

Enhanced Sulfate Formation from Gas-Phase SO2 Oxidation in Non–•OH–Radical Environments

 , and
, and
Abstract
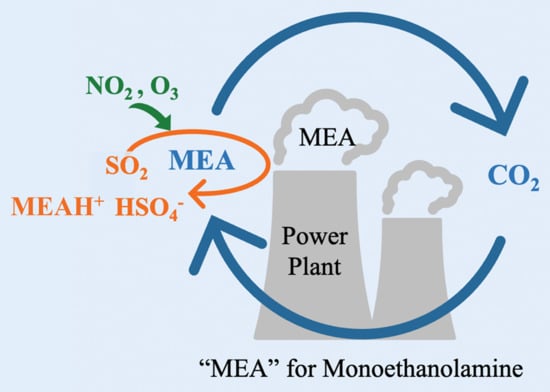
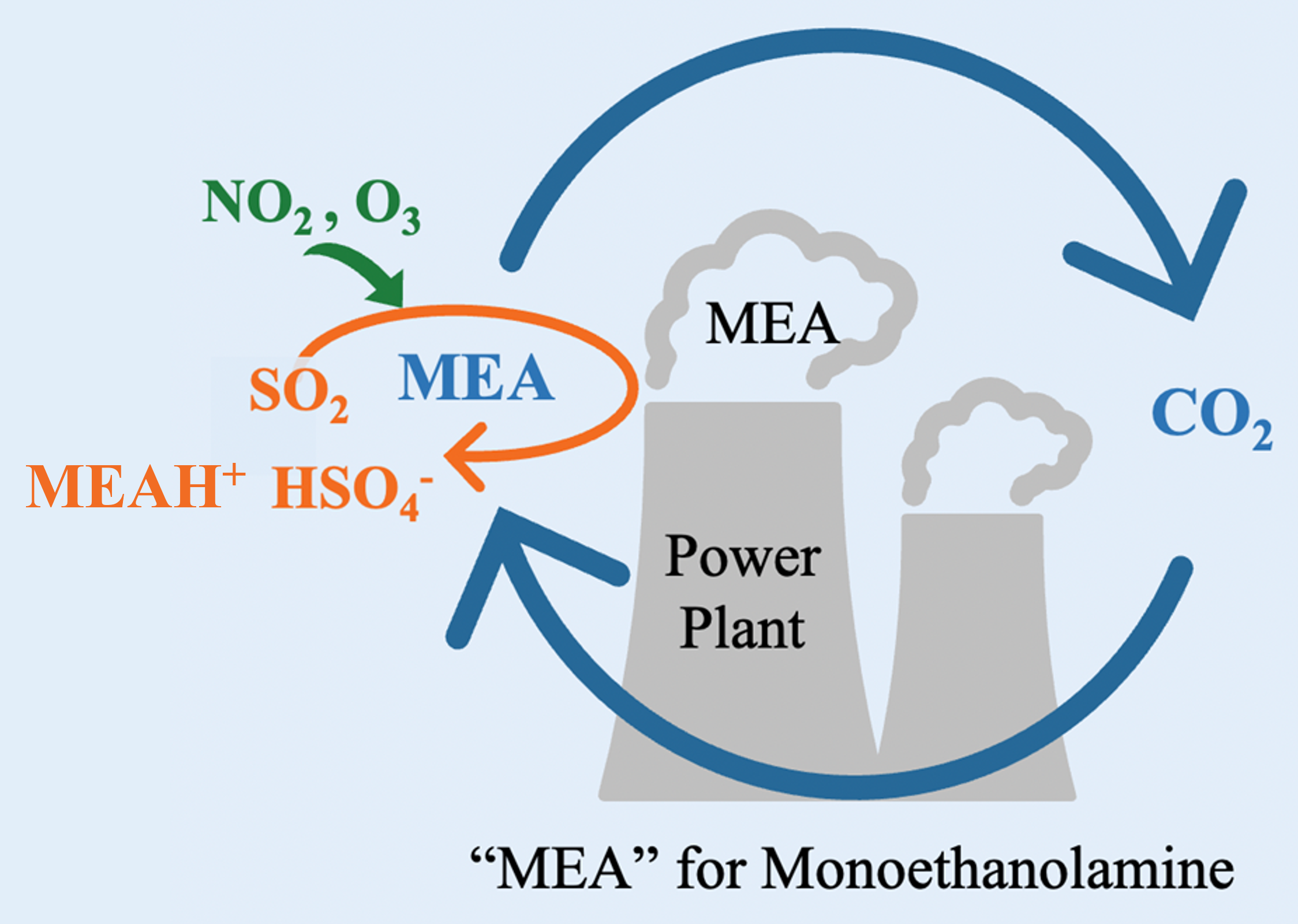
1. Introduction
2. Methods
2.1. Quantum Chemical Calculations
2.2. Reaction Kinetics Computation
3. Results and Discussion
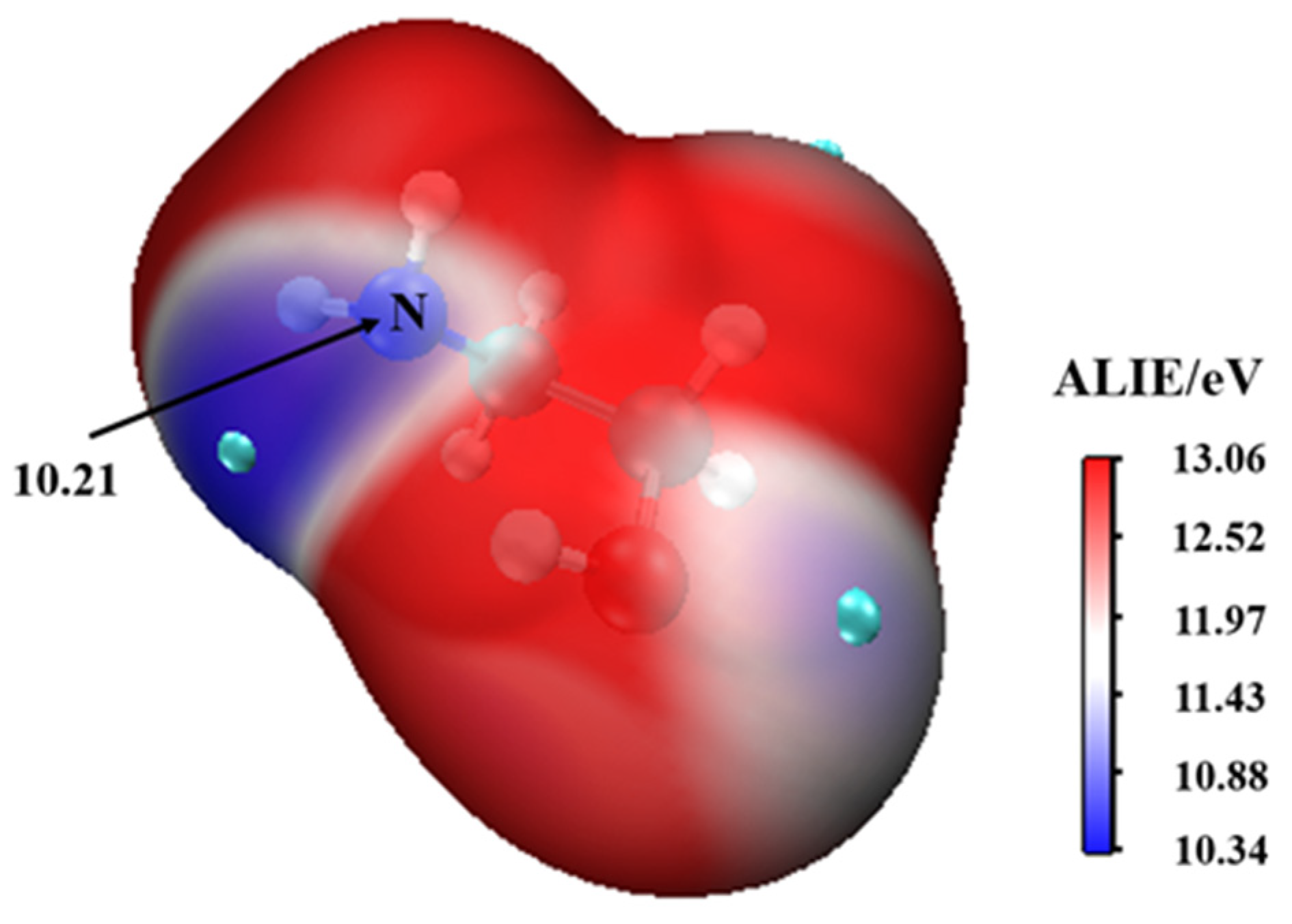
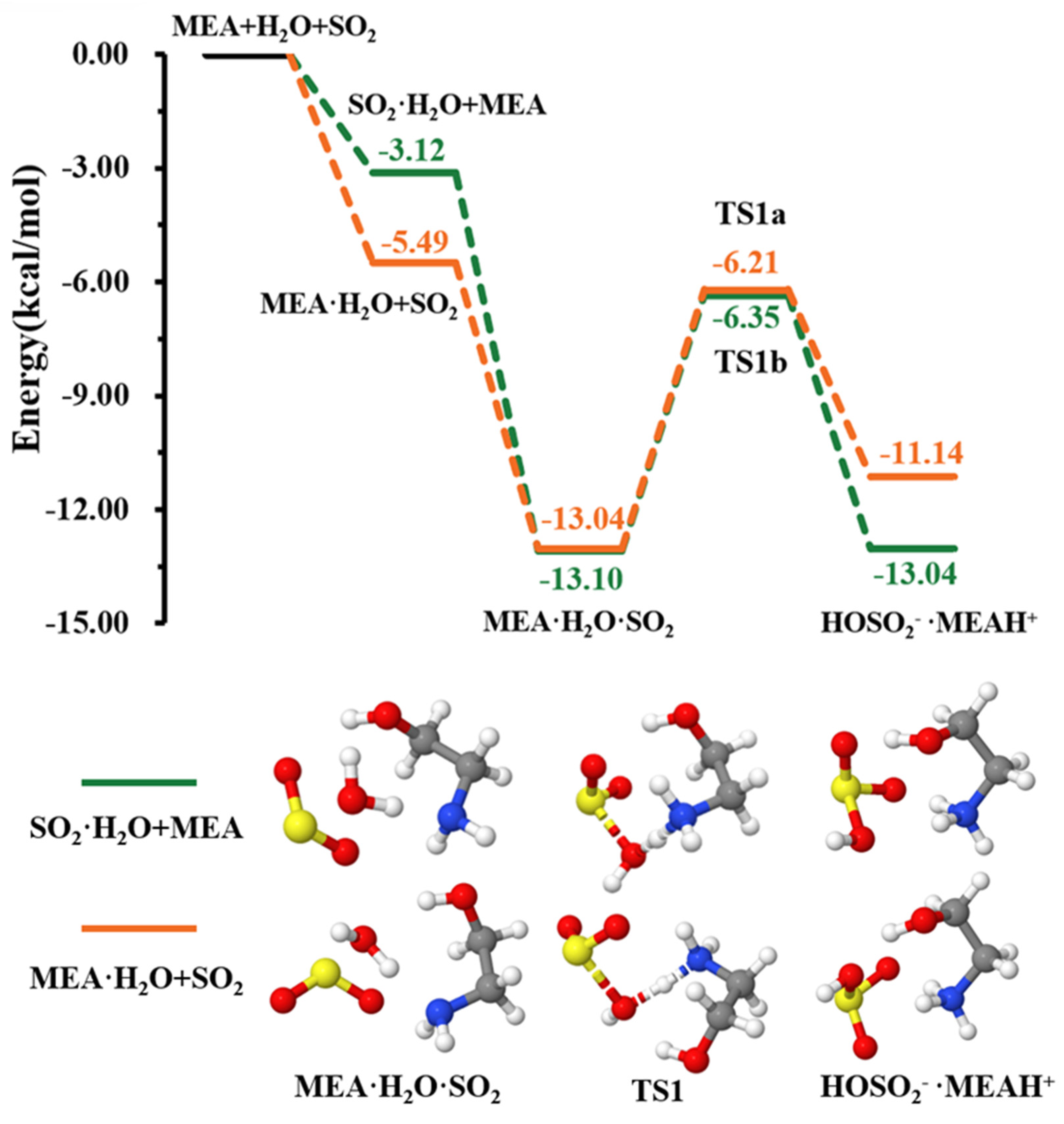
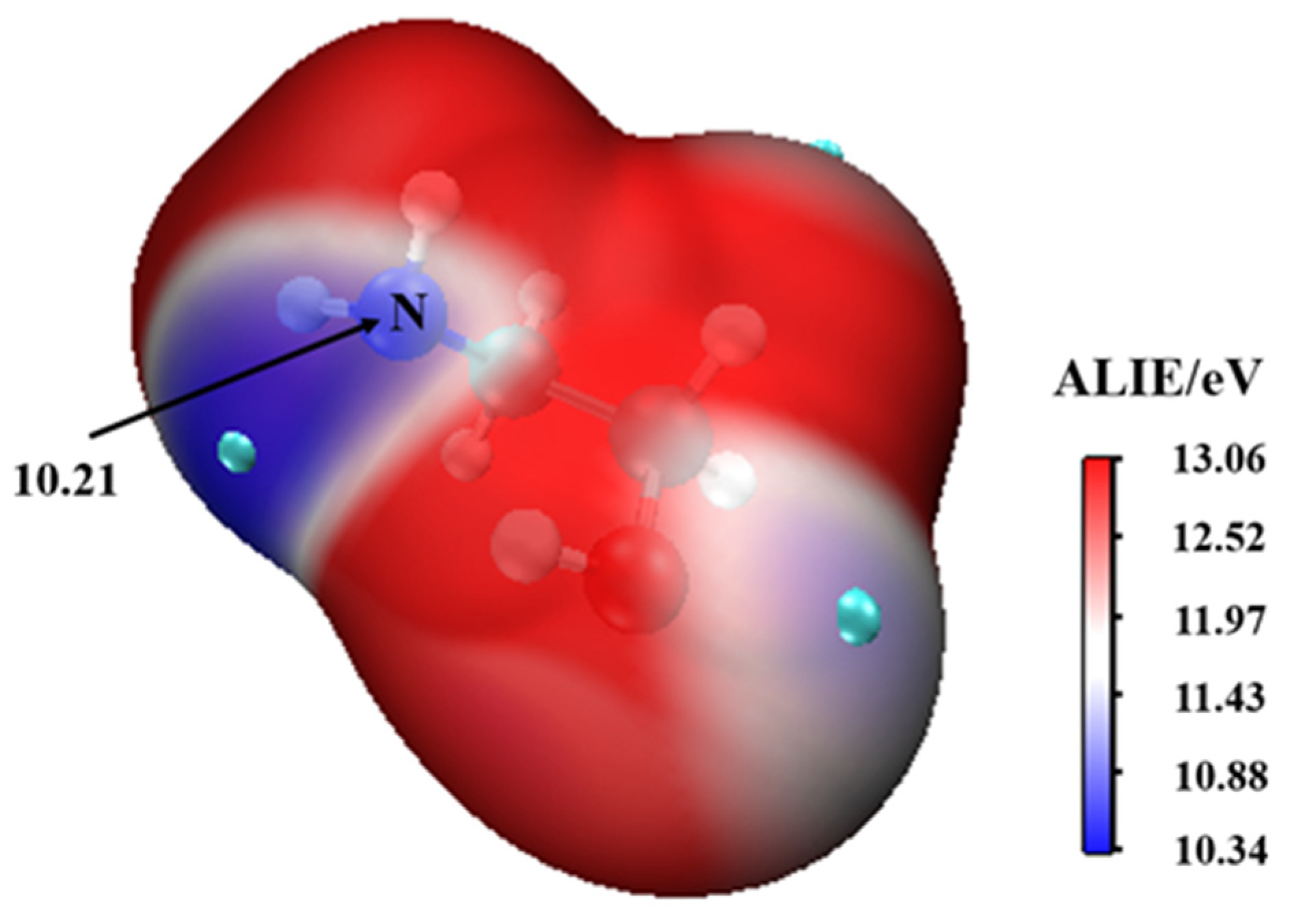
3.1. MEAAssisted Hydrolysis

{kind=link}
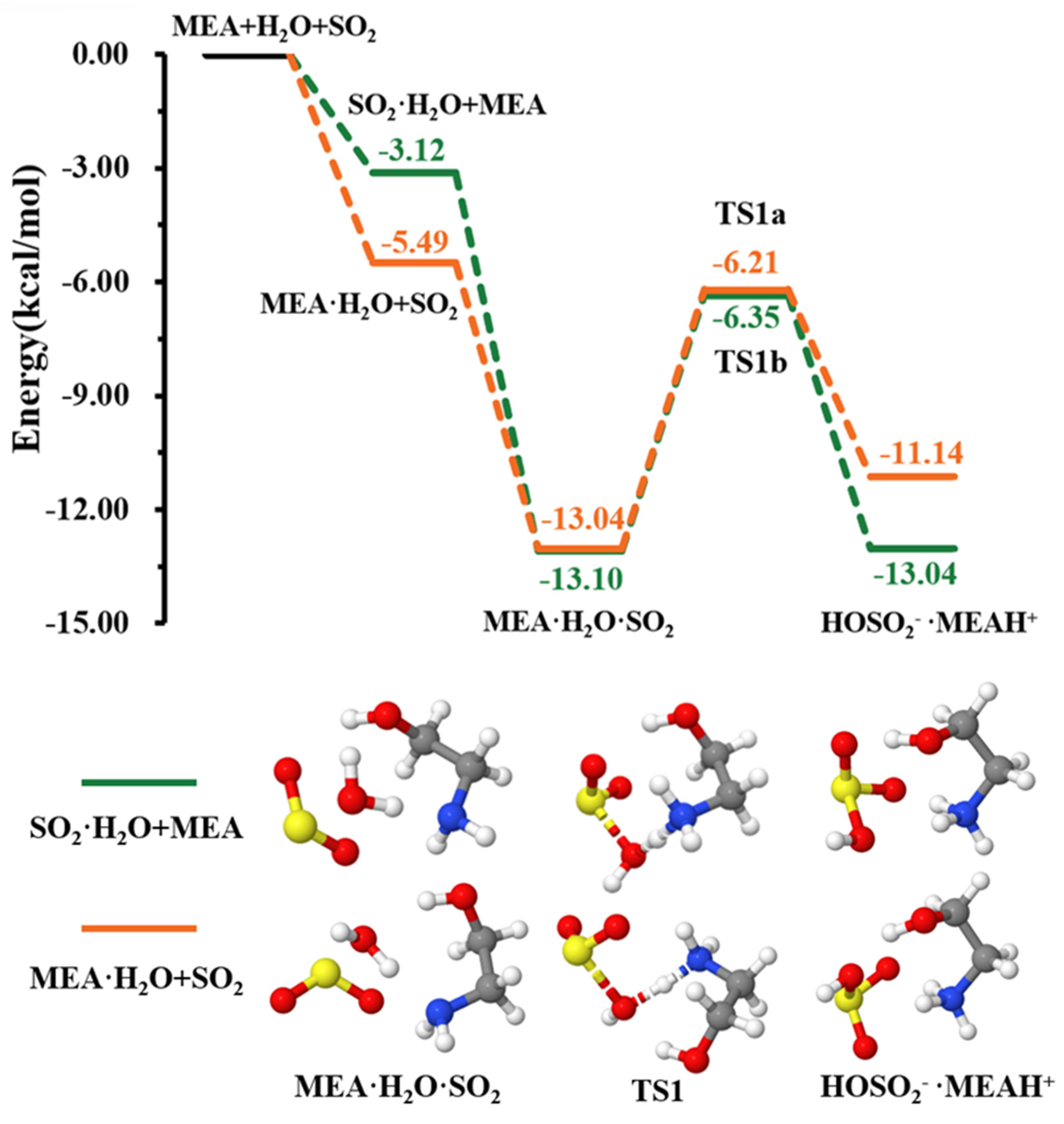{kind=link}
{kind=link}
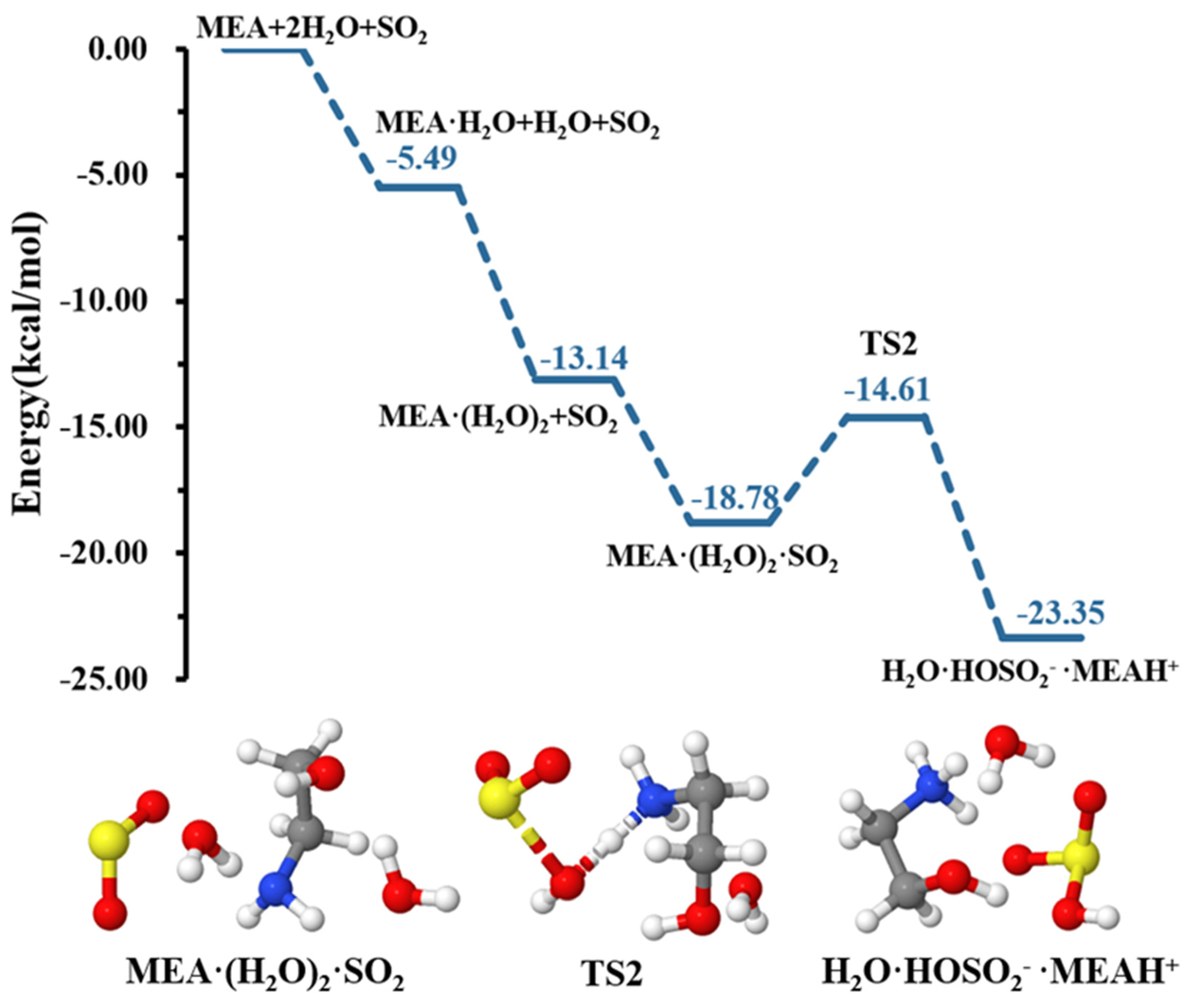{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔE | ΔG | Keq | Concentration | |
|---|---|---|---|---|
| −5.49 | 5.90 | 1.92 10−24 | 3.63 109 | |
| −3.12 | 4.94 | 9.85 10−24 | 7.66 106 | |
| −2.78 | 5.12 | 7.20 10−24 | 4.35 1012 | |
| −4.01 | 6.40 | 8.37 10−25 | 2.03 103 | |
| −13.04 | 8.98 | |||
| TS1a | −6.21 | 16.86 | ||
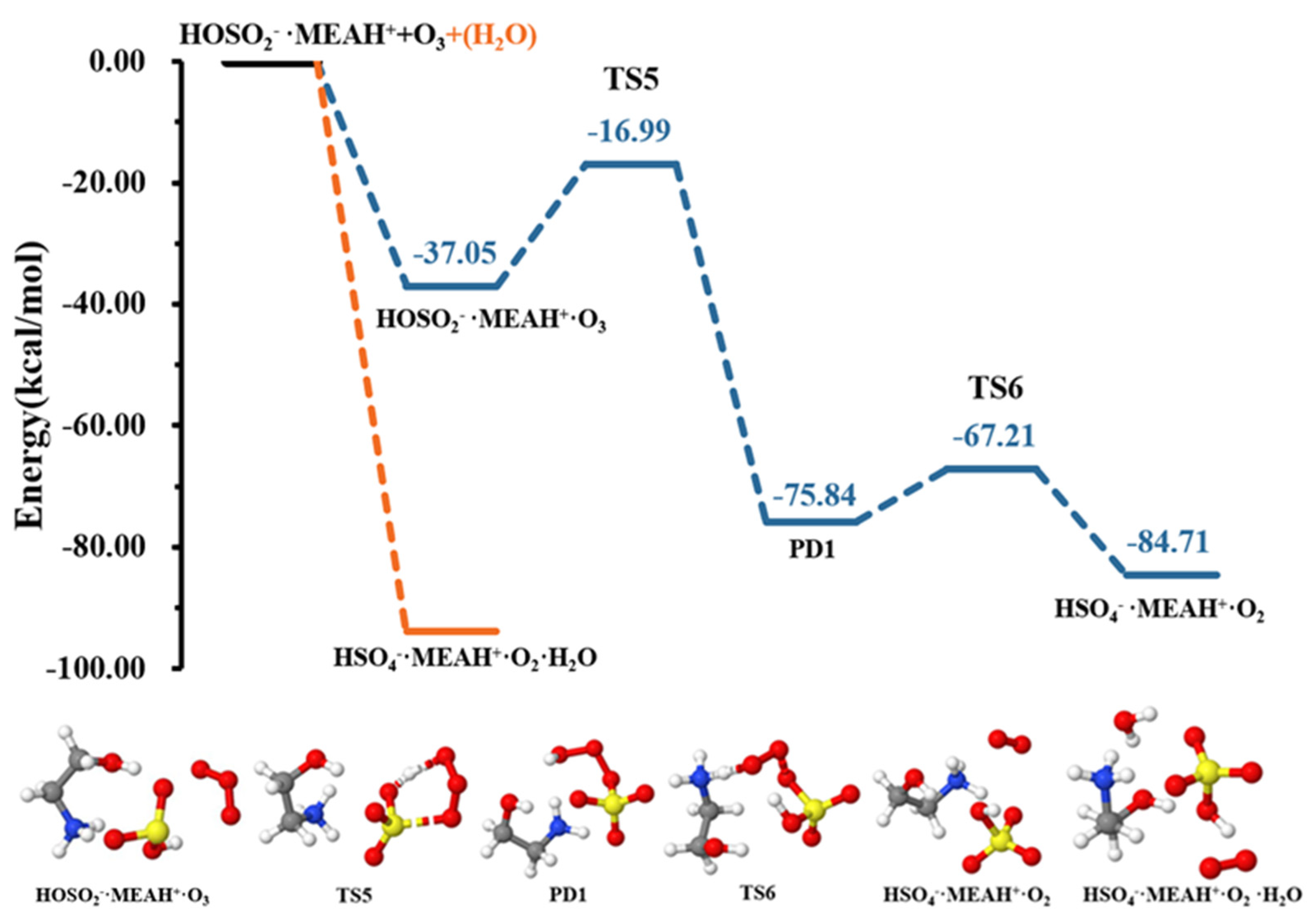
| • | −11.14 | 15.77 | ||
| • | −37.05 | −3.61 | ||
| TS5 | −16.99 | −1.29 | ||
| • (PD1) | −75.84 | −5.85 | ||
| TS6 | −67.21 | −2.78 | ||
| • | −84.71 | −0.76 | ||
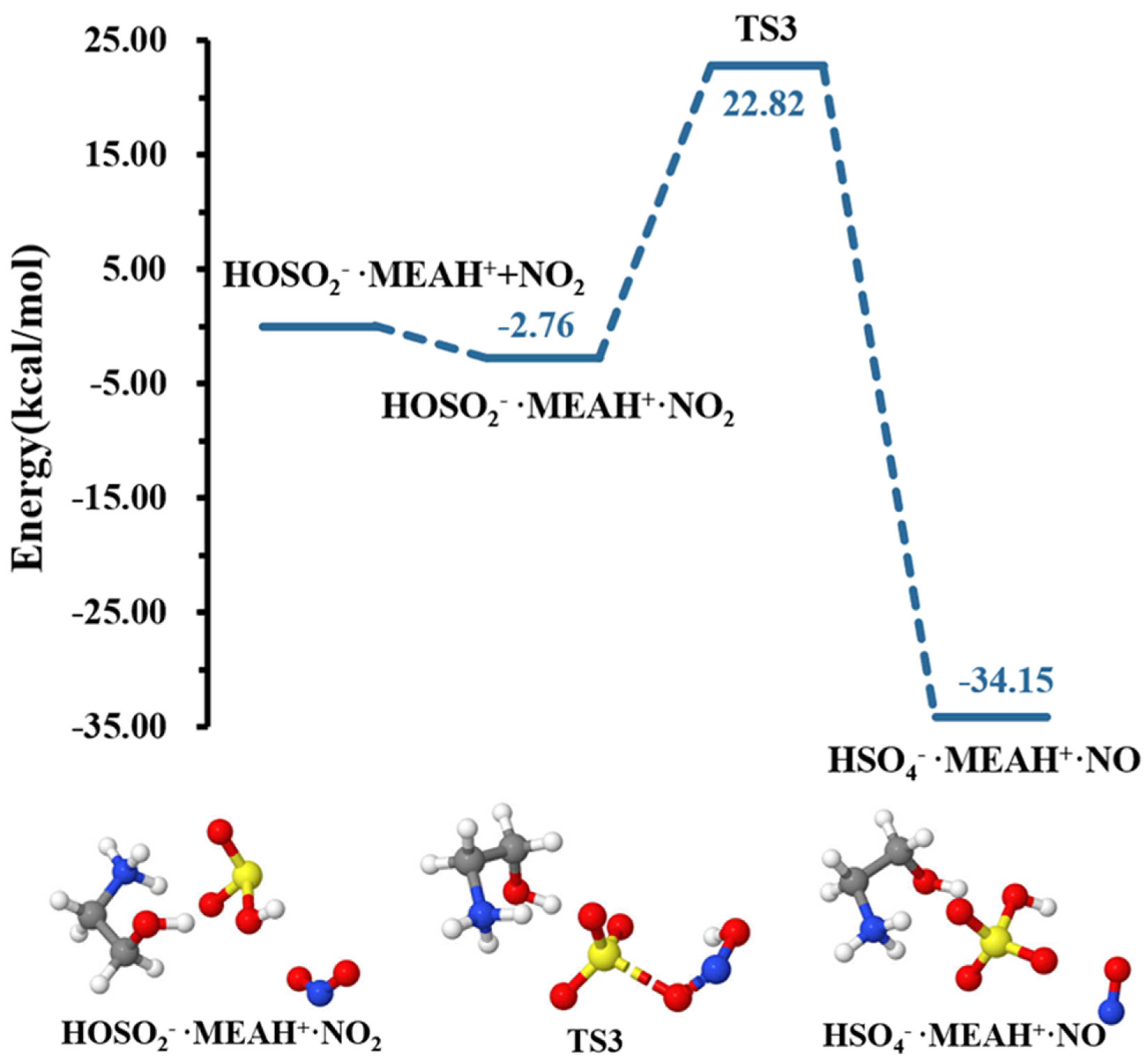
| • | −2.76 | 5.79 | ||
| TS3 | 22.82 | 37.96 | ||
| • | −34.15 | −3.42 | ||
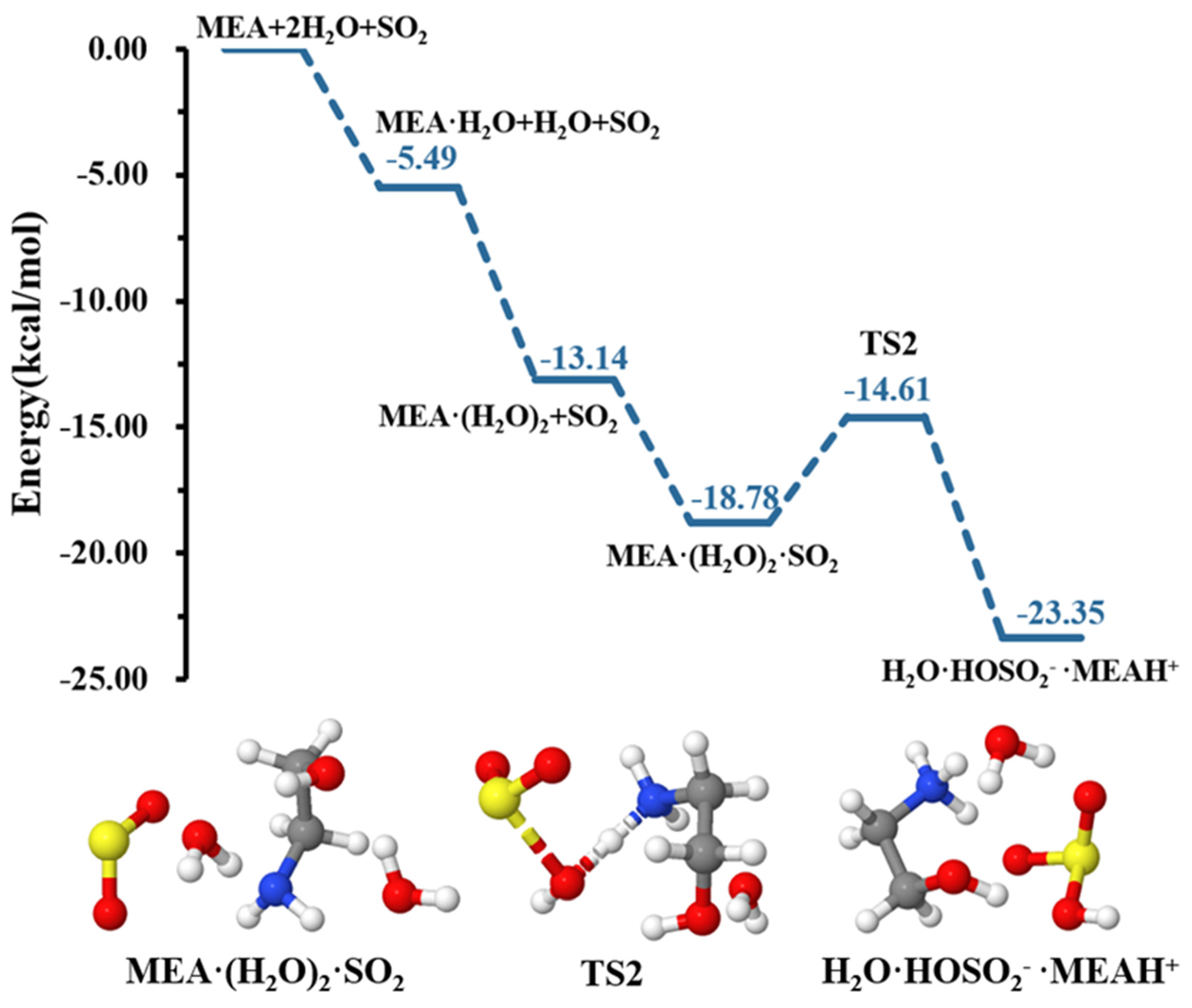
| With an additional water molecule | ||||
| −13.14 | 9.67 | |||
| −18.28 | 15.41 | |||
| TS2 | −14.61 | 18.09 | ||
| −23.35 | 15.47 | |||
| ++ | −93.89 | −6.83 | ||
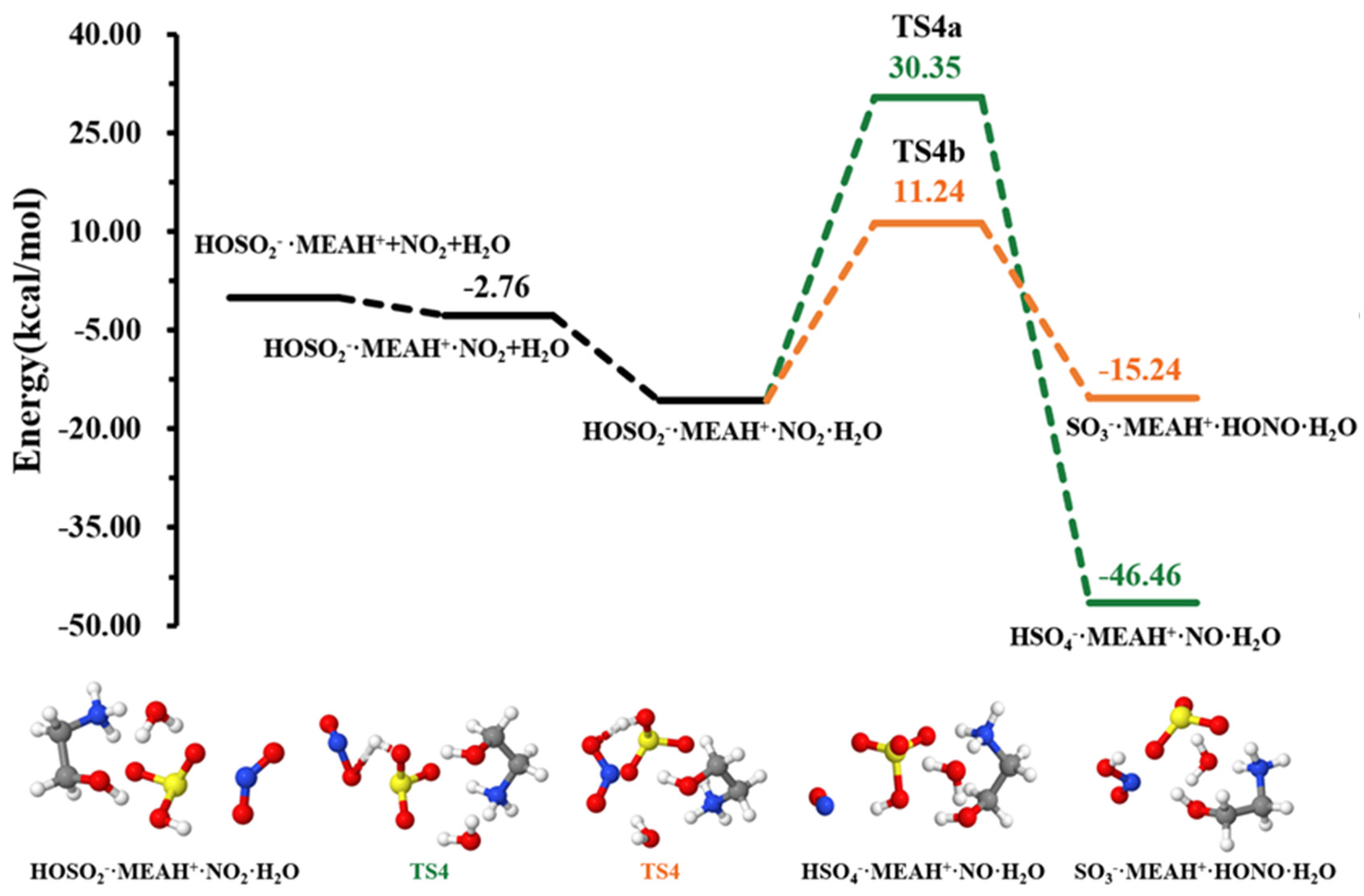
| −15.74 | 5.43 | |||
| TS4a | 30.35 | 51.96 | ||
| −46.46 | −23.30 | |||
| TS4b | 11.24 | 33.33 | ||
| −15.24 | 9.19 | |||
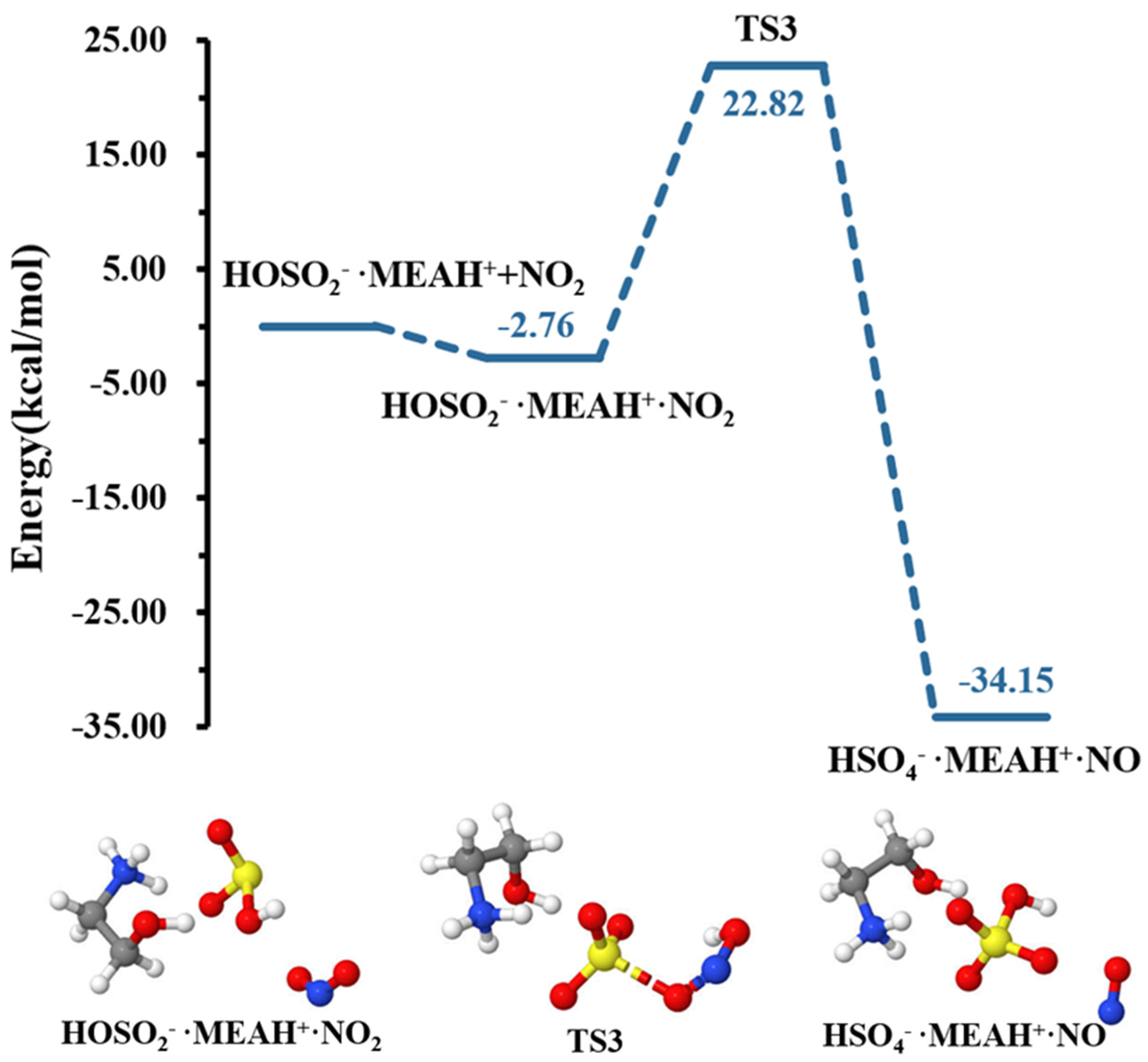
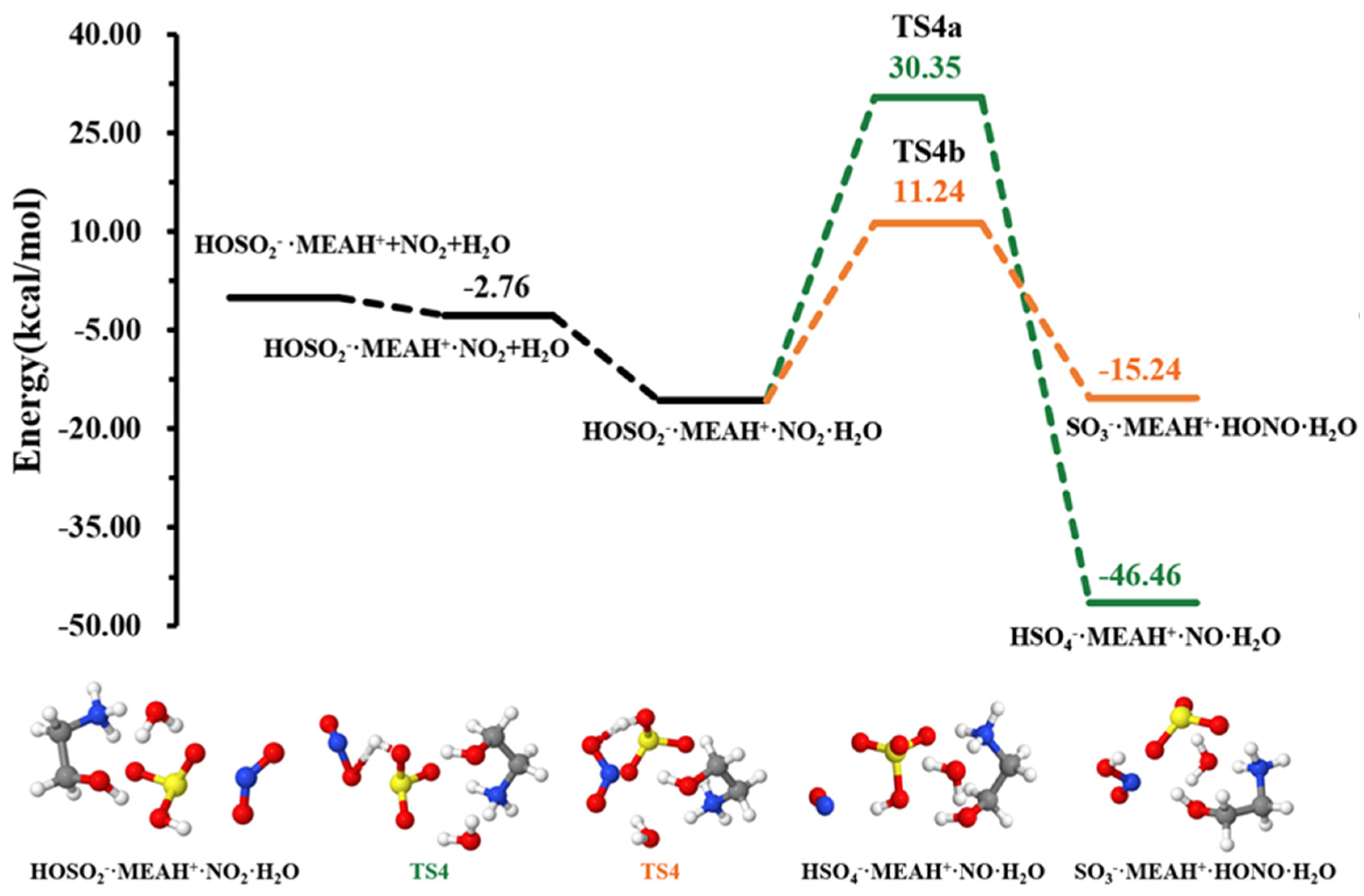
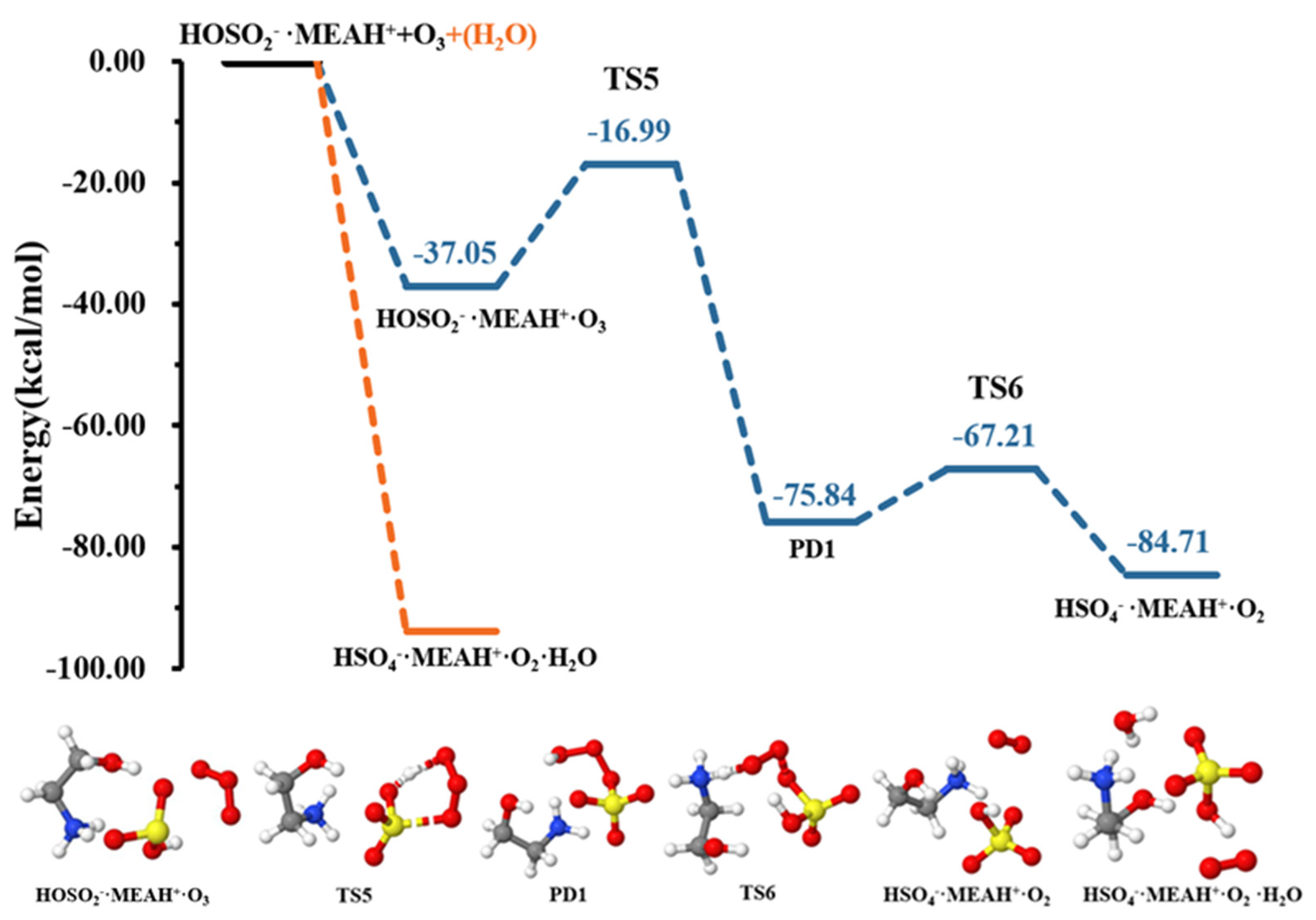
3.2. Further Reaction with
3.3. Further Reaction with
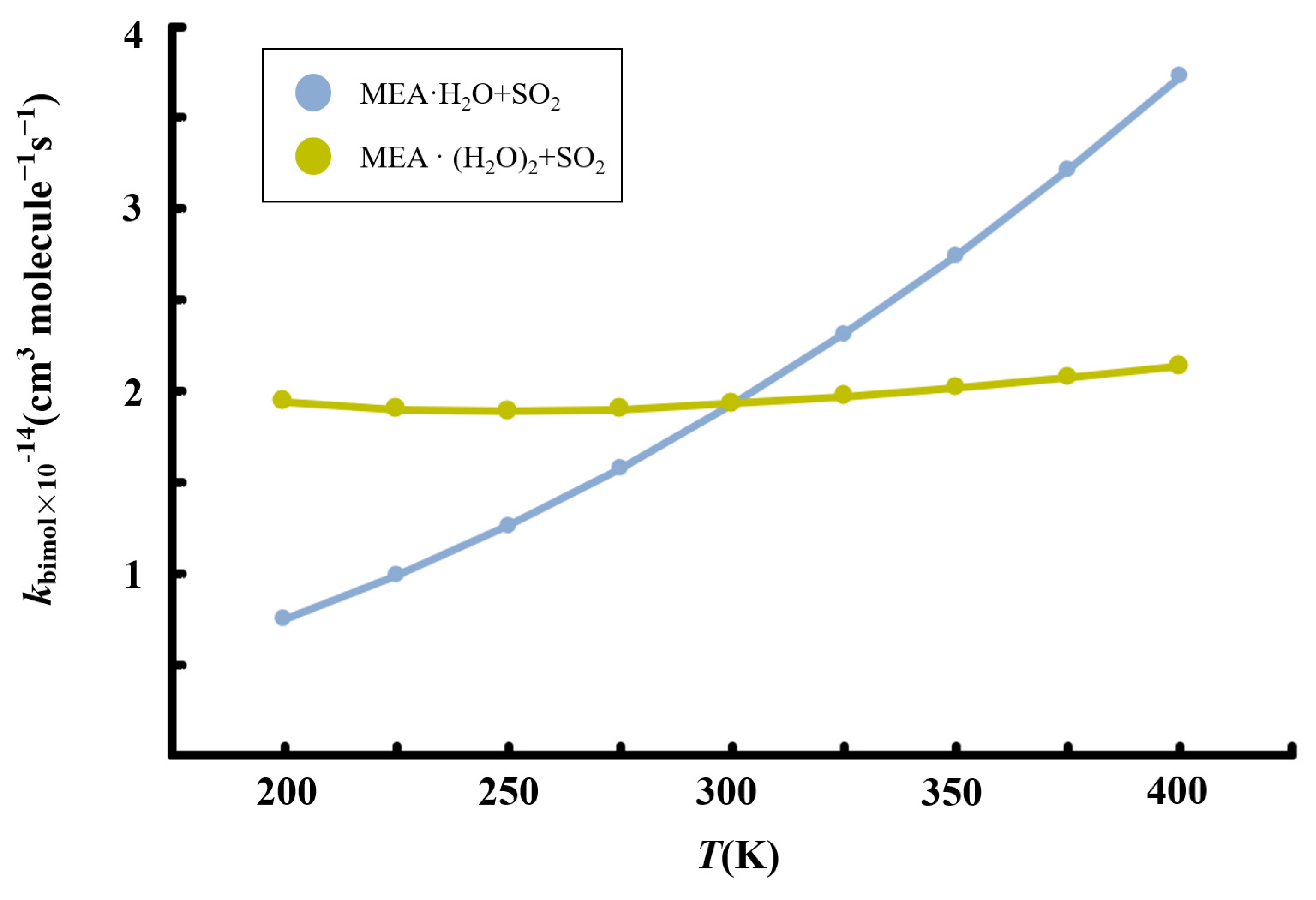
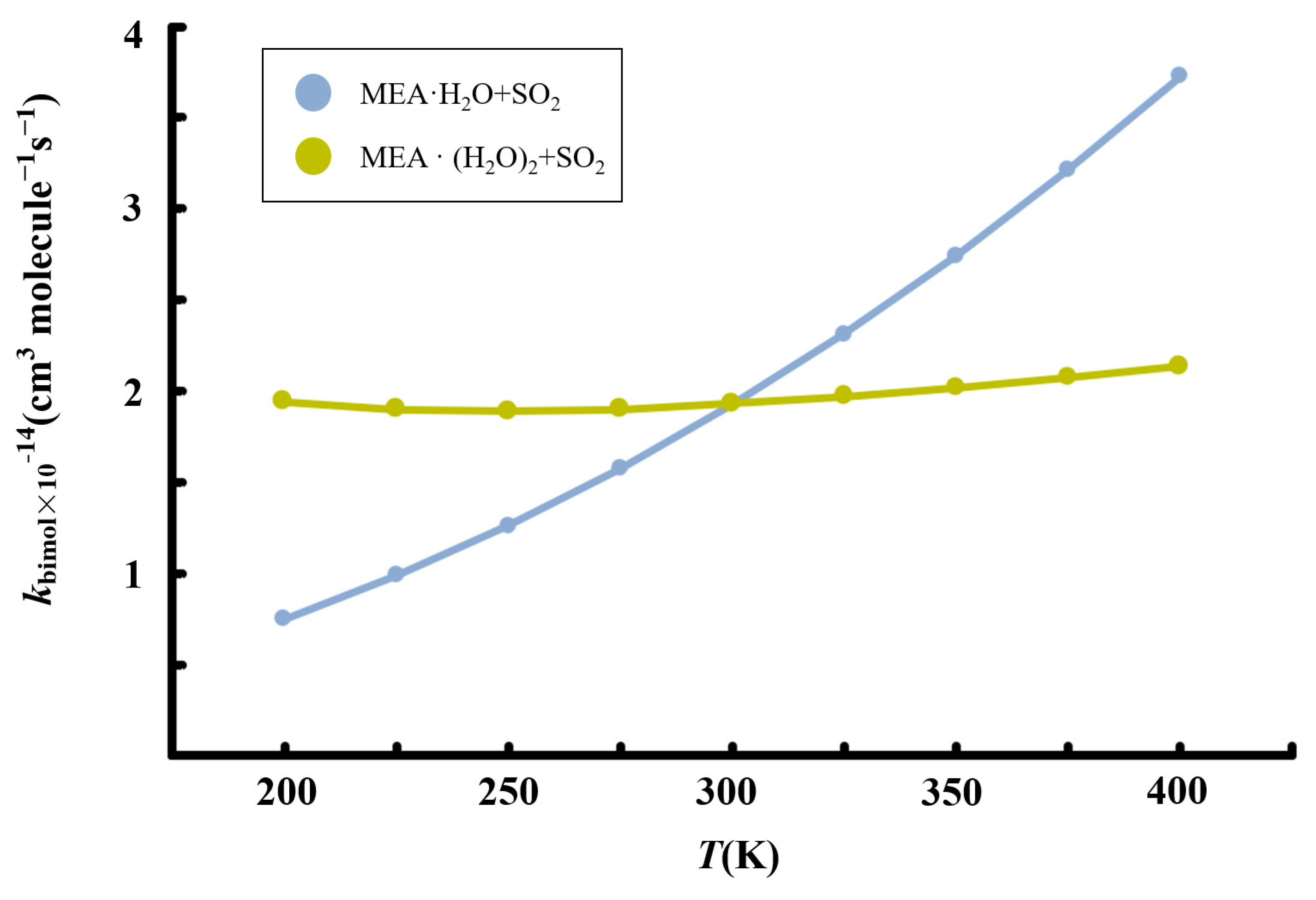
3.4. Kinetics of MEAAssisted Hydrolysis and Implications for Atmospheric Sulfate Formation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, M.; Wu, Z.; Slanina, J.; Lin, P.; Liu, S.; Zeng, L. Acidic gases, ammonia and water-soluble ions in PM2.5 at a coastal site in the Pearl River Delta, China. Atmos. Environ. 2008, 42, 6310–6320. [Google Scholar] [CrossRef]
