
Trisomy 22 Mosaicism from Prenatal to Postnatal Findings: A Case Series and Systematic Review of the Literature
 , , , , , , ,
, , , , , , ,  , , ,
, , ,
Abstract
1. Introduction
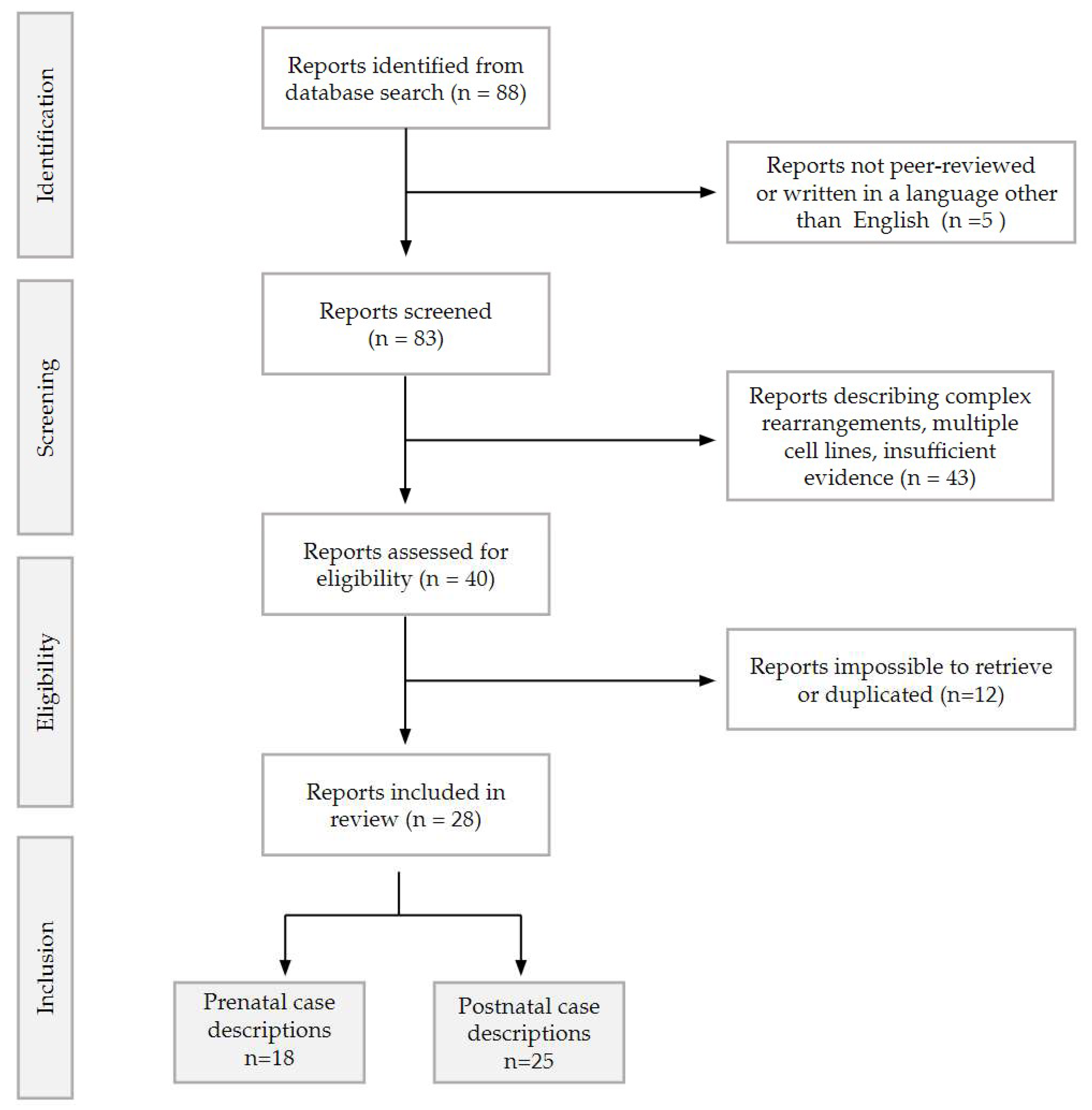
2. Materials and Methods
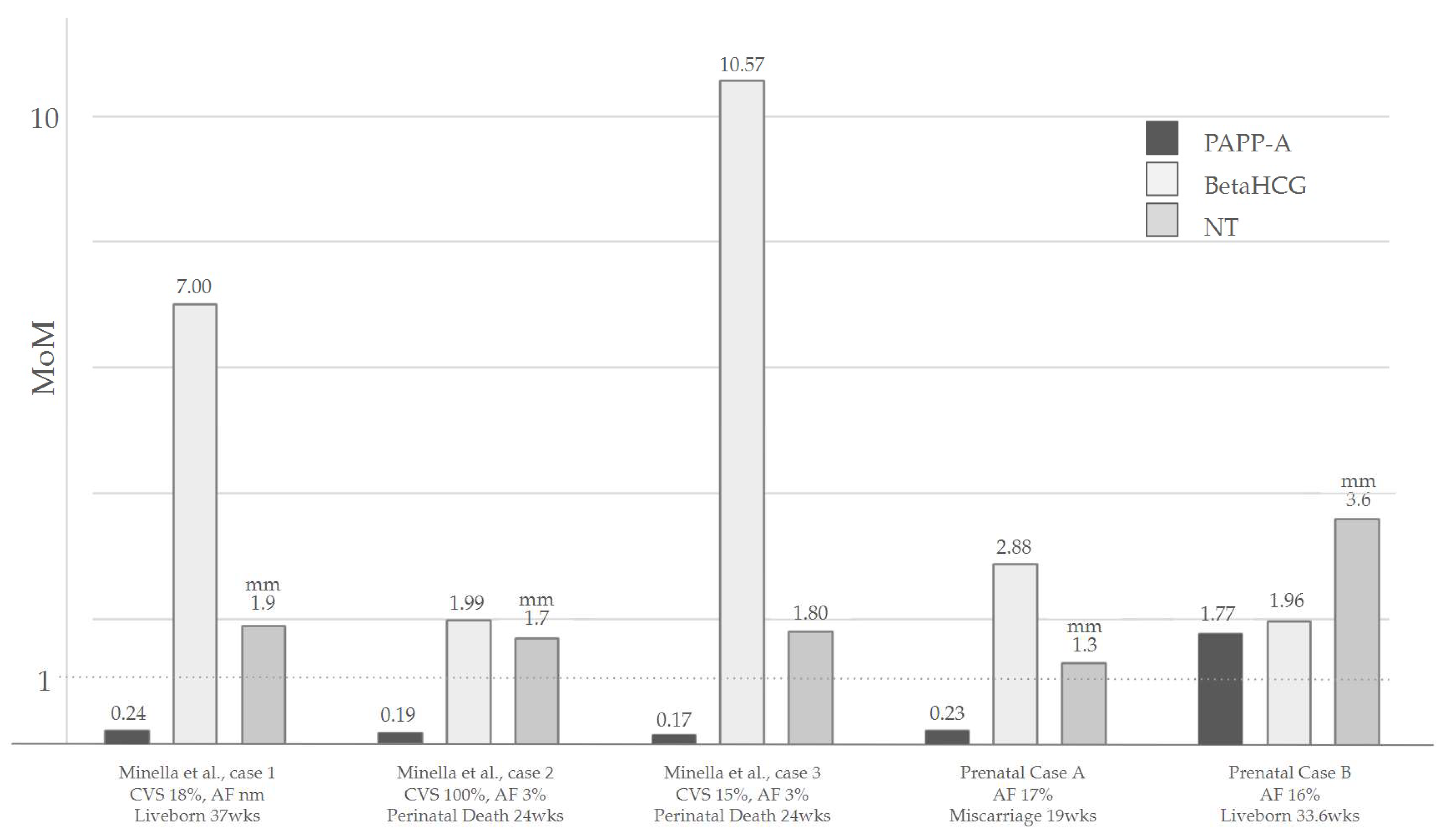
2.1. Prenatal Case A
2.2. Prenatal and Postnatal Case B
2.3. Postnatal Case C
3. Results
3.1. Prenatal Phenotype: General Overview
3.1.1. Intrauterine Growth Restriction (IUGR)
3.1.2. Increased Nuchal Translucency (NT) in the First Trimester
3.1.3. Other Reported Prenatal Abnormalities
3.2. Systematic Review of the Postnatal Phenotype and Shared Features between Prenatal and Postnatal Phenotype
3.3. Craniofacial Dysmorphic Features
3.4. Cardiovascular Malformations
3.5. Urogenital Findings and Puberty
3.6. Ophthalmic Manifestations
3.7. Ears and Auditory Apparatus
3.8. Growth Parameters
3.9. Neuropsychiatric Features
3.10. Skeletal Anomalies
3.11. Skin and Nail Anomalies
3.12. Gastrointestinal Tract Anomalies
3.13. Other Reported Abnormalities
4. Molecular Diagnosis in Prenatal and Postnatal Settings
5. Suggested Recommendations for the Management of Suspected mT22 Cases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Leclercq, S.; Baron, X.; Jacquemont, M.; Cuillier, F.; Cartault, F. Mosaic trisomy 22: Five new cases with variable outcomes. Implications for genetic counselling and clinical management. Prenat. Diagn. 2010, 30, 168–172. [Google Scholar] [CrossRef]
- Martínez-Glez, V.; Tenorio, J.; Nevado, J.; Gordo, G.; Rodríguez-Laguna, L.; Feito, M.; Lapunzina, P. A six-attribute classification of genetic mosaicism. Genet. Med. 2020, 22, 1743–1757. [Google Scholar] [CrossRef]
