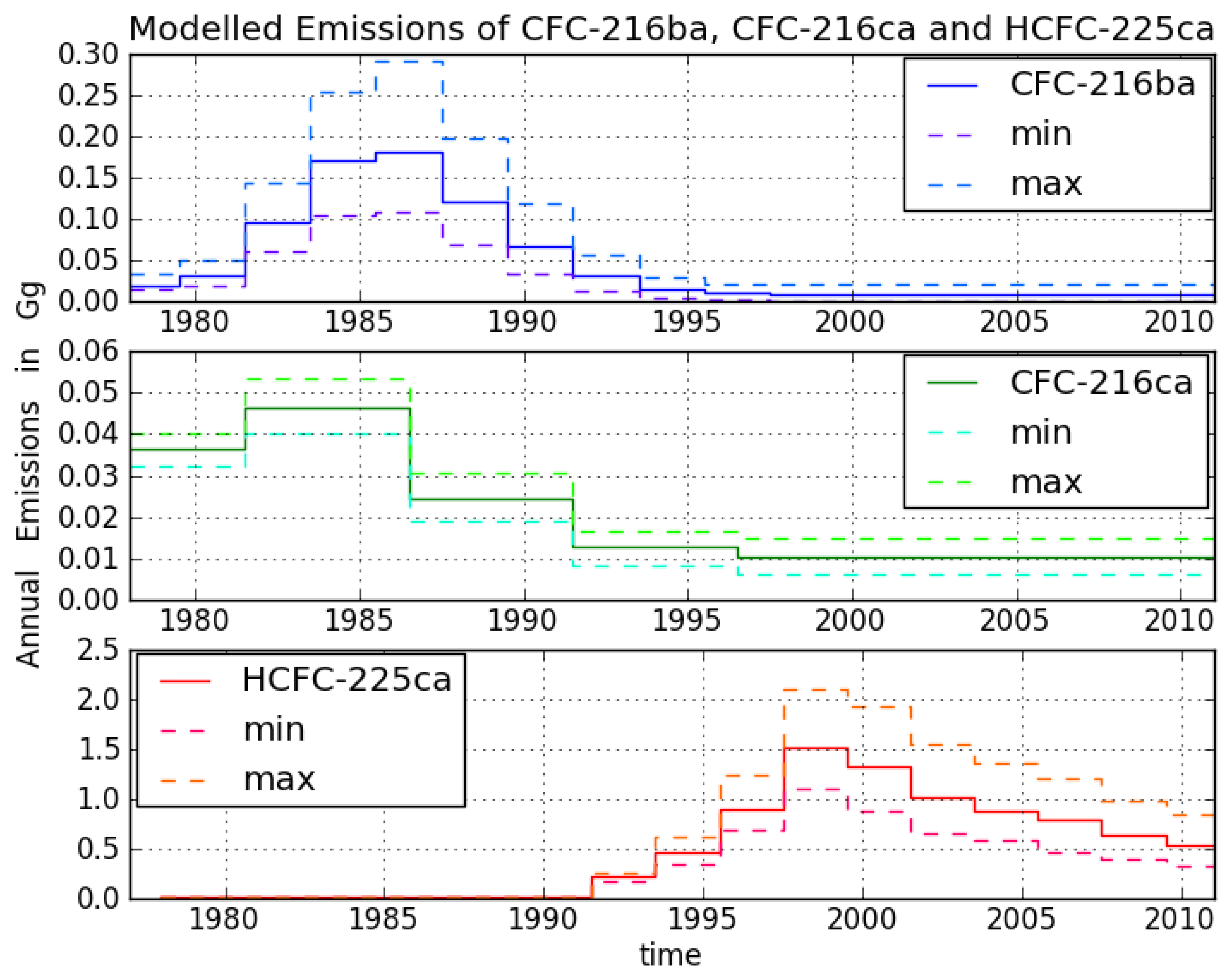
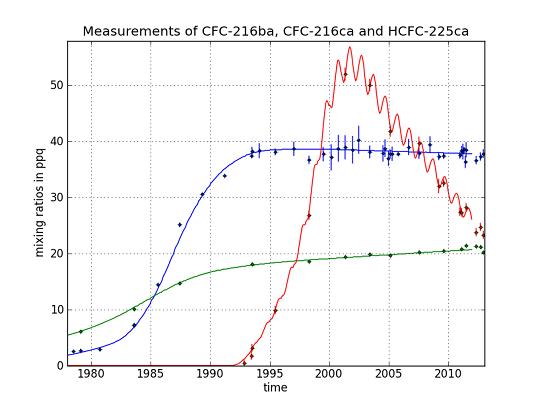
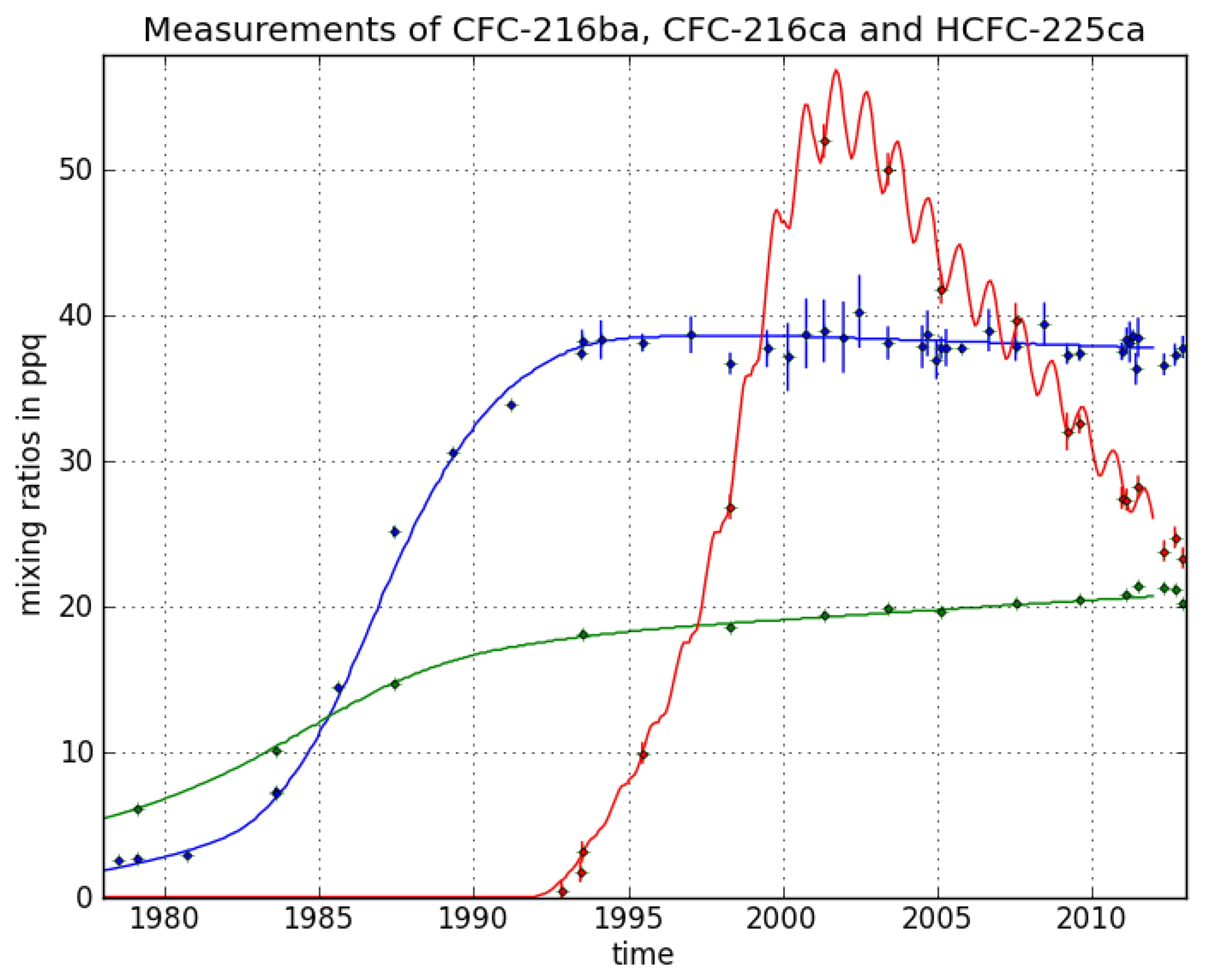
CFC-216ba measurements on 42 different Cape Grim samples give an atmospheric record from 1978–2012 (
Figure 3). The sample collected on 7 July 1978, exhibits the lowest mixing ratio of 2.5 ± 0.04 ppq (parts per quadrillion; 10
15). Abundances reached a maximum of 38.3 ± 0.8 ppq in 1993 and have been approximately constant from 1993 to 2012 at around 38.0 ppq. From 26 September 1980 to 7 June 1993, we found an average increase of 3.4 ppq per year. Respective annual emission estimates are presented in
Figure 4. Emissions sharply increased in the 1980s and peak around 1986 at about 0.18 Gg/year, then decreased and became indistinguishable from zero in the late 1990s.