- Lai, S.C.; Zou, S.C.; Cao, J.J.; Lee, S.C.; Ho, K.F. Characterizing ionic species in PM2.5 and PM10 in four pearl river delta cities, south china. J. Environ. Sci. 2007, 19, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zheng, G.; Wei, C.; Mu, Q.; Zheng, B.; Wang, Z.; Gao, M.; Zhang, Q.; He, K.; Carmichael, G.; et al. Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China. Sci. Adv. 2016, 2, e1601530. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: An analysis of data from the Global Burden of Diseases Study 2015. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Chan, A.W.H.; Abbatt, J.P.D. Multiphase oxidation of sulfur dioxide in aerosol particles: Implications for sulfate formation in polluted environments. Environ. Sci. Technol. 2021, 55, 4227–4242. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Zhang, Y.; Lin, Y.; Li, J.; Cheng, H.; An, N.; Sun, Y.; Qiu, Y.; Cao, F.; Fu, P. Roles of Sulfur Oxidation Pathways in the Variability in Stable Sulfur Isotopic Composition of Sulfate Aerosols at an Urban Site in Beijing, China. Environ. Sci. Technol. Lett. 2020, 7, 883–888. [Google Scholar] [CrossRef]
- Zheng, B.; Zhang, Q.; Zhang, Y.; He, K.B.; Wang, K.; Zheng, G.J.; Duan, F.K.; Ma, Y.L.; Kimoto, T. Heterogeneous chemistry: A mechanism missing in current models to explain secondary inorganic aerosol formation during the January 2013 haze episode in North China. Atmos. Chem. Phys. 2015, 15, 2031–2049. [Google Scholar] [CrossRef]
- Li, Y.; Han, Z.; Song, Y.; Li, J.; Sun, Y.; Wang, T. Impacts of the COVID-19 lockdown on atmospheric oxidizing capacity and secondary aerosol formation over the Beijing-Tianjin-Hebei region in Winter-Spring 2020. Atmos. Environ. 2023, 295, 119540. [Google Scholar] [CrossRef]