- Schinzel, A.; Schmid, W.; Auf der Maur, P.; Moser, H.; Degenhardt, K.H.; Geisler, M.; Grubisic, A. Incomplete trisomy 22. I. Familial 11/22 translocation with 3:1 meiotic disjunction. Delineation of a common clinical picture and report of nine new cases from six families. Hum. Genet. 1981, 56, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, W.; Be, C.; Schnapp, C.; Roy, M.; Wimalasundera, R. Second-trimester sonographic findings in trisomy 22: Report of 3 cases and review of the literature. J. Ultrasound Med. 2003, 22, 1271–1275. [Google Scholar] [CrossRef]
- Woods, C.G.; Bankier, A.; Curry, J.; Sheffield, L.J.; Slaney, S.F.; Smith, K.; Voullaire, L.; Wellesley, D. Asymmetry and skin pigmentary anomalies in chromosome mosaicism. J. Med. Genet. 1994, 31, 694–701. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mazza, V.; Latella, S.; Fenu, V.; Ferrari, P.; Bonilauri, C.; Santucci, S.; Percesepe, A. Prenatal diagnosis and postnatal follow-up of a child with mosaic trisomy 22 with several levels of mosaicism in different tissues. J. Obstet. Gynaecol. Res. 2010, 36, 1116–1120. [Google Scholar] [CrossRef]
- Abdelgadir, D.; Nowaczyk, M.J.; Li, C. Trisomy 22 Mosaicism and Normal Developmental Outcome: Report of Two Patients and Review of the Literature. Am. J. Med. Genet. Part A 2013, 161, 1126–1131. [Google Scholar] [CrossRef]
- Florez, L.; Lacassie, Y. Mosaic trisomy 22: Report of a patient with normal intelligence. Am. J. Med. Genet. Part A 2005, 132A, 223–225. [Google Scholar] [CrossRef] [PubMed]
- De Pater, J.M.; Schuring-Blom, G.H.; Bogaard, R.V.D.; Van Der Sijs-Bos, C.J.M.; Christiaens, G.C.M.L.; Stoutenbeek, P.; Leschot, N.J. Maternal Uniparental Disomy for Chromosome 22 in a Child with Generalized Mosaicism for Trisomy 22. Prenat. Diagn. 1997, 17, 81–86. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ Syst. Rev. 2021, 10, 105906. [Google Scholar] [CrossRef]
- National Library of Medicine (U.S.). National Library of Medicine Programs and Services. 1999. Available online: https://medlineplus.gov/ (accessed on 24 January 2024).