Although the first reduction step of the Montreal Protocol (20% for developed countries) was sought for 1993, emissions had decreased already in the late 1980s. The total phase-out of CFC-216 in developed countries was scheduled for 1 January 1996 (for developing countries on 1 January 2010), which agrees well with our estimated emission trajectory. Constant mixing ratios in the last 20 years indicate the success of the Montreal Protocol in reducing CFC-216ba emissions. The fact that CFC-216ba mixing ratios have not significantly declined since 1993 can mostly be explained by its long atmospheric lifetime. However, using the best lifetime estimate suggests remaining emissions of 0.01 Gg/year between 2000 and 2012. The trends observed during the last decade of the record, when emissions are estimated to be indistinguishable from zero, are consistent with the estimated lifetime range. The corresponding expected decrease in mixing ratios would not be detectable within the derived uncertainties.
For CFC-216ca, the dataset consists of 15 different Cape Grim samples. Compared to its isomer, we derived lower atmospheric abundances throughout most of the record, i.e., between 6.1 ± 0.2 ppq (6 February 1979) and 21.4 ± 0.3 ppq (21 June 2011). However, in contrast to CFC-216ba, we found statistically significant increases in mixing ratios between 1994 and 2012 (around 0.2 ppq/year). Emissions considerably increased to 0.05 Gg/year between 1979 and the mid-1980s. The largest estimated annual emissions are around four times smaller than those of CFC-216ba. From the mid-1980s onwards, emissions decreased until the late 1990s, but have remained constant ever since.
The decline in emissions starting around 1985 preceded the first step of the Montreal Protocol phase-out schedule, which sought a reduction of 20% CFC-216ca production by 1 January 1993. Although a complete production phase-out was sought for 2010, our records suggest continuing emissions of about 0.01 Gg/year up until 2012.
Because of their long lifetimes, both CFCs will continue to provide chlorine to the stratosphere for many decades. However, since the abundances of CFC-216ba and CFC-216ca in the atmosphere are in the range of parts per quadrillion, the amount of chlorine released into the stratosphere by CFC-216 isomers is small compared to the amount of 2.08 ppb (parts per billion) of chlorine available in the form of CFCs in 2008 [
3].
The HCFC-225ca dataset consists of 24 Cape Grim samples. The air sample from 27 April 2001, contains the highest amount at a mixing ratio of 52 ± 1.2 ppq. The mixing ratio for HCFC-225ca was below the detection limit before 13 March 1991. Average growth rates of 6 ppq per year (1992–2001) and —2.6 ppq per year (2001–2012) were observed. Annual variations can be explained by the comparably short lifetime and the seasonal cycle of the reaction with OH. Until 1991, no significant emissions of HCFC-225ca occurred. Annual emissions subsequently increased to about 1.5 Gg/year around 1999, which is about eight times higher than the peak emissions of CFC-216ba.
