


EMAST Type of Microsatellite Instability—A Distinct Entity or Blurred Overlap between Stable and MSI Tumors


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Genomic Instability as a Hallmark of Cancer
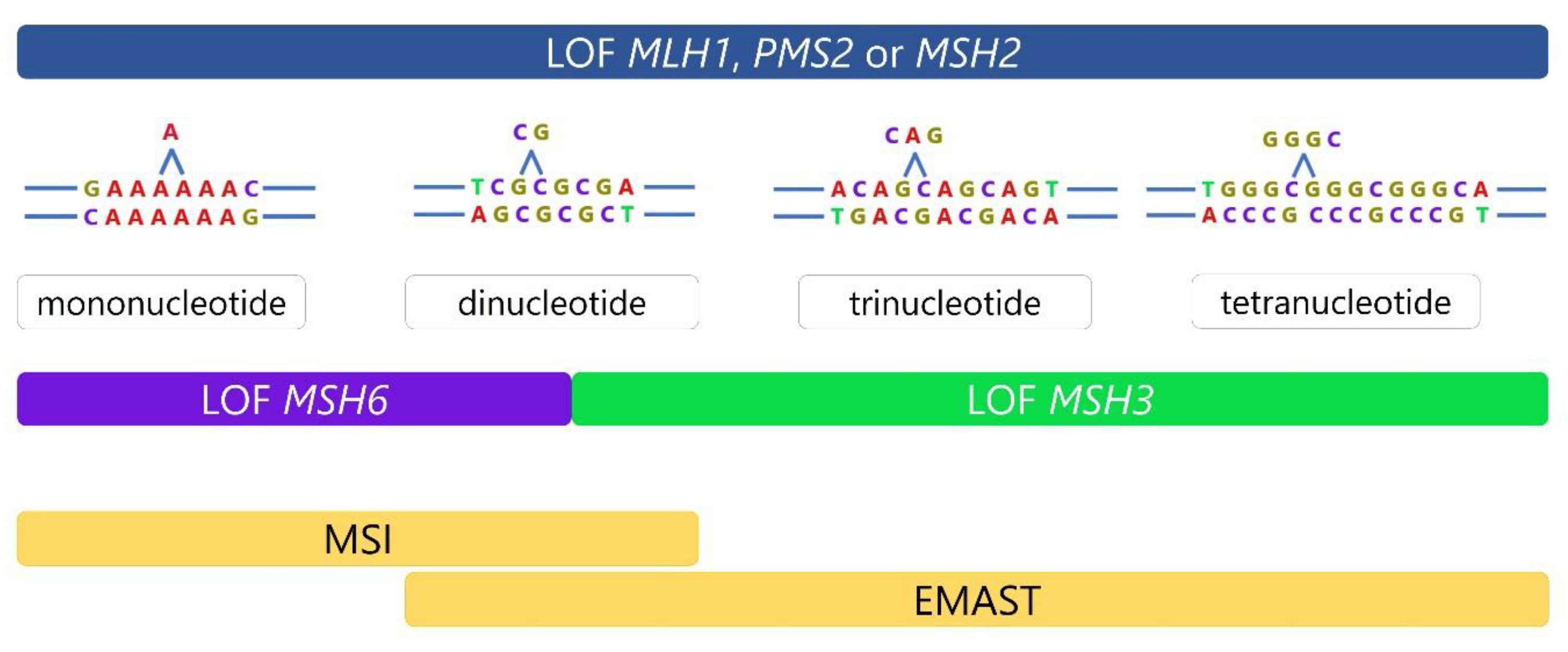
1.2. Two Types of Microsatellite Instability, MSI and EMAST
1.3. DNA Mismatch Repair and Mechanisms Leading to Microsatellite Instability
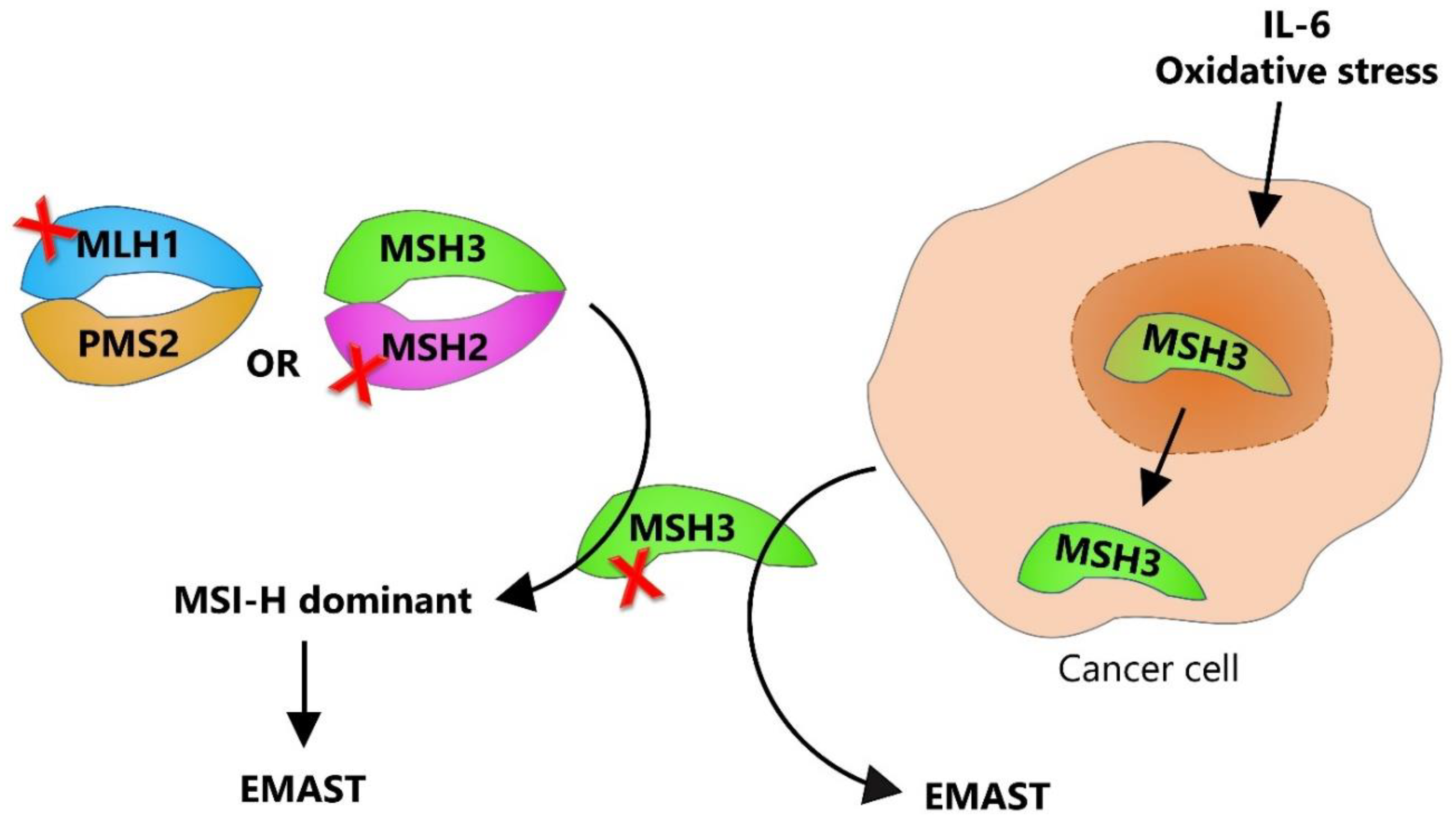
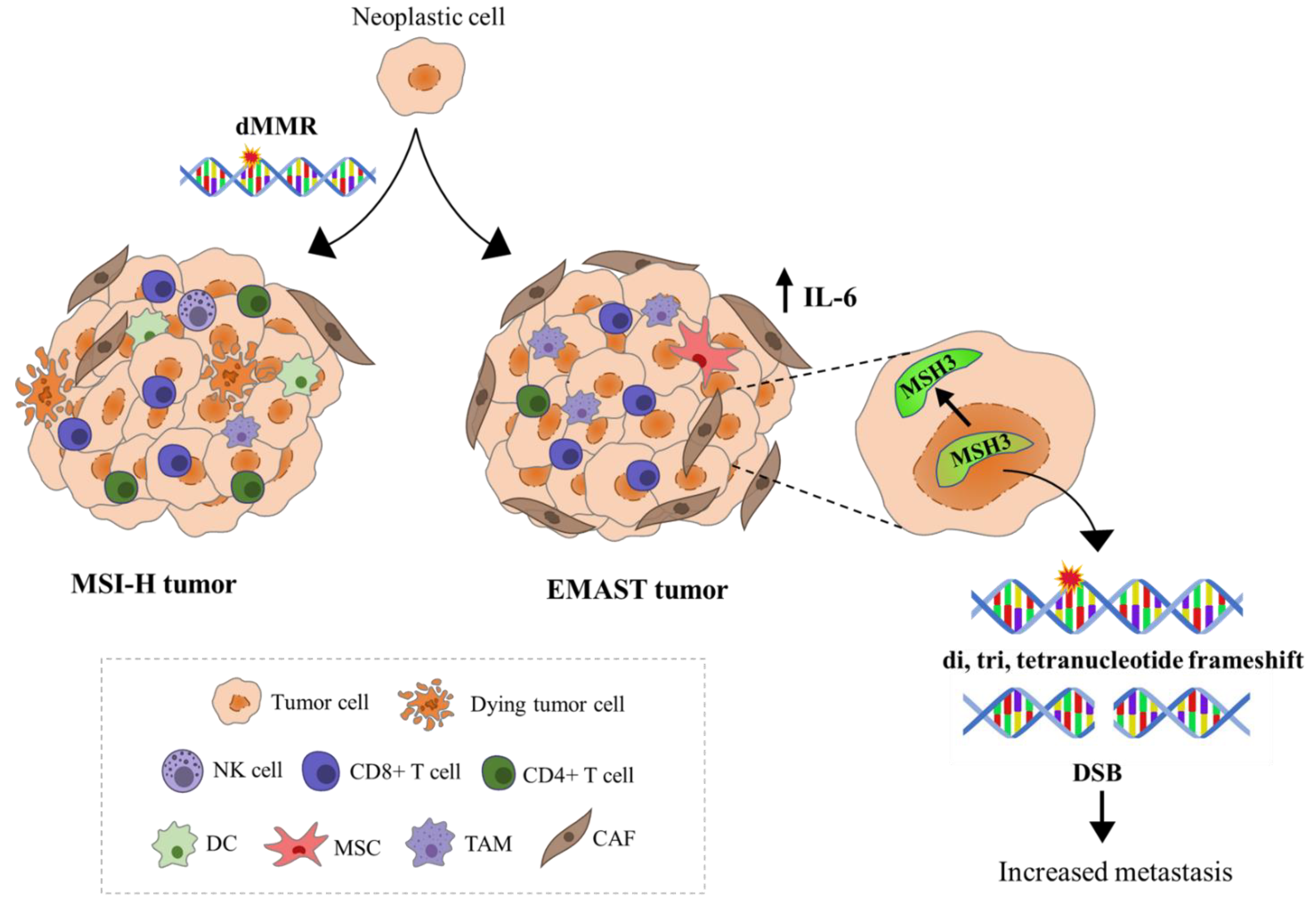
1.4. Dual Etiology of EMAST—Two Sides of the Same Coin
1.5. Immune Response-Related Features of MSI and EMAST Tumors, Their Clinical Implications, and Relevance for Antitumor Therapy
1.6. Challenges of MSI and EMAST Detection
1.7. Why Is the Isolated MSH3 Dysfunction so Difficult to Pinpoint?
1.8. Is EMAST a Distinctive Entity Separate from MSI and MSS and Do We Need to Discriminate between Different EMAST Etiologies with Respect to Patients’ Prognosis and Therapy?
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer—The stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M. Microsatellite Instability Pathway and EMAST in Colorectal Cancer. Curr. Color. Cancer Rep. 2017, 13, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, L.A.; Peltomäki, P.; Leach, F.S.; Sistonen, P.; Pylkkänen, L.; Mecklin, J.-P.; Järvinen, H.; Powell, S.M.; Jen, J.; Hamilton, S.R.; et al. Clues to the Pathogenesis of Familial Colorectal Cancer. Science 1993, 260, 812–816. [Google Scholar] [CrossRef]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef]
- Herman, J.G.; Umar, A.; Polyak, K.; Graff, J.R.; Ahuja, N.; Issa, J.-P.J.; Markowitz, S.; Willson, J.K.V.; Hamilton, S.R.; Kinzler, K.W.; et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 6870–6875. [Google Scholar] [CrossRef]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodri-guez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of mi-crosatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar]
- Thibodeau, S.N.; Bren, G.; Schaid, D. Microsatellite Instability in Cancer of the Proximal Colon. Science 1993, 260, 816–819. [Google Scholar] [CrossRef]
- Fishel, R.; Lescoe, M.K.; Rao, M.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef]
- Leach, F.S.; Nicolaides, N.C.; Papadopoulos, N.; Liu, B.; Jen, J.; Parsons, R.; Peltomäki, P.; Sistonen, P.; Aaltonen, L.A.; Nyström-Lahti, M.; et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993, 75, 1215–1225. [Google Scholar] [CrossRef]
- Schöniger, S.; Rüschoff, J. Mismatch Repair Deficiency and Microsatellite Instability. Encyclopedia 2022, 2, 1559–1576. [Google Scholar] [CrossRef]
- Boland, C.R.; Koi, M.; Chang, D.K.; Carethers, J.M. The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch Syndrome: From bench to bedside. Fam. Cancer 2008, 7, 41–52. [Google Scholar] [CrossRef]