- Wertelecki, W.; Breg, W.R.; Graham, J.M.; Iinuma, K.; Puck, S.M.; Sergovich, F.R.; Opitz, J.M.; Reynolds, J.F. Trisomy 22 mosaicism syndrome and Ullrich-Turner stigmata. Am. J. Med. Genet. 1986, 23, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Naoufal, R.; Legendre, M.; Couet, D.; Gilbert-Dussardier, B.; Kitzis, A.; Bilan, F.; Harbuz, R. Association of structural and numerical anomalies of chromosome 22 in a patient with syndromic intellectual disability. Eur. J. Med. Genet. 2016, 59, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, A.; Laskoski, L.V.; Luiz, A.F.; Silveira, M.A.D.; D’arce, L.P.G. Occurrence of mosaic trisomy 22 and pericentric inversion of chromosome 9 in a patient with a good prognosis. BMC Med. Genom. 2023, 16, 286. [Google Scholar] [CrossRef] [PubMed]
- Dulitzky, F.; Shabtal, F.; Zlotogora, J.; Halbrecht, I.; Elian, E. Unilateral radial aplasia and trisomy 22 mosaicism. J. Med. Genet. 1981, 18, 473–476. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Osztovics, M.; Ivády, G. A case of 22-trisomy mosaic. Acta Paediatr. Acad. Sci. Hung. 1977, 18, 197–200. [Google Scholar]
- Stioui, S.; de Silvestris, M.; Molinari, A.; Stripparo, L.; Ghisoni, L.; Simoni, G. Trisomic 22 placenta in a case of severe intrauterine growth retardation. Prenat. Diagn. 1989, 9, 673–676. [Google Scholar] [CrossRef]
- Phillips, O.P.; Tharapel, A.T.; Lerner, J.L.; Park, V.M.; Wachtel, S.S.; Shulman, L.P. Risk of fetal mosaicism when placental mosaicism is diagnosed by chorionic villus sampling. Am. J. Obstet. Gynecol. 1996, 174, 850–855. [Google Scholar] [CrossRef]
- Berghella, V.; Wapner, R.J.; Yang-Feng, T.; Mahoney, M.J. Prenatal confirmation of true fetal trisomy 22 mosaicism by fetal skin biopsy following normal fetal blood sampling. Prenat. Diagn. 1998, 18, 384–389. [Google Scholar] [CrossRef]
- Bryan, J.; Peters, M.; Pritchard, G.; Healey, S.; Payton, D. A second case of intrauterine growth retardation and primary hypospadias associated with a trisomy 22 placenta but with biparental inheritance of chromosome 22 in the fetus. Prenat. Diagn. 2002, 22, 137–140. [Google Scholar] [CrossRef]
- Wang, J.; Dang, L.; Mondal, T.K.; Khan, A. Prenatally diagnosed mosaic trisomy 22 in a fetus with left ventricular non-compaction cardiomyopathy. Am. J. Med. Genet. Part A 2007, 143A, 2744–2746. [Google Scholar] [CrossRef]
- Chen, C.-P.; Huang, M.-C.; Chern, S.-R.; Wu, P.-S.; Chen, S.-W.; Chuang, T.-Y.; Town, D.-D.; Wang, W. Mosaic trisomy 22 at amniocentesis: Prenatal diagnosis and literature review. Taiwan. J. Obstet. Gynecol. 2019, 58, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Minella, C.; Jeandidier, E.; Koch, A.; Antal, M.C.; Favre, R.; Sananes, N.; Weingertner, A.-S. Trisomy 22: First and Second Trimester Cytogenetic Analysis and Phenotypic Presentation in a Series of Seven Cases. Fetal Diagn. Ther. 2023, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cicero, S.; Bindra, R.; Rembouskos, G.; Spencer, K.; Nicolaides, K.H. Integrated ultrasound and biochemical screening for trisomy 21 using fetal nuchal translucency, absent fetal nasal bone, free beta-hCG and PAPP-A at 11 to 14 weeks. Prenat. Diagn. 2003, 23, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.; Kagan, K.O.; Molina, F.S.; Gazzoni, A.; Nicolaides, K.H. A mixture model of nuchal translucency thickness in screening for chromosomal defects. Ultrasound Obstet. Gynecol. 2008, 31, 376–383. [Google Scholar] [CrossRef]
- Hadlock, F.P.; Harrist, R.B.; Martinez-Poyer, J. In utero analysis of fetal growth: A sonographic weight standard. Radiology 1991, 181, 129–133. [Google Scholar] [CrossRef]
- Bertino, E.; Spada, E.; Occhi, L.; Coscia, A.; Giuliani, F.; Gagliardi, L.; Gilli, G.; Bona, G.; Fabris, C.; De Curtis, M.; et al. Neonatal Anthropometric Charts: The Italian Neonatal Study Compared with Other European Studies. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 353–361. [Google Scholar] [CrossRef]
- Growth Charts. 4 April 2023. Available online: https://www.cdc.gov/growthcharts/index.htm (accessed on 31 January 2024).
