Evolutionary and Medical Consequences of Archaic Introgression into Modern Human Genomes



{kind=link}
{kind=link}
Abstract
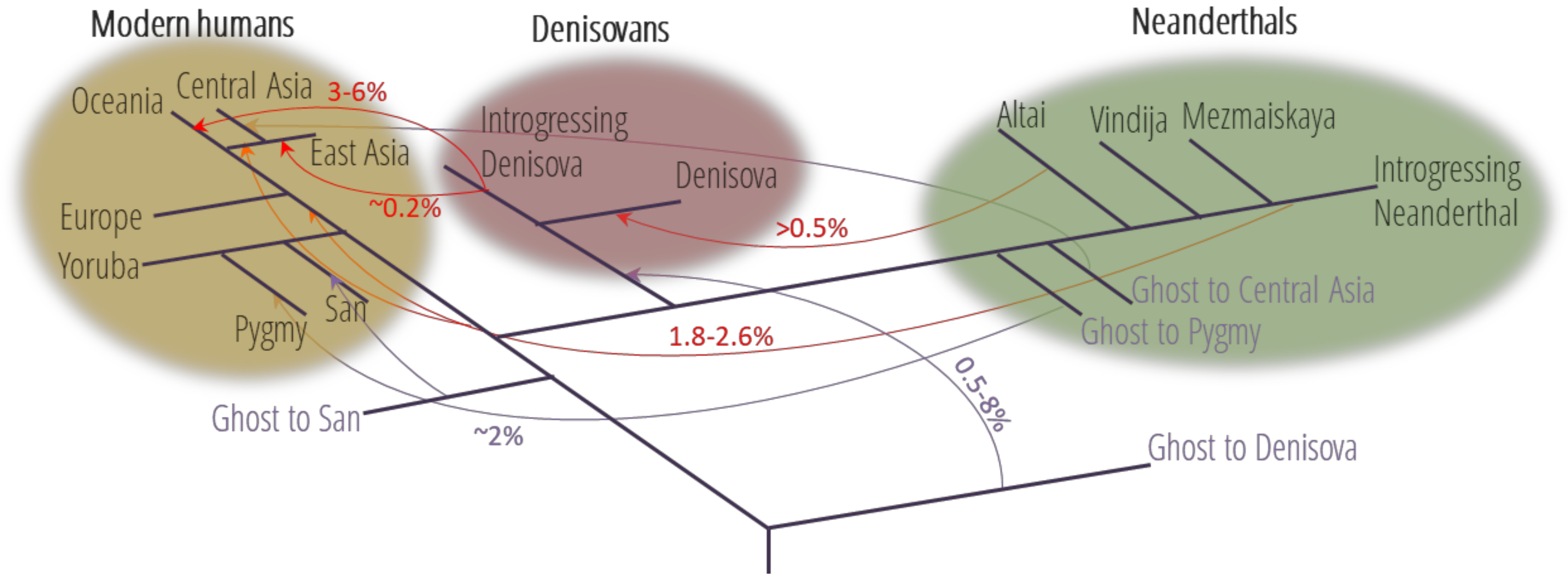
1. Widespread Interbreeding between Hominins
2. Selection against Introgressed Regions at the Level of Genomes and Individual Loci
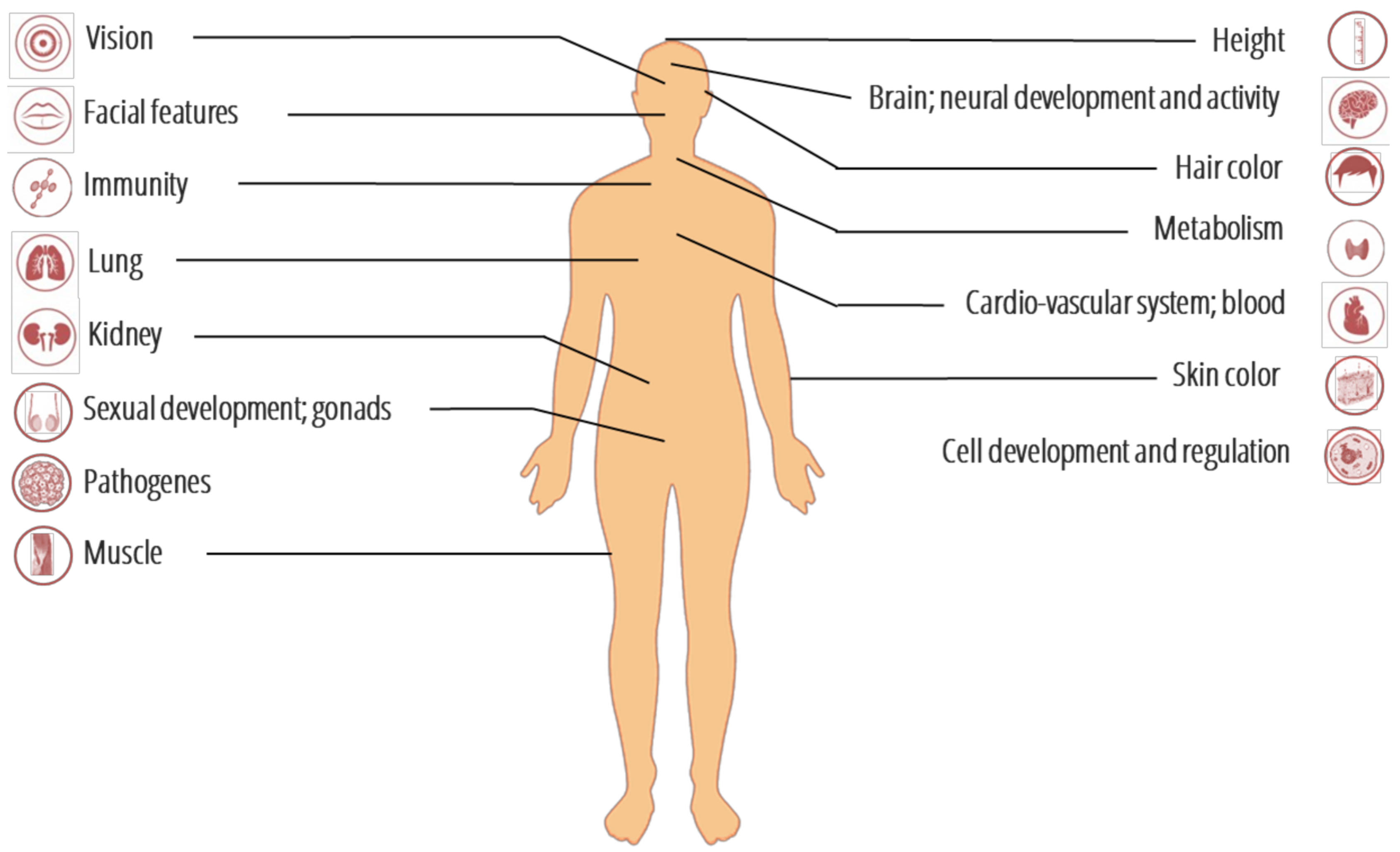
3. Genomic Signatures of Adaptive Introgression from Archaic to Modern Humans
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nielsen, R.; Akey, J.M.; Jakobsson, M.; Pritchard, J.K.; Tishkoff, S.; Willerslev, E. Tracing the peopling of the world through genomics. Nature 2017, 541, 302–310. [Google Scholar] [CrossRef] [PubMed]
- White, T.D.; Asfaw, B.; DeGusta, D.; Gilbert, H.; Richards, G.D.; Suwa, G.; Howell, F.C. Pleistocene Homo sapiens from Middle Awash, Ethiopia. Nature 2003, 423, 742–747. [Google Scholar] [CrossRef] [PubMed]
- McDougall, I.; Brown, F.H.; Fleagle, J.G. Stratigraphic placement and age of modern humans from Kibish, Ethiopia. Nature 2005, 433, 733–736. [Google Scholar] [CrossRef]
- Mercier, N.; Valladas, H.; Bar-Yosef, O.; Vandermeersch, B.; Stringer, C.; Joron, J.-L. Thermoluminescence Date for the Mousterian Burial site of Es-Skhul, Mt. Carmel. J. Archaeol. Sci. 1993, 20, 169–174. [Google Scholar] [CrossRef]
- Stringer, C.B.; Grün, R.; Schwarcz, H.P.; Goldberg, P. ESR dates for the hominid burial site of Es Skhul in Israel. Nature 1989, 338, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, H.P.; Grün, R.; Vandermeersch, B.; Bar-Yosef, O.; Valladas, H.; Tchernov, E. ESR dates for the hominid burial site of Qafzeh in Israel. Hum. Evol. 1988, 17, 733–737. [Google Scholar] [CrossRef]
- Liu, W.; Martinón-Torres, M.; Cai, Y.J.; Xing, S.; Tong, H.W.; Pei, S.W.; Sier, M.J.; Wu, X.H.; Edwards, R.L.; Cheng, H.; et al. The earliest unequivocally modern humans in southern China. Nature 2015, 526, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Hublin, J.J.; Ben-Ncer, A.; Bailey, S.E.; Freidline, S.E.; Neubauer, S.; Skinner, M.M.; Bergmann, I.; Le Cabec, A.; Benazzi, S.; Harvati, K.; et al. New fossils from Jebel Irhoud, Morocco and the pan-African origin of Homo sapiens. Nature 2017, 546, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Richter, D.; Grün, R.; Joannes-Boyau, R.; Steele, T.E.; Amani, F.; Rué, M.; Fernandes, P.; Raynal, J.P.; Geraads, D.; Ben-Ncer, A.; et al. The age of the hominin fossils from Jebel Irhoud, Morocco, and the origins of the Middle Stone Age. Nature 2017, 546, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Hershkovitz, I.; Weber, G.W.; Quam, R.; Duval, M.; Grün, R.; Kinsley, L.; Ayalon, A.; Bar-Matthews, M.; Valladas, H.; Mercier, N.; et al. The earliest modern humans outside Africa. Science 2018, 359, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Lalueza-Fox, C.; Gilbert, M.T.P. Paleogenomics of Archaic Hominins Review. Curr. Biol. 2011, 21, R1002–R1009. [Google Scholar] [CrossRef] [PubMed]