- Carethers, J.M. Differentiating Lynch-Like From Lynch Syndrome. Gastroenterology 2014, 146, 602–604. [Google Scholar] [CrossRef]
- Imai, K.; Yamamoto, H. Carcinogenesis and microsatellite instability: The interrelationship between genetics and epigenetics. Carcinogenesis 2008, 29, 673–680. [Google Scholar] [CrossRef]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.-Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 2017, 1–15. [Google Scholar] [CrossRef]
- Carethers, J.M.; Koi, M.; Tseng-Rogenski, S.S. EMAST is a Form of Microsatellite Instability That is Initiated by Inflammation and Modulates Colorectal Cancer Progression. Genes 2015, 6, 185–205. [Google Scholar] [CrossRef]
- Watson, M.M.C.; Berg, M.; Søreide, K. Prevalence and implications of elevated microsatellite alterations at selected tetranucleotides in cancer. Br. J. Cancer 2014, 111, 823–827. [Google Scholar] [CrossRef]
- Ranjbar, R.; Esfahani, A.T.; Nazemalhosseini-Mojarad, E.; Olfatifar, M.; Aghdaei, H.A.; Mohammadpour, S. EMAST frequency in colorectal cancer: A meta-analysis and literature review. Biomark. Med. 2020, 14, 1021–1030. [Google Scholar] [CrossRef]
- Slattery, M.L.; Curtin, K.; Anderson, K.; Ma, K.-N.; Ballard, L.; Edwards, S.; Schaffer, D.; Potter, J.; Leppert, M.; Samowitz, W.S. Associations Between Cigarette Smoking, Lifestyle Factors, and Microsatellite Instability in Colon Tumors. Gynecol. Oncol. 2000, 92, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Samowitz, W.S.; Slattery, M.L.; Kerber, R.A. Microsatellite instability in human colonic cancer is not a useful clinical indicator of familial colorectal cancer. Gastroenterology 1995, 109, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Haugen, A.C.; Goel, A.; Yamada, K.; Marra, G.; Nguyen, T.-P.; Nagasaka, T.; Kanazawa, S.; Koike, J.; Kikuchi, Y.; Zhong, X.; et al. Genetic Instability Caused by Loss of MutS Homologue 3 in Human Colorectal Cancer. Cancer Res 2008, 68, 8465–8472. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Kanazawa, S.; Koike, J.; Sugiyama, H.; Xu, C.; Funahashi, K.; Boland, C.R.; Koi, M.; Hemmi, H. Mi-crosatellite instability at tetranucleotide repeats in sporadic colorectal cancer in Japan. Oncol. Rep. 2010, 23, 551–561. [Google Scholar]
- Koi, M.; Garcia, M.; Choi, C.; Kim, H.-R.; Koike, J.; Hemmi, H.; Nagasaka, T.; Okugawa, Y.; Toiyama, Y.; Kitajima, T.; et al. Microsatellite Alterations with Allelic Loss at 9p24.2 Signify Less-Aggressive Colorectal Cancer Metastasis. Gastroenterology 2016, 150, 944–955. [Google Scholar] [CrossRef]