- Menasha, J.; Levy, B.; Hirschhorn, K.; Kardon, N.B. Incidence and spectrum of chromosome abnormalities in spontaneous abortions: New insights from a 12-year study. Genet. Med. 2005, 7, 251–263. [Google Scholar] [CrossRef]
- Kehinde, F.I.; Anderson, C.E.; McGowan, J.E.; Jethva, R.N.; Wahab, M.A.; Glick, A.R.; Sterner, M.R.; Pascasio, J.M.; Punnett, H.H.; Liu, J. Co-occurrence of non-mosaic trisomy 22 and inherited balanced t(4;6)(q33;q23.3) in a liveborn female: Case report and review of the literature. Am. J. Med Genet. Part A 2014, 164, 3187–3193. [Google Scholar] [CrossRef]
- Eggenhuizen, G.M.; Go, A.; Koster, M.P.H.; Baart, E.B.; Galjaard, R.J. Confined placental mosaicism and the association with pregnancy outcome and fetal growth: A review of the literature. Hum. Reprod. Update 2021, 27, 885–903. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.; Syngelaki, A.; Bradbury, I.; Akolekar, R.; Nicolaides, K. First-Trimester Screening for Trisomies 21, 18 and 13 by Ultrasound and Biochemical Testing. Fetal Diagn. Ther. 2014, 35, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.L.; Blakemore, K.J. A historical and practical review of first trimester aneuploidy screening. Semin. Fetal Neonatal Med. 2014, 19, 183–187. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Azar, G.; Byrne, D.; Mansur, C.; Marks, K. Fetal nuchal translucency: Ultrasound screening for chromosomal defects in first trimester of pregnancy. BMJ 1992, 304, 867–869. [Google Scholar] [CrossRef] [PubMed]
- Kelley, J.; McGillivray, G.; Meagher, S.; Hui, L. Increased nuchal translucency after low-risk noninvasive prenatal testing: What should we tell prospective parents? Prenat. Diagn. 2021, 41, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Paladini, D.; Donarini, G.; Bottelli, L.; Rossi, A.; Coltri, A.; Fulcheri, E. Isolated, persisting, large choroid plexus cysts should warrant neurosonographic follow-up. Ultrasound Obstet. Gynecol. 2021, 57, 1006–1008. [Google Scholar] [CrossRef] [PubMed]
- Schinzel, A. Incomplete trisomy 22. III. Mosaic-trisomy 22 and the problem of full trisomy 22. Hum. Genet. 1981, 56, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Pridjian, A.K.; Frohlich, E.D.; VanMeter, C.H.; McFadden, P.M.; Ochsner, J.L. Pharmacologic support with high-energy phosphate preservation in the postischemic neonatal heart. Ann. Thorac. Surg. 1995, 59, 1435–1440. [Google Scholar] [CrossRef]
- Kalayinia, S.; Shahani, T.; Biglari, A.; Maleki, M.; Rokni-Zadeh, H.; Razavi, Z.; Mahdieh, N. Mosaic trisomy 22 in a 4-year-old boy with congenital heart disease and general hypotrophy: A case report. J. Clin. Lab. Anal. 2019, 33, e22663. [Google Scholar] [CrossRef]
- Dayasiri, K.C.; De Silva, D.; Weerasekara, K. Confirmation of mosaic trisomy 22 in an infant with failure to thrive. Sri Lanka J. Child Heal. 2018, 47, 174. [Google Scholar] [CrossRef]
- Lin, A.E.; Santoro, S.; High, F.A.; Goldenberg, P.; Gutmark-Little, I. Congenital heart defects associated with aneuploidy syndromes: New insights into familiar associations. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 53–63. [Google Scholar] [CrossRef]
- Abuhamad, A.Z.; Chaoui, R. A Practical Guide to Fetal Echocardiography: Normal and Abnormal Hearts; Lippincott Williams & Wilkins: Philadelphia, PE, USA, 2012. [Google Scholar]
- Patey, O.; Carvalho, J.S.; Thilaganathan, B. Perinatal changes in cardiac geometry and function in growth-restricted fetuses at term. Ultrasound Obstet. Gynecol. 2019, 53, 655–662. [Google Scholar] [CrossRef]
- Ruiter, E.M.; Toorman, J.; Hochstenbach, R.; de Vries, B.B. Mosaic trisomy 22 in a boy with a terminal transverse limb reduction defect. Clin. Dysmorphol. 2004, 13, 99–102. [Google Scholar] [CrossRef]
- Lessick, M.L.; Szego, K.; Wong, P.W.K. Trisomy 22 Mosaicism with Normal Blood Chromosomes. Case report with literature review. Clin. Pediatr. 1988, 27, 451–454. [Google Scholar] [CrossRef]
- Fruhman, G.; El-Hattab, A.W.; Belmont, J.W.; Patel, A.; Cheung, S.W.; Sutton, V.R. Suspected trisomy 22: Modification, clarification, or confirmation of the diagnosis by aCGH. Am. J. Med. Genet. Part A 2011, 155, 434–438. [Google Scholar] [CrossRef]