- Wang, J.; Xing, J.; Wang, S.; Mathur, R.; Wang, J.; Zhang, Y.; Liu, C.; Pleim, J.; Ding, D.; Chang, X.; et al. The pathway of impacts of aerosol direct effects on secondary inorganic aerosol formation. Atmos. Chem. Phys. 2022, 22, 5147–5156. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, K.; Liu, H.; Lv, W.; Aikawa, M.; Liu, B.; Wang, J. Pollution sources of atmospheric fine particles and secondary aerosol characteristics in Beijing. J. Environ. Sci. 2020, 95, 91–98. [Google Scholar] [CrossRef]
- Shao, J.; Chen, Q.; Wang, Y.; Lu, X.; He, P.; Sun, Y.; Shah, V.; Martin, R.V.; Philip, S.; Song, S.; et al. Heterogeneous sulfate aerosol formation mechanisms during wintertime Chinese haze events: Air quality model assessment using observations of sulfate oxygen isotopes in Beijing. Atmos. Chem. Phys. 2019, 19, 6107–6123. [Google Scholar] [CrossRef]
- Gen, M.; Zhang, R.; Huang, D.D.; Li, Y.; Chan, C.K. Heterogeneous SO2 Oxidation in Sulfate Formation by Photolysis of Particulate Nitrate. Environ. Sci. Technol. Lett. 2019, 6, 86–91. [Google Scholar] [CrossRef]
- Xue, J.; Yu, X.; Yuan, Z.; Griffith, S.M.; Lau, A.K.H.; Seinfeld, J.H.; Yu, J.Z. Efficient control of atmospheric sulfate production based on three formation regimes. Nat. Geosci. 2019, 12, 977–982. [Google Scholar] [CrossRef]
- Itahashi, S.; Hattori, S.; Ito, A.; Sadanaga, Y.; Yoshida, N.; Matsuki, A. Role of Dust and Iron Solubility in Sulfate Formation during the Long-Range Transport in East Asia Evidenced by 17O-Excess Signatures. Environ. Sci. Technol. 2022, 56, 13634–13643. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.; Allman, D.J.; Amos, H.M.; Fairlie, T.D.; Dachs, J.; Hegg, D.A.; Sletten, R.S. Isotopic constraints on the formation pathways of sulfate aerosol in the marine boundary layer of the subtropical northeast Atlantic Ocean. J. Geophys. Res. Atmos. 2012, 117, D06304. [Google Scholar] [CrossRef]
- Chen, Q.; Sherwen, T.; Evans, M.; Alexander, B. DMS oxidation and sulfur aerosol formation in the marine troposphere: A focus on reactive halogen and multiphase chemistry. Atmos. Chem. Phys. 2018, 18, 13617–13637. [Google Scholar] [CrossRef]
- Harris, E.; Sinha, B.; van Pinxteren, D.; Tilgner, A.; Fomba, K.W.; Schneider, J.; Roth, A.; Gnauk, T.; Fahlbusch, B.; Mertes, S.; et al. Enhanced role of transition metal ion catalysis during in-cloud oxidation of SO2. Science 2013, 340, 727–730. [Google Scholar] [CrossRef]