- Prüfer, K.; Racimo, F.; Patterson, N.; Jay, F.; Sankararaman, S.; Sawyer, S.; Heinze, A.; Renaud, G.; Sudmant, P.H.; De Filippo, C.; et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 2014, 505, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Prüfer, K.; de Filippo, C.; Grote, S.; Mafessoni, F.; Korlević, P.; Hajdinjak, M.; Vernot, B.; Skov, L.; Hsieh, P.; Peyrégne, S.; et al. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science 2017, 505, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Mondal, M.; Casals, F.; Xu, T.; Dall’Olio, G.M.; Pybus, M.; Netea, M.G.; Comas, D.; Laayouni, H.; Li, Q.; Majumder, P.P.; et al. Genomic analysis of Andamanese provides insights into ancient human migration into Asia and adaptation. Nat. Genet. 2016, 48, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.; Woerner, A.E.; Wall, J.D.; Lachance, J.; Tishkoff, S.A.; Gutenkunst, R.N.; Hammer, M.F. Model-based analyses of whole-genome data reveal a complex evolutionary history involving archaic introgression in Central African Pygmies. Genome Res. 2016, 26, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Medina-Gomez, C.; Lao, O.; Rivadeneira, F. Evolution of complex traits in human populations. In Evolutionary Biology: Self/Nonself Evolution, Species and Complex Traits Evolution, Methods and Concepts; Pontarotti, P., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 165–186. ISBN 978-3-319-61568-4. [Google Scholar]
- Sankararaman, S.; Patterson, N.; Li, H.; Pääbo, S.; Reich, D. The date of interbreeding between Neandertals and modern humans. PLoS Genet. 2012, 8, e1002947. [Google Scholar] [CrossRef] [PubMed]
- Wall, J.D.; Yang, M.A.; Jay, F.; Kim, S.K.; Durand, E.Y.; Stevison, L.S.; Gignoux, C.; Woerner, A.; Hammer, M.F.; Slatkin, M. Higher levels of Neanderthal ancestry in east Asians than in Europeans. Genetics 2013. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Li, H.; Moorjani, P.; Jay, F.; Slepchenko, S.M.; Bondarev, A.-P.; Pääbo, S. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 2014, 514, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Seguin-Orlando, A.; Korneliussen, T.S.; Sikora, M.; Malaspinas, A.S.; Manica, A.; Moltke, I.; Albrechtsen, A.; Ko, A.; Margaryan, A.; Moiseyev, V.; et al. Genomic structure in Europeans dating back at least 36,200 years. Science 2014, 346, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Sankararaman, S.; Mallick, S.; Patterson, N.; Reich, D. The Combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Curr. Boil. 2016, 26, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.; et al. A draft sequence of the Neandertal genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Li, H.; Lipson, M.; Mathieson, I.; Gymrek, M.; Racimo, F.; Zhao, M.; Chennagiri, N.; Nordenfelt, S.; Tandon, A.; et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 2016, 538, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Browning, S.R.; Zhou, Y.; Tucci, S.; Akey, J.M. Analysis of Human Sequence Data Reveals Two Pulses of Archaic Denisovan Admixture. Cell 2018, 178, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Labuda, D.; Zietkiewicz, E.; Yotova, V. Archaic lineages in the history of modern humans. Genetics 2000, 156, 799–808. [Google Scholar] [PubMed]