- Pridjian, G.; Gill, W.L.; Shapira, E. Goldenhar sequence and mosaic trisomy 22. Am. J. Med. Genet. 1995, 59, 411–413. [Google Scholar] [CrossRef]
- Thomas, S.; Parker, M.; Tan, J.; Duckett, D.; Woodruff, G. Ocular manifestations of mosaic trisomy 22: A case report and review of the literature. Ophthalmic Genet. 2004, 25, 53–56. [Google Scholar] [CrossRef]
- Lund, H.T.; Tranebæerg, L. Trisomy 22 Mosaicism Limited to Skin Fibroblasts in a Mentally Retarded, Dysmorphic Girl. Acta Paediatr. Scand. 1990, 79, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Crowe, C.A.; Schwartz, S.; Black, C.J.; Jaswaney, V. Mosaic trisomy 22: A case presentation and literature review of trisomy 22 phenotypes. Am. J. Med. Genet. 1997, 71, 406–413. [Google Scholar] [CrossRef]
- Allotey, J.; Lacaille, F.; Lees, M.M.; Strautnieks, S.; Thompson, R.J.; Davenport, M. Congenital bile duct anomalies (biliary atresia) and chromosome 22 aneuploidy. J. Pediatr. Surg. 2008, 43, 1736–1740. [Google Scholar] [CrossRef]
- Mollica, F.; Sorge, G.; Pavone, L. Trisomy 22 mosaicism. J. Med. Genet. 1977, 14, 224–225. [Google Scholar] [CrossRef] [PubMed]
- Basaran, N.; Berkil, H.; Ay, N.; Durak, B.; Ataman, C.; Ozdemir, M.; Ozon, Y.H.; Kaya, I. A rare case: Mosaic trisomy 22. Ann. Genet. 2001, 44, 183–186. [Google Scholar] [CrossRef]
- Hall, T.; Samuel, M.; Brain, J. Mosaic trisomy 22 associated with total colonic aganglionosis and malrotation. J. Pediatr. Surg. 2009, 44, e9–e11. [Google Scholar] [CrossRef] [PubMed]
- Battle, D.E. Diagnostic and Statistical Manual of Mental Disorders (DSM). Codas 2013, 25, 191–192. [Google Scholar] [PubMed]
- Khan, I.; Leventhal, B.L. Developmental Delay. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Jansen, S.; Vissers, L.E.L.M.; de Vries, B.B.A. The Genetics of Intellectual Disability. Brain Sci. 2023, 13, 231. [Google Scholar] [CrossRef] [PubMed]
- Merks, J.H.; Ceelie, N.; Caron, H.N.; Hennekam, R.C. Neuroblastoma, maternal valproic acid use, in-vitro fertilization and family history of mosaic chromosome 22: Coincidence or causal relationship? Clin. Dysmorphol. 2004, 13, 197–198. [Google Scholar] [CrossRef]
- Beedgen, B.; Querfeld, U.; Weiss-Wichert, P.; Nützenadel, W. “Partial trisomy 22 and 11” due to a paternal 11;22 translocation associated with hirschsprung disease. Eur. J. Pediatr. 1986, 145, 229–232. [Google Scholar] [CrossRef]
- dos Santos, J.L.; Quelhas, P.; Cerski, C. Update on Etiology and Pathogenesis of Biliary Atresia. Curr. Pediatr. Rev. 2022, 19, 48–67. [Google Scholar] [CrossRef]
- Gangbo, E.; Lacombe, D.; Alberti, E.M.; Taine, L.; Saura, R.; Carles, D. Trisomy 22 with thyroid isthmus agenesis and absent gall bladder. Genet. Couns. 2004, 15, 311–315. [Google Scholar]
- Biesecker, L.G.; Spinner, N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013, 14, 307–320. [Google Scholar] [CrossRef]
- Ledbetter, D.; Engel, E. Uniparental disomy in humans: Development of an imprinting map and its implications for prenatal diagnosis. Hum. Mol. Genet. 1995, 4, 1757–1764. [Google Scholar] [CrossRef]
- Sdano, M.R.; Vanzo, R.J.; Martin, M.M.; Baldwin, E.E.; South, S.T.; Rope, A.F.; Allen, W.P.; Kearney, H. Clinical Utility of Chromosomal Microarray Analysis of DNA from Buccal Cells: Detection of Mosaicism in Three Patients. J. Genet. Couns. 2014, 23, 922–927. [Google Scholar] [CrossRef]
- Ballif, B.C.; Rorem, E.A.; Sundin, K.; Lincicum, M.; Gaskin, S.; Coppinger, J.; Kashork, C.D.; Shaffer, L.G.; Bejjani, B.A. Detection of low-level mosaicism by array CGH in routine diagnostic specimens. Am. J. Med. Genet. Part A 2006, 140, 2757–2767. [Google Scholar] [CrossRef] [PubMed]