- Zhao, D.; Song, X.; Zhu, T.; Zhang, Z.; Liu, Y.; Shang, J. Multiphase oxidation of SO2 by NO2 on CaCO3 particles. Atmos. Chem. Phys. 2018, 18, 2481–2493. [Google Scholar] [CrossRef]
- Gao, J.; Shi, G.; Zhang, Z.; Wei, Y.; Tian, X.; Feng, Y.; Russell, A.G.; Nenes, A. Targeting Atmospheric Oxidants Can Better Reduce Sulfate Aerosol in China: H2O2 Aqueous Oxidation Pathway Dominates Sulfate Formation in Haze. Environ. Sci. Technol. 2022, 56, 10608–10618. [Google Scholar] [CrossRef]
- Liu, T.; Clegg, S.L.; Abbatt, J.P.D. Fast oxidation of sulfur dioxide by hydrogen peroxide in deliquesced aerosol particles. Proc. Natl. Acad. Sci. USA 2020, 117, 1354–1359. [Google Scholar] [CrossRef]
- Karl, M.; Wright, R.F.; Berglen, T.F.; Denby, B. Worst case scenario study to assess the environmental impact of amine emissions from a CO2 capture plant. Int. J. Greenh. Gas Control 2011, 5, 439–447. [Google Scholar] [CrossRef]
- Tian, X.; Chu, Y.; Chan, C.K. Reactive Uptake of Monoethanolamine by Sulfuric Acid Particles and Hygroscopicity of Monoethanolaminium Salts. Environ. Sci. Technol. Lett. 2022, 9, 16–21. [Google Scholar] [CrossRef]
- Xie, H.; Elm, J.; Halonen, R.; Myllys, N.; Kurtén, T.; Kulmala, M.; Vehkamäki, H. Atmospheric Fate of Monoethanolamine: Enhancing New Particle Formation of Sulfuric Acid as an Important Removal Process. Environ. Sci. Technol. 2017, 51, 8422–8431. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Han, H.; Li, W.; Ma, X.; Xu, L. Experimental study on the absorption enhancement of CO2 by MDEA-MEA based nanofluids. Can. J. Chem. Eng. 2022, 100, 3335–3344. [Google Scholar] [CrossRef]
- Zhang, T.; Yu, Y.; Zhang, Z. Effects of non-aqueous solvents on CO2 absorption in monoethanolamine: Ab initio calculations. Mol. Simul. 2018, 44, 815–825. [Google Scholar] [CrossRef]
- Feron, P.; Conway, W.; Puxty, G.; Wardhaugh, L.; Green, P.; Maher, D.; Fernandes, D.; Cousins, A.; Shiwang, G.; Lianbo, L.; et al. Amine Based Post-Combustion Capture Technology Advancement for Application in Chinese Coal Fired Power Stations. Energy Procedia 2014, 63, 1399–1406. [Google Scholar] [CrossRef]