- Garcia, M.; Choi, C.; Kim, H.; Daoud, Y.; Toiyama, Y.; Takahashi, M.; Goel, A.; Boland, C.R.; Koi, M. Association Between Recurrent Metastasis from Stage II and III Primary Colorectal Tumors and Moderate Microsatellite Instability. Gastroenterology 2012, 143, 48–50.e1. [Google Scholar] [CrossRef]
- Li, S.K.; Martin, A. Mismatch Repair and Colon Cancer: Mechanisms and Therapies Explored. Trends Mol. Med. 2016, 22, 274–289. [Google Scholar] [CrossRef]
- Kantelinen, J.; Kansikas, M.; Korhonen, M.K.; Ollila, S.; Heinimann, K.; Kariola, R.; Nyström, M. MutSβ exceeds MutSα in dinucleotide loop repair. Br. J. Cancer 2010, 102, 1068–1073. [Google Scholar] [CrossRef]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cell Int. 2020, 20, 16. [Google Scholar] [CrossRef]
- Grady, W.M.; Carethers, J.M. Genomic and Epigenetic Instability in Colorectal Cancer Pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef]
- Tseng-Rogenski, S.S.; Hamaya, Y.; Choi, D.Y.; Carethers, J.M. Interleukin 6 Alters Localization of hMSH3, Leading to DNA Mismatch Repair Defects in Colorectal Cancer Cells. Gastroenterology 2015, 148, 579–589. [Google Scholar] [CrossRef]
- Tseng-Rogenski, S.S.; Chung, H.; Wilk, M.B.; Zhang, S.; Iwaizumi, M.; Carethers, J.M. Oxidative Stress Induces Nuclear-to-Cytosol Shift of hMSH3, a Potential Mechanism for EMAST in Colorectal Cancer Cells. PLoS ONE 2012, 7, e50616. [Google Scholar] [CrossRef]
- Plaschke, J.; Krüger, S.; Jeske, B.; Theissig, F.; Kreuz, F.R.; Pistorius, S.; Saeger, H.D.; Iaccarino, I.; Marra, G.; Schackert, H.K. Loss of MSH3 Protein Expression Is Frequent in MLH1-Deficient Colorectal Cancer and Is Associated with Disease Progression. Cancer Res 2004, 64, 864–870. [Google Scholar] [CrossRef]
- Devaraj, B.; Lee, A.; Cabrera, B.L.; Miyai, K.; Luo, L.; Ramamoorthy, S.; Keku, T.; Sandler, R.S.; McGuire, K.L.; Carethers, J.M. Relationship of EMAST and Microsatellite Instability Among Patients with Rectal Cancer. J. Gastrointest. Surg. 2010, 14, 1521–1528. [Google Scholar] [CrossRef]
- Munakata, K.; Koi, M.; Kitajima, T.; Tseng-Rogenski, S.; Uemura, M.; Matsuno, H.; Kawai, K.; Sekido, Y.; Mizushima, T.; Toiyama, Y.; et al. Inflammation-Associated Microsatellite Alterations Caused by MSH3 Dysfunction Are Prevalent in Ulcerative Colitis and Increase with Neoplastic Advancement. Clin. Transl. Gastroenterol. 2019, 10, e00105. [Google Scholar] [CrossRef]
- Tseng-Rogenski, S.S.; Munakata, K.; Choi, D.Y.; Martin, P.K.; Mehta, S.; Koi, M.; Zheng, W.; Zhang, Y.; Carethers, J.M. The Human DNA Mismatch Repair Protein MSH3 Contains Nuclear Localization and Export Signals That Enable Nuclear-Cytosolic Shuttling in Response to Inflammation. Mol. Cell. Biol. 2020, 40, e00029-20. [Google Scholar] [CrossRef]
- Bodmer, W.; Bishop, T.; Karran, P. Genetic steps in colorectal cancer. Nat. Genet. 1994, 6, 217–219. [Google Scholar] [CrossRef]