- Conlin, L.K.; Thiel, B.D.; Bonnemann, C.G.; Medne, L.; Ernst, L.M.; Zackai, E.H.; Deardorff, M.A.; Krantz, I.D.; Hakonarson, H.; Spinner, N.B. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet. 2010, 19, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Papavassiliou, P.; York, T.P.; Gursoy, N.; Hill, G.; Nicely, L.V.; Sundaram, U.; Jackson-Cook, C. The phenotype of persons having mosaicism for trisomy 21/Down syndrome reflects the percentage of trisomic cells present in different tissues. Am. J. Med. Genet. A 2009, 149A, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Akolekar, R.; Beta, J.; Picciarelli, G.; Ogilvie, C.; D’Antonio, F. Procedure-related risk of miscarriage following amniocentesis and chorionic villus sampling: A systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2015, 45, 16–26. [Google Scholar] [CrossRef]
- Schinzel, A.A.; Basaran, S.; Bernasconi, F.; Karaman, B.; Yüksel-Apak, M.; Robinson, W.P. Maternal uniparental disomy 22 has no impact on the phenotype. Am. J. Hum. Genet. 1994, 54, 21–24. [Google Scholar]
- Li, M.; Hao, N.; Jiang, Y.; Xue, H.; Dai, Y.; Wang, M.; Bai, J.; Lv, Y.; Qi, Q.; Zhou, X. Contribution of uniparental disomy to fetal growth restriction: A whole-exome sequencing series in a prenatal setting. Sci. Rep. 2024, 14, 238. [Google Scholar] [CrossRef]
- Yong, P.J.; Langlois, S.; von Dadelszen, P.; Robinson, W. The association between preeclampsia and placental trisomy 16 mosaicism. Prenat. Diagn. 2006, 26, 956–961. [Google Scholar] [CrossRef]
- Ream, M.A.; Lehwald, L. Neurologic Consequences of Preterm Birth. Curr. Neurol. Neurosci. Rep. 2018, 18, 48. [Google Scholar] [CrossRef]
- Smith, A.E.; Shah, M.; Badireddy, M. Failure to Thrive. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Heinrich, T.; Nanda, I.; Rehn, M.; Zollner, U.; Frieauff, E.; Wirbelauer, J.; Grimm, T.; Schmid, M. Live-Born Trisomy 22: Patient Report and Review. Mol. Syndr. 2012, 3, 262–269. [Google Scholar] [CrossRef]
- Romaniello, R.; Arrigoni, F.; De Salvo, P.; Bonaglia, M.C.; Panzeri, E.; Bassi, M.T.; Parazzini, C.; Righini, A.; Borgatti, R. Long-term follow-up in a cohort of children with isolated corpus callosum agenesis at fetal MRI. Ann. Clin. Transl. Neurol. 2021, 8, 2280–2288. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Sex | Phenotype | Mosaic | Fate | |
|---|---|---|---|---|
| Schinzel et al., 1981 [3] | Female | Hypertelorism, retrognathia, low set ears with poorly formed auricles, anal atresia, tapering fingers, hypoplastic thumbs, partial cutaneous syndactyly (2nd and 3rd toes), VSD, ASD, RV hypoplasia, tricuspid atresia, ARSA, IUGR | Amniotic fluid 20%, Cord blood 4%, Blood 90%. No UPD | IUD 36 weeks |
| Stioui et al., 1989 [17] | Female | Normal morphology, late IUGR | Amniotic fluid 28–32%, CVS 100%. Blood, cord blood and skin: non-mosaic | Liveborn 33 weeks |
| Phillips et al., 1996 [18] | Male | Normal morphology, early IUGR | Placental mesenchymal cells 42%, Cytotrophoblast 40%, Kidney cells 4%; placental membranes, amniotic fluid, lungs, fibroblast from umbilical cord: non-mosaic. No UPD | TOP |
| De Pater et al., 1997 [9] | Female | Late IUGR, VSD, clinodactyly, micrognathia, low set ears | Amniotic fluid 20%, CVS 100%, skin 22%, blood: non-mosaic. Maternal UPD | Liveborn 39.6 weeks |
| Berghella et al., 1998 [19] | Male | Hypertelorism, bilateral pulmonary polylobation, large low set ears, choroid plexus cyst | Amniotic fluid 21%, skin 47%, lungs 3%, blood: non-mosaic | TOP |
| Bryan et al., 2002 [20] | Male | Early IUGR, oligohydramnios, hypospadias | CVS 100%, amniotic fluid and blood: non-mosaic. Maternal UPD | Liveborn 32 weeks |
| Wang et al., 2007 [21] | Female | IUGR, ASD, ductal aneurysm, hydrothorax, pericardial effusion, tricuspid regurgitation, LVNC, crumpled helix, low set ears, hypertelorism, tapered fingers | Amniotic fluid 35%, skin 76%, blood: non-mosaic | Liveborn 35.6 weeks |
| Leclercq et al., 2010 [1] | Female | Normal morphology | Amniotic fluid 16%, skin 6%, CVS and blood: non-mosaic | Liveborn |
| Female | Dysmorphic ears, late IUGR | Amniotic fluid 14% | IUD 34 weeks | |
| Female | Nuchal fold thickening, hydrothorax | Amniotic fluid 26%, blood 4% | TOP | |
| Male | Early IUGR, multiple abnormalities | Blood 14%, amniotic fluid: non-mosaic | IUD 33 weeks | |
| Mazza et al., 2010 [6] | Male | Late IUGR, low set ears, anteriorly displaced anus, pLSVC, ASD | Amniotic fluid 33%, CVS 95%, blood 33%, skin 4%, cord blood: non-mosaic. No UPD | Liveborn 37 weeks |