- Reynolds, A.J.; Verheyen, T.V.; Adeloju, S.B.; Chaffee, A.L.; Meuleman, E. Primary sources and accumulation rates of inorganic anions and dissolved metals in a MEA absorbent during PCC at a brown coal-fired power station. Int. J. Greenh. Gas Control 2015, 41, 239–248. [Google Scholar] [CrossRef]
- Veltman, K.; Singh, B.; Hertwich, E.G. Human and Environmental Impact Assessment of Postcombustion CO2 Capture Focusing on Emissions from Amine-Based Scrubbing Solvents to Air. Environ. Sci. Technol. 2010, 44, 1496–1502. [Google Scholar] [CrossRef]
- Miller, D.D.; Chuang, S.S.C. Experimental and Theoretical Investigation of SO2 Adsorption over the 1,3-Phenylenediamine/SiO2 System. J. Phys. Chem. C 2015, 119, 6713–6727. [Google Scholar] [CrossRef]
- You, Y.; Kanawade, V.P.; de Gouw, J.A.; Guenther, A.B.; Madronich, S.; Sierra-Hernández, M.R.; Lawler, M.; Smith, J.N.; Takahama, S.; Ruggeri, G.; et al. Atmospheric amines and ammonia measured with a chemical ionization mass spectrometer (CIMS). Atmos. Chem. Phys. 2014, 14, 12181–12194. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, D.; Shen, Y.; Gao, Y.; Gao, H.; Yao, X. Mapping gaseous dimethylamine, trimethylamine, ammonia, and their particulate counterparts in marine atmospheres of China’s marginal seas—Part 2: Spatiotemporal heterogeneity, causes, and hypothesis. Atmos. Chem. Phys. 2022, 22, 1515–1528. [Google Scholar] [CrossRef]
- Zhong, Q.E.; Cheng, C.; Wang, Z.; Li, L.; Li, M.; Ge, D.; Wang, L.; Li, Y.; Nie, W.; Chi, X.; et al. Diverse mixing states of amine-containing single particles in Nanjing, China. Atmos. Chem. Phys. 2021, 21, 17953–17967. [Google Scholar] [CrossRef]
- Chai, J.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Elm, J.; Jen, C.N.; Kurtén, T.; Vehkamäki, H. Strong Hydrogen Bonded Molecular Interactions between Atmospheric Diamines and Sulfuric Acid. J. Phys. Chem. A 2016, 120, 3693–3700. [Google Scholar] [CrossRef] [PubMed]