- Maby, P.; Tougeron, D.; Hamieh, M.; Mlecnik, B.; Kora, H.; Bindea, G.; Angell, H.K.; Fredriksen, T.; Elie, N.; Fauquembergue, E.; et al. Correlation between Density of CD8+ T-cell Infiltrate in Microsatellite Unstable Colorectal Cancers and Frameshift Mutations: A Rationale for Personalized Immunotherapy. Cancer Res. 2015, 75, 3446–3455. [Google Scholar] [CrossRef]
- Dolcetti, R.; Viel, A.; Doglioni, C.; Russo, A.; Guidoboni, M.; Capozzi, E.; Vecchiato, N.; Macrì, E.; Fornasarig, M.; Boiocchi, M. High Prevalence of Activated Intraepithelial Cytotoxic T Lymphocytes and Increased Neoplastic Cell Apoptosis in Colorectal Carcinomas with Microsatellite Instability. Am. J. Pathol. 1999, 154, 1805–1813. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The Vigorous Immune Microenvironment of Microsatellite Instable Colon Cancer Is Balanced by Multiple Counter-Inhibitory Checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef]
- Phillips, S.M.; Banerjea, A.; Feakins, R.; Li, S.R.; Bustin, S.A.; Dorudi, S. Tumour-infiltrating lymphocytes in colorectal cancer with microsatellite instability are activated and cytotoxic. Br. J. Surg. 2004, 91, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Nakayama, K.; Ishikawa, M.; Ishibashi, T.; Nakamura, K.; Sawada, K.; Yoshimura, Y.; Tatsumi, N.; Kurose, S.; Minamoto, T.; et al. Relationship between Microsatellite Instability, Immune Cells Infiltration, and Expression of Immune Checkpoint Molecules in Ovarian Carcinoma: Immunotherapeutic Strategies for the Future. Int. J. Mol. Sci. 2019, 20, 5129. [Google Scholar] [CrossRef]
- Eso, Y.; Shimizu, T.; Takeda, H.; Takai, A.; Marusawa, H. Microsatellite instability and immune checkpoint inhibitors: Toward precision medicine against gastrointestinal and hepatobiliary cancers. J. Gastroenterol. 2019, 55, 15–26. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Hu, W.; Yang, Y.; Qi, L.; Chen, J.; Ge, W.; Zheng, S. Subtyping of microsatellite instability-high colorectal cancer. Cell Commun. Signal. 2019, 17, 79. [Google Scholar] [CrossRef]
- Sui, Q.; Zhang, X.; Chen, C.; Tang, J.; Yu, J.; Li, W.; Han, K.; Jiang, W.; Liao, L.; Kong, L.; et al. Inflammation promotes resistance to immune checkpoint inhibitors in high microsatellite instability colorectal cancer. Nat. Commun. 2022, 13, 7316. [Google Scholar] [CrossRef]
- Mandal, R.; Samstein, R.M.; Lee, K.-W.; Havel, J.J.; Wang, H.; Krishna, C.; Sabio, E.Y.; Makarov, V.; Kuo, F.; Blecua, P.; et al. Genetic diversity of tumors with mismatch repair deficiency influences anti–PD-1 immunotherapy response. Science 2019, 364, 485–491. [Google Scholar] [CrossRef]