| Abdelgadir et al., 2013 [7] | Female | Tricuspid regurgitation, ASD, pericardial effusion, VNC. Hypertelorism, low set ears, dysmorphic left ear, thick nuchal fold, bilateral 5th finger clinodactyly, IUGR | Amniotic fluid 35%, skin 76%, blood: non-mosaic | Liveborn 35 weeks |
| Female | TOF, thickened nuchal fold, short femur, single umbilical artery, bilateral 5th finger clino-brachydactyly, broad thumbs, anteriorly displaced anus, IUGR | Fibroblast from umbilical cord 50%, skin 3%, cardiomyocytes 17%, blood: non-mosaic | Liveborn 39 weeks | |
| Chen et al., 2019 [22] | Female | Median facial cleft, oligohydramnios, IUGR, hypertelorism, low set ears | Amniotic fluid 50–60% skin 82% | TOP |
| Minella et al., 2023 [23] | Female | Pelvic kidney, IUGR | CVS 18%, amniotic fluid: non-mosaic | Liveborn 37 weeks |
| Female | Early IUGR + Severe preeclampsia. Rhizomelic long bone shortening, hypertelorism, retrognathia, RV predominance, ARSA, low set ears, bilateral 5th finger clinodactyly | Amniotic fluid 3%, CVS 100%, kidney cells 14% | Perinatal death 24 weeks | |
| Female | Early IUGR, oligohydramnios, TOF, retrognathia, dysmorphic ears | Amniotic fluid (postnatal) 3%, blood 5%, CVS 15% | Perinatal death 24 weeks | |
| Our cases | Female | Early IUGR, oligohydramnios, frontal bossing, flat nose, upslanting eyelids, wide mouth, micro-retrognathia, low set ears with dysmorphic helices, thickened nuchal fold, DORV, hypoplastic thymus, hands flexed, talipes, fixed limbs, single umbilical artery | Amniotic fluid 17% | Miscarriage 19 weeks |
| Male | pLSVC, Late IUGR, nuchal fold thickening | Amniotic fluid 16% | Liveborn 33.6 weeks |
| District | Cases on Total (n = 20) | Feature |
|---|---|---|
| No abnormalities | 3 | - |
| Systemic (17/20) | 17 | IUGR |
| 5 | Nuchal fold thickening | |
| 4 | Oligohydramnios | |
| Facial (10/20) | 10 | Low set ears |
| 7 | Dysmorphic ears | |
| 5 | Hypertelorism | |
| 6 | Micro/Retrognathia | |
| 1 | Facial cleft | |
| 1 | Frontal bossing, flat nose, up-slanting eyelids, wide mouth | |
| Cardiac (8/20) | 4 | ASD |
| 2 | TOF | |
| 2 | VSD | |
| 2 | ARSA | |
| 2 | Tricuspid regurgitation | |
| 2 | Pericardial effusion | |
| 2 | VNC | |
| 2 | pLSVC | |
| 1 | RV hypoplasia | |
| 1 | RV predominance | |
| 1 | Tricuspid atresia | |
| 1 | Ductal aneurysm | |
| 1 | DORV | |
| Hands (6/20) | 4 | Clinodactyly 5th fingers |
| 2 | Tapered fingers | |
| 1 | Hypoplastic thumbs | |
| 1 | Broad thumbs | |
| 1 | Syndactyly | |
| Urogenital (4/20) | 2 | Anteriorized anus |
| 1 | Anal atresia | |
| 1 | Pelvic kidney | |
| Miscellaneous | 2 | Short femur |
| 2 | Hydrothorax | |
| 3 | Single umbilical artery | |
| 1 | Pulmonary polylobation | |
| 1 | Choroid plexus cyst | |
| 1 | Hypoplastic thymus, talipes, hands flexed, fixed limbs | |
| 1 | Abnormal unspecified |
| Clinical Features | Specific Feature | Cases on Total (n = 25) | Pt.1 | Pt.2 |
|---|---|---|---|---|
| Sex | F/M | 15/10 | M | M |
| Status at report | Alive/deceased | 22/3 | Alive | Alive |
| Growth pattern (15/25) | Delayed | 15 | − | − |
| Failure to thrive | 8 | − | − | |
| Facial Dysmorphisms (14/25) | Frontal bossing | 8 | − | − |
| Microcephaly | 6 | − | − | |
| Hypertelorism | 10 | − | − | |
| Epicanthal folds | 9 | + | − | |
| Ptosis (mono-bilateral) | 6 | + | − | |
| Flattened nasal bridge | 9 | − | − | |
| Anteverted nostrils | 5 | − | − | |
| Long philtrum | 5 | − | − | |
| Thin lips | 3 | + | − | |
| Micro/retrognathia | 6 | − | − | |
| Low set ears | 11 | − | − | |
| Dysmorphic ears | 10 | + | − | |
| Pre-auricolar pits/tags | 15 | − | − | |
| Low posterior airline | 7 | − | − | |
| Cleft palate | 3 | − | − | |
| Webbed neck | 7 | + | − | |
| Congenital heart disease (14/25) | Isolated defects | 5 | − | + |
| Complex cardiac defect | 5 | − | − | |
| Combined cardiac and vascular defects | 4 | + | − | |
| ASD | 7 | + | − | |
| PVS | 4 | − | − | |
| VSD | 6 | − | − | |
| TOF | 2 | − | − | |
| PDA | 3 | − | − | |