- Elm, J.; Myllys, N.; Hyttinen, N.; Kurtén, T. Computational Study of the Clustering of a Cyclohexene Autoxidation Product C6H8O7 with Itself and Sulfuric Acid. J. Phys. Chem. A 2015, 119, 8414–8421. [Google Scholar] [CrossRef]
- Elm, J.; Myllys, N.; Luy, J.; Kurtén, T.; Vehkamäki, H. The effect of water and bases on the clustering of a cyclohexene autoxidation product C6H8O7 with sulfuric acid. J. Phys. Chem. A 2016, 120, 2240–2249. [Google Scholar] [CrossRef]
- Li, H.; Zhong, J.; Vehkamäki, H.; Kurtén, T.; Wang, W.; Ge, M.; Zhang, S.; Li, Z.; Zhang, X.; Francisco, J.S.; et al. Self-Catalytic Reaction of SO3 and NH3 To Produce Sulfamic Acid and Its Implication to Atmospheric Particle Formation. J. Am. Chem. Soc. 2018, 140, 11020–11028. [Google Scholar] [CrossRef]
- Myllys, N.; Olenius, T.; Kurtén, T.; Vehkamäki, H.; Riipinen, I.; Elm, J. Effect of Bisulfate, Ammonia, and Ammonium on the Clustering of Organic Acids and Sulfuric Acid. J. Phys. Chem. A 2017, 121, 4812–4824. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system.Wires. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Duchovic, R.J.; Pettigrew, J.D.; Welling, B.; Shipchandler, T. Conventional transition state theory/Rice–Ramsperger–Kassel–Marcus theory calculations of thermal termolecular rate coefficients for H(D)+O2+M. J. Chem. Phys. 1996, 105, 10367–10379. [Google Scholar] [CrossRef]
- Elm, J.; Jørgensen, S.; Bilde, M.; Mikkelsen, K.V. Ambient reaction kinetics of atmospheric oxygenated organics with the oh radical: A computational methodology study. Phys. Chem. Chem. Phys. 2013, 15, 9636–9645. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Pan, S.; Li, Y.; Wang, L. Atmospheric oxidation mechanism of toluene. J. Phys. Chem. A 2014, 118, 4533–4547. [Google Scholar] [CrossRef] [PubMed]
- Buszek, R.J.; Barker, J.R.; Francisco, J.S. Water Effect on the OH + HCl Reaction. J. Phys. Chem. A 2012, 116, 4712–4719. [Google Scholar] [CrossRef]
- Wine, P.H.; Thompson, R.J.; Ravishankara, A.R.; Semmes, D.H.; Gump, C.A.; Torabi, A.; Nicovich, J.M. Kinetics of the Reaction OH + SO2 + M→ HOSO2 + M. Temperature and Pressure Dependence in the Falloff Region. J. Chem. Phys. 1984, 88, 2095–2104. [Google Scholar] [CrossRef]