The turning point in emissions around 1999 and the following downward trend are in line with the requirements of the Copenhagen (1992) and the Vienna Amendments (1995) of the Montreal Protocol [
12]. The first phase-out step for HCFC-225ca was sought for 1 January 2004 (reducing of 35% of production, 75% of consumption). However, the turning point around 1999 indicates that HCFC-225ca emissions were reduced beforehand. The rapid downward trend in the last decade can be explained by decreasing emissions in combination with its short lifetime. The total phase-out in production and consumption for HCFC-225ca for developed countries is sought for 1 January 2030 (for developing countries, 1 January 2040), and the currently observed atmospheric trend looks promising for the fulfillment of the phase-out schedule. HCFCs accounted for 251 ppt or 7.5% of the total chlorine in the troposphere in 2008 [
3]. Mixing ratios of 23.3 ppq of HCFC-225ca in 2012 represent a fraction of 9 × 10
−3% of total chlorine. Owing to its low abundances and its shorter lifetime (as compared to CFCs), HCFC-225ca has a negligible effect on past and future stratospheric chlorine and ozone depletion.
3.3. CARIBIC Measurements
Fifty-six upper tropospheric samples collected during 2013 CARIBIC flights between Frankfurt, Germany, and Bangkok, Thailand, have also been analyzed. These samples were collected exclusively over Asia (latitude range: 17° to 32°N; longitude range: 70° to 97°E; altitude range: 11 to 12 km) with no influence from stratospheric air masses. Results were generally found to support the Cape Grim abundances, and averages are listed in
Table 1. One exception is the mixing ratios of CFC-216ca, which are, on average, 21% higher than those observed at Cape Grim in 2012, supporting a continuation of the observed increases. Due to its long lifetime, a large interhemispheric difference is not expected for this compound, unless there are significant sources, predominantly located in one hemisphere. The observed higher abundances and the variability between flights could therefore indicate a new onset of emissions and/or that CFC-216ca emission sources are situated in the vicinity of the CARIBIC flight track.
4. Conclusions
We achieved quantifications, trend analyses and emission estimates for the chlorofluorocarbons, CFC-216ba and CFC-216ca, and the hydrochlorofluorocarbon, HCFC-225ca, in the atmosphere. A stratospheric air sample analysis was utilized to infer atmospheric lifetimes of 135 years (range 85–472 years) for both CFCs, meaning that CFC-216ba/ca will be present in the atmosphere for the coming centuries. To reconstruct trends, clean marine air samples from the Cape Grim baseline observatory from 1978 to 2012 were analyzed and supported with recent measurements of CARIBIC upper tropospheric samples.
The observed time series for CFC-216ba abundances shows a slowdown of the increases in the early/mid-1990s and no further significant changes since then. A similar behavior is observed for CFC-216ca until the 1990s, but in contrast, abundances have continued to increase up until 2012. Indications for ongoing emissions and likely further increases of CFC-216ca come from CARIBIC samples collected in the upper troposphere above Asia in 2013, which are well above (21%) the abundances observed at Cape Grim in 2012. Respective annual emission estimates for both CFCs show decreasing values in the mid-1980s prior to the first phase-out step of the Montreal Protocol, scheduled for 1993. However, our best estimates indicate that small emissions of both CFCs could persist up until 2012. For HCFC-225ca, we found a first appearance in the 1990s, followed by a sharp increase. Decreasing mixing ratios were observed from the early 2000s onwards, corresponding to decreasing emissions after the late 1990s. Hence, emissions were reduced shortly after the Vienna Amendments to the Montreal Protocol (1995), before the first phase-out step was sought in 2004.
The examined substances, CFC-216ba/ca and HCFC-225ca, both exhibit mixing ratios in the range of parts per quadrillion and, therefore, do not represent a substantial contribution to the amount of chlorine transported to the stratosphere and, consequentially, depletion of ozone.
The determination of quantities, trends and emissions of CFC-216ba, CFC-216ca and HCFC-225ca improve the current knowledge of present and past CFC and HCFC abundances in the atmosphere and extend the current CFC/HCFC time series record by three substances.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}