- Hammer, M.F.; Woerner, A.E.; Mendez, F.L.; Watkins, J.C.; Wall, J.D. Genetic evidence for archaic admixture in Africa. Proc. Natl. Acad. Sci. USA 2011, 108, 15123–15128. [Google Scholar] [CrossRef] [PubMed]
- Lachance, J.; Vernot, B.; Elbers, C.C.; Ferwerda, B.; Froment, A.; Bodo, J.M.; Lema, G.; Fu, W.; Nyambo, T.B.; Rebbeck, T.R.; et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell 2012, 150, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Pavlidis, P.; Taskent, R.O.; Alachiotis, N.; Flanagan, C.; Degiorgio, M.; Blekhman, R.; Ruhl, S.; Gokcumen, O. Archaic Hominin Introgression in Africa Contributes to Functional Salivary MUC7 Genetic Variation. Mol. Biol. Evol. 2017, 34, 2704–2715. [Google Scholar] [CrossRef] [PubMed]
- Zanolli, C.; Hourset, M.; Esclassan, R.; Mollereau, C. Neanderthal and Denisova tooth protein variants in present-day humans. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Mallet, J.; Besansky, N.; Hahn, M.W. How reticulated are species? BioEssays 2016, 38, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.H.; Jiggins, C.D. Interpreting the genomic landscape of introgression. Curr. Opin. Genet. Dev. 2017, 47, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Juric, I.; Aeschbacher, S.; Coop, G. The strength of selection against Neanderthal introgression. PLoS Genet. 2016, 12, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.; Nielsen, R. The genetic cost of neanderthal introgression. Genetics 2016, 203, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Presgraves, D.C. Sex chromosomes and speciation in Drosophila. Trends Genet. 2008, 24, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Qvarnström, A.; Bailey, R.I. Speciation through evolution of sex-linked genes. Heredity 2009, 102, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Sankararaman, S.; Mallick, S.; Dannemann, M.; Prüfer, K.; Kelso, J.; Pääbo, S.; Patterson, N.; Reich, D. The landscape of Neandertal ancestry in present-day humans. Nature 2014, 20, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Garrigan, D.; Mobasher, Z.; Severson, T.; Wilder, J.A.; Hammer, M.F. Evidence for archaic Asian ancestry on the human X chromosome. Mol. Biol. Evol. 2005, 22, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Garrigan, D.; Kingan, S.B.; Geneva, A.J.; Andolfatto, P.; Clark, A.G.; Thornton, K.R.; Presgraves, D.C. Genome sequencing reveals complex speciation in the Drosophila simulans clade. Genome Res. 2012, 22, 1499–1511. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.; Bengtsson, B.O. The barrier to genetic exchange between hydridizing populations. Heredity 1986, 57, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, M.C.; Pease, J.B.; Steele, A.; Waterhouse, R.M.; Neafsey, D.E.; Sharakhov, I.V.; Jiang, X.; Hall, A.B.; Catteruccia, F.; Kakani, E.; et al. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 2015, 347, 1258524. [Google Scholar] [CrossRef] [PubMed]
- Janoušek, V.; Munclinger, P.; Wang, L.; Teeter, K.C.; Tucker, P.K. Functional organization of the genome may shape the species boundary in the house mouse. Mol. Boil. Evol. 2015, 32, 1208–1220. [Google Scholar] [CrossRef] [PubMed]