- Liu, J.; Fang, S.; Liu, W.; Wang, M.; Tao, F.; Liu, J. Mechanism of the Gaseous Hydrolysis Reaction of SO2: Effects of NH3 versus H2O. J. Phys. Chem. A 2015, 119, 102–111. [Google Scholar] [CrossRef]
- Buszek, R.J.; Torrent-Sucarrat, M.; Anglada, J.M.; Francisco, J.S. Effects of a Single Water Molecule on the OH + H2O2 Reaction. Phys. Chem. A 2012, 116, 5821–5829. [Google Scholar] [CrossRef]
- Wang, S.; Zeng, X.C.; Li, H.; Francisco, J.S. A possible unaccounted source of atmospheric sulfate formation: Amine-promoted hydrolysis and non-radical oxidation of sulfur dioxide. Chem. Sci. 2020, 11, 2093–2102. [Google Scholar] [CrossRef]
- Hall, H.K., Jr. Correlation of base strengths of amines. J. Am. Chem. Soc. 1957, 79, 5441–5444. [Google Scholar] [CrossRef]
- Franchin, A.; Ehrhart, S.; Leppä, J.; Nieminen, T.; Gagné, S.; Schobesberger, S.; Wimmer, D.; Duplissy, J.; Riccobono, F.; Dunne, E.M.; et al. Experimental investigation of ion–ion recombination under atmospheric conditions. Atmos. Chem. Phys. 2015, 15, 7203–7216. [Google Scholar] [CrossRef]
- Jara-Toro, R.A.; Hernandez, F.J.; Garavagno, M.; Taccone, R.A.; Pino, G.A. Water catalysis of the reaction between hydroxyl radicals and linear saturated alcohols (ethanol and n-propanol) at 294 k. Phys. Chem. Chem. Phys. 2018, 20, 27885–27896. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Tsona, N.T.; Tang, S.; Li, J.; Du, L. Role of (H2O)n (n = 1–2) in the Gas-Phase Reaction of Ethanol with Hydroxyl Radical: Mechanism, Kinetics, and Products. ACS Omega 2019, 4, 5805–5817. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.B.; Rubin, E.S. A Technical, Economic, and Environmental Assessment of Amine-Based CO2 Capture Technology for Power Plant Greenhouse Gas Control. Environ. Sci. Technol. 2002, 36, 4467–4475. [Google Scholar] [CrossRef]
- Sharma, S.D.; Azzi, M. A critical review of existing strategies for emission control in the monoethanolamine-based carbon capture process and some recommendations for improved strategies. Fuel 2014, 121, 178–188. [Google Scholar] [CrossRef]