- Lee, S.; Chung, H.; Devaraj, B.; Iwaizumi, M.; Han, H.S.; Hwang, D.; Seong, M.K.; Jung, B.H.; Carethers, J.M. Microsatellite Alterations at Selected Tetranucleotide Repeats Are Associated with Morphologies of Colorectal Neoplasias. Gastroenterology 2010, 139, 1519–1525. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Miyai, K.; Han, H.S.; Hwang, D.-Y.; Seong, M.K.; Chung, H.; Jung, B.H.; Devaraj, B.; McGuire, K.L.; Carethers, J.M. Microsatellite Instability, EMAST, and Morphology Associations with T Cell Infiltration in Colorectal Neoplasia. Dig. Dis. Sci. 2011, 57, 72–78. [Google Scholar] [CrossRef]
- Chen, M.-H.; Chang, S.-C.; Lin, P.-C.; Yang, S.-H.; Lin, C.-C.; Lan, Y.-T.; Lin, H.-H.; Lin, C.-H.; Lai, J.-I.; Liang, W.-Y.; et al. Combined Microsatellite Instability and Elevated Microsatellite Alterations at Selected Tetranucleotide Repeats (EMAST) Might Be a More Promising Immune Biomarker in Colorectal Cancer. Oncology 2019, 24, 1534–1542. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Tahara, T.; Yamamoto, E.; Suzuki, H.; Maruyama, R.; Chung, W.; Garriga, J.; Jelinek, J.; Yamano, H.-O.; Sugai, T.; An, B.; et al. Fusobacterium in Colonic Flora and Molecular Features of Colorectal Carcinoma. Cancer Res 2014, 74, 1311–1318. [Google Scholar] [CrossRef]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatumin colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, Y.; Yu, J.; Chen, T.; Wu, Y.; Shi, L.; Li, Q.; Wu, J.; Fu, X. Invasive Fusobacterium nucleatum activates β-catenin signaling in colorectal cancer via a TLR4/P-PAK1 cascade. Oncotarget 2017, 8, 31802–31814. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Chen, T.; Li, Q.; Wu, J.; Wu, Y.; Peng, W.; Li, H.; Wang, J.; Tang, X.; Peng, Y.; Fu, X. Fusobacterium nucleatum promotes M2 polarization of macrophages in the microenvironment of colorectal tumours via a TLR4-dependent mechanism. Cancer Immunol. Immunother. 2018, 67, 1635–1646. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Okita, Y.; Koi, M.; Takeda, K.; Ross, R.; Mukherjee, B.; Koeppe, E.; Stoffel, E.M.; Galanko, J.A.; McCoy, A.N.; Keku, T.O.; et al. Fusobacterium nucleatum infection correlates with two types of microsatellite alterations in colorectal cancer and triggers DNA damage. Gut Pathog. 2020, 12, 46. [Google Scholar] [CrossRef]
- Ros, J.; Balconi, F.; Baraibar, I.; Gonzalez, N.S.; Salva, F.; Tabernero, J.; Elez, E. Advances in immune checkpoint inhibitor combination strategies for microsatellite stable colorectal cancer. Front. Oncol. 2023, 13, 1112276. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.L. Current Microsatellite Instability Testing in Management of Colorectal Cancer. Clin. Color. Cancer 2021, 20, e12–e20. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M. Hereditary, sporadic and metastatic colorectal cancer are commonly driven by specific spectrums of defective dna mismatch repair components. Trans. Am. Clin. Clim. Assoc. 2016, 127, 81–97. [Google Scholar]