- Runemark, A.; Trier, C.N.; Eroukhmanoff, F.; Hermansen, J.S.; Matschiner, M.; Ravinet, M.; Elgvin, T.O.; Sætre, G.P. Variation and constraints in hybrid genome formation. Nat. Ecol. Evol. 2018, 2, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Schumer, M.; Cui, R.; Powell, D.L.; Rosenthal, G.G.; Andolfatto, P. Ancient hybridization and genomic stabilization in a swordtail fish. Mol. Ecol. 2016, 25, 2661–2679. [Google Scholar] [CrossRef] [PubMed]
- Jégou, B.; Sankararaman, S.; Rolland, A.D.; Reich, D.; Chalmel, F. Meiotic genes are enriched in regions of reduced archaic ancestry. Mol. Boil. Evol. 2017, 34, 1974–1980. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ho, W.C.S.; Boon, S.S.; Law, P.T.Y.; Chan, M.C.W.; DeSalle, R.; Burk, R.D.; Chan, P.K.S. Ancient Evolution and Dispersion of Human Papillomavirus Type 58 Variants. J. Virol. 2017, JVI.01285-17. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Posth, C.; Hajdinjak, M.; Petr, M.; Mallick, S.; Fernandes, D.; Furtwängler, A.; Haak, W.; Meyer, M.; Mittnik, A.; et al. The genetic history of Ice Age Europe. Nature 2016, 534, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Vernot, B.; Akey, J.M. Human evolution: Genomic gifts from Archaic hominins. Curr. Biol. 2014, 24, R845–R848. [Google Scholar] [CrossRef] [PubMed]
- Vernot, B.; Tucci, S.; Kelso, J.; Schraiber, J.G.; Wolf, A.B.; Gittelman, R.M.; Dannemann, M.; Grote, S.; McCoy, R.C.; Norton, H.; et al. Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science 2016. [Google Scholar] [CrossRef] [PubMed]
- Konopka, G.; Roberts, T.F. Insights into the neural and genetic basis of vocal communication. Cell 2016, 164, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.H.; Dominy, N.J.; Claw, K.G.; Lee, A.S.; Fiegler, H.; Redon, R.; Werner, J.; Villanea, F.A.; Mountain, J.L.; Misra, R.; et al. Diet and the evolution of human amylase gene copy number variation. Nat. Genet. 2007, 39, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.L.; Saus, E.; Smalley, S.V.; Cataldo, L.R.; Alberti, G.; Parada, J.; Gratacòs, M.; Estivill, X. Copy number polymorphism of the salivary amylase gene: Implications in human nutrition research. Lifestyle Genom. 2012, 5, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.H.; Kistler, L.; Kelaita, M.A.; Sams, A.J. Insights into hominin phenotypic and dietary evolution from ancient DNA sequence data. J. Hum. Evol. 2015, 79, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Chintalapati, M.; Dannemann, M.; Prüfer, K. Using the Neandertal genome to study the evolution of small insertions and deletions in modern humans. BMC Evol. Biol. 2017, 17, 179. [Google Scholar] [CrossRef] [PubMed]
- Dannemann, M.; Kelso, J. The Contribution of Neanderthals to Phenotypic Variation in Modern Humans. Am. J. Hum. Genet. 2017, 101, 578–589. [Google Scholar] [CrossRef] [PubMed]