- Long, B.; Bao, J.L.; Truhlar, D.G. Reaction of SO2 with OH in the atmosphere. Phys. Chem. Chem. Phys. 2017, 19, 8091–8100. [Google Scholar] [CrossRef]
- Nielsen, C.J.; Herrmannb, H.; Wellerb, C. Atmospheric chemistry and environmental impact of the use of amines in carbon capture and storage (CCS). Chem. Soc. Rev. 2012, 41, 6684–6704. [Google Scholar] [CrossRef]
- Borduas, N.; Abbatt, J.P.D.; Murphy, J.G. Gas Phase Oxidation of Monoethanolamine (MEA) with OH Radical and Ozone: Kinetics, Products, and Particles. Environ. Sci. Technol. 2013, 47, 6377–6383. [Google Scholar] [CrossRef]
- Xie, H.; Ma, F.; Wang, Y.; He, N.; Yu, Q.; Chen, J. Quantum Chemical Study on ·Cl-Initiated Atmospheric Degradation of Monoethanolamine. Environ. Sci. Technol. 2015, 49, 13246–13255. [Google Scholar] [CrossRef]
| Reaction | kuni | kbimol |
|---|---|---|
| + • (R2) | 8.33 107 | 1.90 10−14 |
| + •• (R3) | 7.64 109 | 1.93 10−14 |
| + • ••NO (R4) | 1.29 10−6 | |
| + • + ••NO• (R5) | 1.70 10−21 | |
| + • + ••HONO• (R6) | 2.41 10−7 | |
| + • •• (R7) | 1.35 10−2 | |
| •• •• (R8) | 4.74 106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, X.; Lily, M.; Tasheh, S.N.; Ghogomu, J.N.; Du, L.; Tsona Tchinda, N. Enhanced Sulfate Formation from Gas-Phase SO2 Oxidation in Non–•OH–Radical Environments. Atmosphere 2024, 15, 64. https://doi.org/10.3390/atmos15010064
Lv X, Lily M, Tasheh SN, Ghogomu JN, Du L, Tsona Tchinda N. Enhanced Sulfate Formation from Gas-Phase SO2 Oxidation in Non–•OH–Radical Environments. Atmosphere. 2024; 15(1):64. https://doi.org/10.3390/atmos15010064
Chicago/Turabian StyleLv, Xiaofan, Makroni Lily, Stanley Numbonui Tasheh, Julius Numbonui Ghogomu, Lin Du, and Narcisse Tsona Tchinda. 2024. "Enhanced Sulfate Formation from Gas-Phase SO2 Oxidation in Non–•OH–Radical Environments" Atmosphere 15, no. 1: 64. https://doi.org/10.3390/atmos15010064
APA StyleLv, X., Lily, M., Tasheh, S. N., Ghogomu, J. N., Du, L., & Tsona Tchinda, N. (2024). Enhanced Sulfate Formation from Gas-Phase SO2 Oxidation in Non–•OH–Radical Environments. Atmosphere, 15(1), 64. https://doi.org/10.3390/atmos15010064