- Kim, H.G.; Lee, S.; Kim, D.Y.; Ryu, S.Y.; Joo, J.K.; Kim, J.C.; Lee, K.H.; Lee, J.H. Aberrant methylation of DNA mismatch repair genes in elderly patients with sporadic gastric carcinoma: A comparison with younger patients. J. Surg. Oncol. 2010, 101, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.; Spier, I.; Zhao, B.; Kloth, M.; Marquez, J.; Hinrichsen, I.; Kirfel, J.; Tafazzoli, A.; Horpaopan, S.; Uhlhaas, S.; et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am. J. Hum. Genet. 2016, 99, 337–351. [Google Scholar] [CrossRef]
- Venderbosch, S.; Vliet, S.v.L.; de Haan, A.F.J.; Ligtenberg, M.J.; Goossens, M.; Punt, C.J.A.; Koopman, M.; Nagtegaal, I.D. EMAST Is Associated with a Poor Prognosis in Microsatellite Instable Metastatic Colorectal Cancer. PLoS ONE 2015, 10, e0124538. [Google Scholar] [CrossRef]
- Watson, M.M.; Lea, D.; Hagland, H.R.; Søreide, K. Elevated Microsatellite Alterations at Selected Tetranucleotides (EMAST) Is Not Attributed to MSH3 Loss in Stage I-III Colon cancer: An Automated, Digitalized Assessment by Immunohistochemistry of Whole Slides and Hot Spots. Transl. Oncol. 2019, 12, 1583–1588. [Google Scholar] [CrossRef]
- Laycock, A.; Kang, A.; Ang, S.; Texler, M.; Bentel, J. Lack of correlation between MSH3 immunohistochemistry and microsatellite analysis for the detection of elevated microsatellite alterations at selected tetranucleotide repeats (EMAST) in colorectal cancers. Hum. Pathol. 2021, 118, 9–17. [Google Scholar] [CrossRef]
- Meessen, S.; Currey, N.; Jahan, Z.; Parker, H.W.; Jenkins, M.A.; Buchanan, D.D.; Hopper, J.L.; Segelov, E.; Dahlstrom, J.E.; Kohonen-Corish, M.R.J. Tetranucleotide and Low Microsatellite Instability Are Inversely Associated with the CpG Island Methylator Phenotype in Colorectal Cancer. Cancers 2021, 13, 3529. [Google Scholar] [CrossRef]
- Shin, G.; Greer, S.U.; Hopmans, E.; Grimes, S.M.; Lee, H.; Zhao, L.; Miotke, L.; Suarez, C.; Almeda, A.F.; Haraldsdottir, S.; et al. Profiling diverse sequence tandem repeats in colorectal cancer reveals co-occurrence of microsatellite and chromosomal instability involving Chromosome 8. Genome Med. 2021, 13, 145. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor Microsatellite-Instability Status as a Predictor of Benefit from Fluorouracil-Based Adjuvant Chemotherapy for Colon Cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef]
- Halford, S.; Sasieni, P.; Rowan, A.; Wasan, H.; Bodmer, W.; Talbot, I.; Hawkins, N.; Ward, R.; Tomlinson, I. Low-level microsatellite instability occurs in most colorectal cancers and is a nonrandomly distributed quantitative trait. Cancer Res 2002, 62, 53–57. [Google Scholar]