- McCoy, R.C.; Wakefield, J.; Akey, J.M. Impacts of Neanderthal-introgressed sequences on the landscape of human gene expression. Cell 2017, 168, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Racimo, F.; Sankararaman, S.; Nielsen, R.; Huerta-Sánchez, E. Evidence for archaic adaptive introgression in humans. Nat. Rev. Genet. 2015, 16, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Gittelman, R.M.; Schraiber, J.G.; Vernot, B.; Mikacenic, C.; Wurfel, M.M.; Akey, J.M. Archaic Hominin Admixture Facilitated Adaptation to Out-of-Africa Environments. Curr. Biol. 2016, 26, 3375–3382. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W. Adaptive introgression in animals: Examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol. Ecol. 2013, 22, 4606–4618. [Google Scholar] [CrossRef] [PubMed]
- Kelso, J.; Prüfer, K. Ancient humans and the origin of modern humans. Curr. Opin. Genet. Dev. 2014, 29, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Vattathil, S.; Akey, J.M. Small Amounts of Archaic Admixture Provide Big Insights into Human History. Cell 2015, 163, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Pittman, A.; Myers, A.; Gwinn-Hardy, K.; Fung, H. C.; de Silva, R.; Hutton, M.; Duckworth, J. Evidence suggesting that Homo neanderthalensis contributed the H2 MAPT haplotype to Homo sapiens. Biochem. Soc. Trans. 2005, 33, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Setó-Salvia, N.; Sánchez-Quinto, F.; Carbonell, E.; Lorenzo, C.; Comas, D.; Clarimón, J. Using the Neanderthal and Denisova Genetic Data to Understand the Common MAPT 17q21 Inversion in Modern Humans. Hum. Biol. 2012, 84, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.D.; Mekel-Bobrov, N.; Vallender, E.J.; Hudson, R.R.; Lahn, B.T. Evidence that the adaptive allele of the brain size gene microcephalin introgressed into Homo sapiens from an archaic Homo lineage. PNAS 2006, 103, 18178–18183. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.D.; Kippenhan, J.Sh.; Eisenberg, D.P.; Kohn, Ph.D.; Dickinson, D.; Mattay, V.S.; Chen, Q.; Weinberger, D.R.; Saad, Z.S.; Berman, K.F. Neanderthal-Derived Genetic Variation Shapes Modern Human Cranium and Brain. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Racimo, F.; Gokhman, D.; Fumagalli, M.; Ko, A.; Hansen, T.; Moltke, I.; Albrechtsen, A.; Carmel, L.; Huerta-Sanchez, E.; Nielsen, R. Archaic adaptive introgression in TBX15/WARS2. Mol. Biol. Evol. 2017, 34, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Hu, Y.; Xu, S.; Wang, J.; Jin, L. Neanderthal introgression at chromosome 3p21.31 was under positive natural selection in east asians. Mol. Boil. Evol. 2014, 31, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Frost, P.; Kleisner, K.; Flegr, J. Health status by gender, hair color, and eye color: Red-haired women are the most divergent. PLoS ONE 2017, 12, e0190238. [Google Scholar] [CrossRef] [PubMed]