| Neurodevelopment (10/25) | Developmental delay | 11 | mild | no |
| ID assessment | 11 | + | no | |
| Severe ID | 1 | |||
| Moderate ID | 3 | |||
| Mild ID | 5 | |||
| Normal–borderline ID | 3 | 81 WISC−IV | ||
| Hypotonia | 8 | + | no | |
| Audiological aspects (6/25) | Hearing loss | 6 | − | − |
| Skin and nails aspects (16/25) | Hypoplastic nails | 12 | − | − |
| Skin Pigmentary changes | 8 | − | − | |
| Skeletal (19/25) | Body asymmetry | 11 | + | − |
| Craniofacial Symmetry | 7 | − | − | |
| Midface hypoplasia | 6 | + | − | |
| Radial anomalies | 2 | − | − | |
| 5th finger Clinodactyly | 11 | + | − | |
| Syndactyly (partial/cutaneous) | 6 | − | − | |
| Radial anomalies | 2 | − | − | |
| 5th finger Clinodactyly | 11 | + | − | |
| Syndactyly (partial/cutaneous) | 6 | − | − | |
| 5th finger Clinodactyly | 11 | + | − | |
| Vertebral anomalies | 4 | + | − | |
| Relevant anatomic anomalies (14/25) | G.i malrotation | 2 | − | − |
| Anal malformation | 4 | + | − | |
| Genital anomalies | 11 | + | + | |
| Dental anomalies | 6 | + | − | |
| Palatal anomalies | 4 | − | − |
| DNA Substrate for Karyotyping | N. of Procedures Reported | % of Cells Detected in the Analyzed Substrate |
|---|---|---|
| Amniotic epithelium | 6 | 1–46% |
| Chorionic villi sampling | 2 | 100% (CPM) |
| Lymphocytes from Cordocentesis | 1 | mT22 not detected |
| Fibroblasts from umbilical cord tissue | 1 | 50% |
| Peripheral blood lymphocytes | 9 | 1–68% |
| Skin fibroblasts | 16 | 1.6–100 (SCM)% |
| Assessment | Aim |
|---|---|
| Genetic counseling | Discuss the genetic results, evaluate family history, and define recurrence risk. |
| Cardiovascular | Define potential cardiovascular defects by cardiological examination, including echocardiography and ECG. |
| ENT | Otorhinolaryngology consultation, along with auditory screening, to promptly detect hearing loss and palatal defects. |
| Ophthalmic | Assess presence of ophthalmic anomalies (ptosis, coloboma) and periodic evaluation of visual acuity. |
| Orthopedic | Assessment of potential scoliosis, vertebral anomalies, need for orthoses or surgical indication at time of diagnosis, and follow-up. |
| Dermatological | Estimate presence and extension of pigmentary changes. |
| General pediatric | Periodically, to assess general wellbeing, growth, and to evaluate the need of endocrinological investigations. |
| Surgical | Evaluation of surgical indication in case of detection of anatomic malformations (anal anomalies, external genital malformations). |
| Nc | Assess neurodevelopmental and behavioral aspects and intellectual functioning. Longitudinal observation is highly recommended. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trevisan, V.; Meroni, A.; Leoni, C.; Sirchia, F.; Politano, D.; Fiandrino, G.; Giorgio, V.; Rigante, D.; Limongelli, D.; Perri, L.; et al. Trisomy 22 Mosaicism from Prenatal to Postnatal Findings: A Case Series and Systematic Review of the Literature. Genes 2024, 15, 346. https://doi.org/10.3390/genes15030346
Trevisan V, Meroni A, Leoni C, Sirchia F, Politano D, Fiandrino G, Giorgio V, Rigante D, Limongelli D, Perri L, et al. Trisomy 22 Mosaicism from Prenatal to Postnatal Findings: A Case Series and Systematic Review of the Literature. Genes. 2024; 15(3):346. https://doi.org/10.3390/genes15030346
Chicago/Turabian StyleTrevisan, Valentina, Anna Meroni, Chiara Leoni, Fabio Sirchia, Davide Politano, Giacomo Fiandrino, Valentina Giorgio, Donato Rigante, Domenico Limongelli, Lucrezia Perri, and et al. 2024. "Trisomy 22 Mosaicism from Prenatal to Postnatal Findings: A Case Series and Systematic Review of the Literature" Genes 15, no. 3: 346. https://doi.org/10.3390/genes15030346
APA StyleTrevisan, V., Meroni, A., Leoni, C., Sirchia, F., Politano, D., Fiandrino, G., Giorgio, V., Rigante, D., Limongelli, D., Perri, L., Sforza, E., Leonardi, F., Viscogliosi, G., Contaldo, I., Orteschi, D., Proietti, L., Zampino, G., & Onesimo, R. (2024). Trisomy 22 Mosaicism from Prenatal to Postnatal Findings: A Case Series and Systematic Review of the Literature. Genes, 15(3), 346. https://doi.org/10.3390/genes15030346