- Laiho, P.; Launonen, V.; Lahermo, P.; Esteller, M.; Guo, M.; Herman, J.G.; Mecklin, J.P.; Jarvinen, H.; Sistonen, P.; Kim, K.M.; et al. Low-level microsatellite instability in most colorectal carci-nomas. Cancer Res. 2002, 62, 1166–1170. [Google Scholar]
- Esfahani, A.T.; Seyedna, S.Y.; Mojarad, E.N.; Majd, A.; Aghdaei, H.A. MSI-L/EMAST is a predictive biomarker for metastasis in colorectal cancer patients. J. Cell. Physiol. 2019, 234, 13128–13136. [Google Scholar] [CrossRef]
- Herz, A.; Wisser, S.; Kohlruss, M.; Slotta-Huspenina, J.; Jesinghaus, M.; Grosser, B.; Steiger, K.; Novotny, A.; Hapfelmeier, A.; Schmidt, T.; et al. Elevated microsatellite instability at selected tetranucleotide (EMAST) repeats in gastric cancer: A distinct microsatellite instability type with potential clinical impact? J. Pathol. Clin. Res. 2022, 8, 233–244. [Google Scholar] [CrossRef]
- Ahrendt, S.A.; Decker, P.A.; Doffek, K.; Wang, B.; Xu, L.; Demeure, M.J.; Jen, J.; Sidransky, D. Microsatellite in-stability at selected tetranucleotide repeats is associated with p53 mutations in non-small cell lung cancer. Cancer Res. 2000, 60, 2488–2491. [Google Scholar]
- Woenckhaus, M.; Stoehr, R.; Dietmaier, W.; Wild, P.J.; Zieglmeier, U.; Foerster, J.; Merk, J.; Blaszyk, H.; Pfeifer, M.; Hofstaedter, F.; et al. Microsatellite instability at chromosome 8p in non-small cell lung cancer is as-sociated with lymph node metastasis and squamous differentiation. Int. J. Oncol. 2003, 23, 1357–1363. [Google Scholar]
- Arai, H.; Okudela, K.; Oshiro, H.; Komitsu, N.; Mitsui, H.; Nishii, T.; Tsuboi, M.; Nozawa, A.; Noishiki, Y.; Ohashi, K.; et al. Elevated microsatellite alterations at selected tetra-nucleotide (EMAST) in non-small cell lung cancers--a potential determinant of susceptibility to multiple malignancies. Int. J. Clin. Exp. Pathol. 2013, 6, 395–410. [Google Scholar]
- Mori, T.; Hamaya, Y.; Uotani, T.; Yamade, M.; Iwaizumi, M.; Furuta, T.; Miyajima, H.; Osawa, S.; Sugimoto, K. Prevalence of elevated microsatellite alterations at selected tetranucleotide repeats in pancreatic ductal adenocarcinoma. PLoS ONE 2018, 13, e0208557. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.; Burger, S.J.; Denzinger, S.; Wild, P.J.; Wieland, W.F.; Blaszyk, H.; Obermann, E.C.; Stoehr, R.; Hartmann, A.; von Knobloch, R. Elevated Microsatellite Instability at Selected Tetranucleotide Repeats does not Correlate with Clinicopathologic Features of Bladder Cancer. Eur. Urol. 2006, 50, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Singer, G.; Kallinowski, T.; Hartmann, A.; Dietmaier, W.; Wild, P.J.; Schraml, P.; Sauter, G.; Mihatsch, M.J.; Moch, H. Different types of microsatellite instability in ovarian carcinoma. Int. J. Cancer 2004, 112, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Hile, S.E.; Shabashev, S.; Eckert, K.A. Tumor-specific microsatellite instability: Do distinct mechanisms underlie the MSI-L and EMAST phenotypes? Mutat. Res. Mol. Mech. Mutagen. 