- Mendez, F.L.; Watkins, J.C.; Hammer, M.F. Global genetic variation at OAS1 provides evidence of archaic admixture in Melanesian populations. Mol. Biol. Evol. 2012, 29, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Mendez, F.L.; Watkins, J.C.; Hammer, M.F. Neandertal origin of genetic variation at the cluster of OAS immunity genes. Mol. Biol. Evol. 2013, 30, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, M.; Quintana-Murci, L. Immunité innée et maladies chez l’homme. Med. Sci. 2016, 32, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Nédélec, Y.; Sanz, J.; Baharian, G.; Szpiech, Z.A.; Pacis, A.; Dumaine, A.; Grenier, J.C.; Freiman, A.; Sams, A.J.; Hebert, S.; et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 2016, 167, 657–669.e21. [Google Scholar] [CrossRef] [PubMed]
- Quach, H.; Rotival, M.; Pothlichet, J.; Loh, Y.H.E.; Dannemann, M.; Zidane, N.; Laval, G.; Patin, E.; Harmant, C.; Lopez, M.; et al. Genetic adaptation and Neandertal admixture shaped the immune system of human populations. Cell 2016, 167, 643–656.e17. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, M.; Laval, G.; Fagny, M.; Itan, Y.; Abel, L.; Casanova, J.L.; Patin, E.; Quintana-Murci, L. Genomic Signatures of Selective Pressures and Introgression from Archaic Hominins at Human Innate Immunity Genes. Am. J. Hum. Genet. 2016, 1, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Pimenoff, V.N.; Houldcroft, C.J.; Rifkin, R.F.; Underdown, S. The role of aDNA in understanding the coevolutionary patterns of human sexually transmitted infections. Genes 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Petousi, N.; Glusman, G.; Yu, Y.; Bohlender, R.; Tashi, T.; Downie, J.M.; Roach, J.C.; Cole, A.M.; Lorenzo, F.R.; et al. Evolutionary history of Tibetans inferred from whole-genome sequencing. PLoS Genet. 2017, 13, e1006675. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Sánchez, E.; Jin, X.; Bianba, Z.; Peter, B.M.; Vinckenbosch, N.; Liang, Y.; Yi, X.; He, M.; Somel, M.; Ni, P.; et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 2014, 512, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Tashi, T.; Scott Reading, N.; Wuren, T.; Zhang, X.; Moore, L.G.; Hu, H.; Tang, F.; Shestakova, A.; Lorenzo, F.; Burjanivova, T.; et al. Gain-of-function EGLN1 prolyl hydroxylase (PHD2 D4E:C127S) in combination with EPAS1 (HIF-2α) polymorphism lowers hemoglobin concentration in Tibetan highlanders. J. Mol. Med. 2017, 95, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Quach, H.; Quintana-Murci, L. Living in an adaptive world: Genomic dissection of the genus Homo and its immune response. J. Exp. Med. 2017, 214, 877–894. [Google Scholar] [CrossRef] [PubMed]
- Andrés, A.M.; Hubisz, M.J.; Indap, A.; Torgerson, D.G.; Degenhardt, J.D.; Boyko, A.R.; Gutenkunst, R.N.; White, T.J.; Green, E.D.; Bustamante, C.D.; et al. Targets of balancing selection in the human genome. Mol. Biol. Evol. 2009, 26, 2755–2764. [Google Scholar] [CrossRef] [PubMed]
- DeGiorgio, M.; Lohmueller, K.E.; Nielsen, R. A Model-Based Approach for Identifying Signatures of Ancient Balancing Selection in Genetic Data. PLoS Genet. 2014, 10, e1004561. [Google Scholar] [CrossRef] [PubMed]
- Leffler, E.M.; Gao, Z.; Pfeifer, S.; Ségurel, L.; Auton, A.; Venn, O.; Bowden, R.; Bontrop, R.; Wall, J.D.; Sella, G.; et al. Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science 2013, 340, 1578–1582. [Google Scholar] [CrossRef] [PubMed]
- Norman, P.J.; Abi-Rached, L.; Gendzekhadze, K.; Korbel, D.; Gleimer, M.; Rowley, D.; Bruno, D.; Carrington, C.V.F.; Chandanayingyong, D.; Chang, Y.H.; et al. Unusual selection on the KIR3DL1/S1 natural killer cell receptor in Africans. Nat. Genet. 2007, 39, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Single, R.M.; Martin, M.P.; Gao, X.; Meyer, D.; Yeager, M.; Kidd, J.R.; Kidd, K.K.; Carrington, M. Global diversity and evidence for coevolution of KIR and HLA. Nat. Genet. 2007, 39, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Abi-Rached, L.; Jobin, M.J.; Kulkarni, S.; McWhinnie, A.; Dalva, K.; Gragert, L.; Babrzadeh, F.; Gharizadeh, B.; Luo, M.; Plummer, F.A.; et al. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 2011, 334, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Yasukochi, Y.; Ohashi, J. Elucidating the origin of HLA-B*73 allelic lineage: Did modern humans benefit by archaic introgression? Immunogenetics 2017, 69, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.B. Gene expression drives local adaptation in humans. Genome Res. 2013, 23, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet. 2014, 94, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Schaub, M.A.; Boyle, A.P.; Kundaje, A.; Batzoglou, S.; Snyder, M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012, 22, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Dannemann, M.; Prüfer, K.; Kelso, J. Functional implications of Neandertal introgression in modern humans. Genome Biol. 2017, 18, 61. [Google Scholar] [CrossRef] [PubMed]
- Sams, A.J.; Dumaine, A.; Nédélec, Y.; Yotova, V.; Alfieri, C.; Tanner, J.E.; Messer, P.W.; Barreiro, L.B. Adaptively introgressed Neandertal haplotype at the OAS locus functionally impacts innate immune responses in humans. Genome Biol. 2016, 17, 246. [Google Scholar] [CrossRef] [PubMed]
- Mozzi, A.; Forni, D.; Cagliani, R.; Pozzoli, U.; Clerici, M.; Sironi, M. Distinct selective forces and Neanderthal introgression shaped genetic diversity at genes involved in neurodevelopmental disorders. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Simonti, C.N.; Vernot, B.; Bastarache, L.; Bottinger, E.; Carrell, D.S.; Chisholm, R.L.; Crosslin, D.R.; Hebbring, S.J.; Jarvik, G.P.; Kullo, I.J.; et al. The phenotypic legacy of admixture between modern humans and Neandertals. Science 2016, 351, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Taskent, R.O.; Alioglu, N.D.; Fer, E.; Melike Donertas, H.; Somel, M.; Gokcumen, O. Variation and functional impact of Neanderthal ancestry in Western Asia. Genome Boil. Evol. 2017, 9, 3516–3524. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, L.B.; Quintana-Murci, L. From evolutionary genetics to human immunology: How selection shapes host defence genes. Nat. Rev. Genet. 2010, 11, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Brinkworth, J.F.; Barreiro, L.B. The contribution of natural selection to present-day susceptibility to chronic inflammatory and autoimmune disease. Curr. Opin. Immunol. 2014, 31, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Corbett, S.; Courtiol, A.; Lummaa, V.; Moorad, J.; Stearns, S. The transition to modernity and chronic disease: Mismatch and natural selection. Nat. Rev. Genet. 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Clerici, M. The hygiene hypothesis: An evolutionary perspective. Microbes Infect. 2010, 12, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Stearns, S.C. Evolutionary medicine: Its scope, interest and potential. Proc. R. Soc. B Biol. Sci. 2012, 279, 4305–4321. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.A.; Zhernakova, A.; Turner, G.; Heap, G.A.R.; Franke, L.; Bruinenberg, M.; Romanos, J.; Dinesen, L.C.; Ryan, A.W.; Panesar, D.; et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat. Genet. 2008, 40, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Zhernakova, A.; Elbers, C.C.; Ferwerda, B.; Romanos, J.; Trynka, G.; Dubois, P.C.; de Kovel, C.G.F.; Franke, L.; Oosting, M.; Barisani, D.; et al. Evolutionary and Functional Analysis of Celiac Risk Loci Reveals SH2B3 as a Protective Factor against Bacterial Infection. Am. J. Hum. Genet. 2010, 86, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Dannemann, M.; Andrés, A.M.; Kelso, J. Introgression of Neandertal-and Denisovan-like haplotypes contributes to adaptive variation in human Toll-like receptors. Am. J. Hum. Genet. 2016, 98, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, B.P.; Knight, J.C. Genetics of gene expression in immunity to infection. Curr. Opin. Immunol. 2014, 30, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Aguet, F.; Brown, A.A.; Castel, S.E.; Davis, J.R.; He, Y.; Jo, B.; Mohammadi, P.; Park, Y.S.; Parsana, P.; Segrè, A.V.; et al. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Ardlie, K.G.; DeLuca, D.S.; Segrè, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T.; Lek, M.; et al. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Field, Y.; Boyle, E.A.; Telis, N.; Gao, Z.; Gaulton, K.J.; Golan, D.; Yengo, L.; Rocheleau, G.; Froguel, P.; McCarthy, M.I.; et al. Detection of human adaptation during the past 2000 years. Science 2016, 354, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Tishkoff, S.A.; Reed, F.A.; Ranciaro, A.; Voight, B.F.; Babbitt, C.C.; Silverman, J.S.; Powell, K.; Mortensen, H.M.; Hirbo, J.B.; Osman, M.; et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nat. Genet. 2007. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolgova, O.; Lao, O. Evolutionary and Medical Consequences of Archaic Introgression into Modern Human Genomes. Genes 2018, 9, 358. https://doi.org/10.3390/genes9070358
Dolgova O, Lao O. Evolutionary and Medical Consequences of Archaic Introgression into Modern Human Genomes. Genes. 2018; 9(7):358. https://doi.org/10.3390/genes9070358
Chicago/Turabian StyleDolgova, Olga, and Oscar Lao. 2018. "Evolutionary and Medical Consequences of Archaic Introgression into Modern Human Genomes" Genes 9, no. 7: 358. https://doi.org/10.3390/genes9070358
APA StyleDolgova, O., & Lao, O. (2018). Evolutionary and Medical Consequences of Archaic Introgression into Modern Human Genomes. Genes, 9(7), 358. https://doi.org/10.3390/genes9070358