2013, 743–744, 67–77. [Google Scholar] [CrossRef]
- De Salins, A.G.D.; Tachon, G.; Cohen, R.; Karayan-Tapon, L.; Junca, A.; Frouin, E.; Godet, J.; Evrard, C.; Randrian, V.; Duval, A.; et al. Discordance between immunochemistry of mismatch repair proteins and molecular testing of microsatellite instability in colorectal cancer. ESMO Open 2021, 6, 100120. [Google Scholar] [CrossRef]
- Chen, J.; Yan, Q.; Sun, J.; Wang, Q.; Tao, Y.; Xiao, D.; Xie, B. Microsatellite Status Detection of Colorectal Cancer: Evaluation of Inconsistency between PCR and IHC. J. Cancer 2023, 14, 1132–1140. [Google Scholar] [CrossRef]
- McCarthy, A.J.; Capo-Chichi, J.-M.; Spence, T.; Grenier, S.; Stockley, T.; Kamel-Reid, S.; Serra, S.; Sabatini, P.; Chetty, R. Heterogenous loss of mismatch repair (MMR) protein expression: A challenge for immunohistochemical interpretation and microsatellite instability (MSI) evaluation. J. Pathol. Clin. Res. 2018, 5, 115–129. [Google Scholar] [CrossRef]
- Van Oers, J.M.M.; Edwards, Y.; Chahwan, R.; Zhang, W.; Smith, C.; Pechuan, X.; Schaetzlein, S.; Jin, B.; Wang, Y.; Bergman, A.; et al. The MutSβ complex is a modulator of p53-driven tumorigenesis through its functions in both DNA double-strand break repair and mismatch repair. Oncogene 2013, 33, 3939–3946. [Google Scholar] [CrossRef]
- Fang, W.-L.; Chen, M.-H.; Huang, K.-H.; Chang, S.-C.; Lin, C.-H.; Chao, Y.; Lo, S.-S.; Li, A.F.-Y.; Wu, C.-W.; Shyr, Y.-M. The Clinicopathological Features and Genetic Mutations in Gastric Cancer Patients According to EMAST and MSI Status. Cancers 2020, 12, 551. [Google Scholar] [CrossRef]
- Yamamoto, H.; Imai, K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin. Oncol. 2019, 46, 261–270. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vuković Đerfi, K.; Salar, A.; Cacev, T.; Kapitanović, S. EMAST Type of Microsatellite Instability—A Distinct Entity or Blurred Overlap between Stable and MSI Tumors. Genes 2023, 14, 1474. https://doi.org/10.3390/genes14071474
Vuković Đerfi K, Salar A, Cacev T, Kapitanović S. EMAST Type of Microsatellite Instability—A Distinct Entity or Blurred Overlap between Stable and MSI Tumors. Genes. 2023; 14(7):1474. https://doi.org/10.3390/genes14071474
Chicago/Turabian StyleVuković Đerfi, Kristina, Anamarija Salar, Tamara Cacev, and Sanja Kapitanović. 2023. "EMAST Type of Microsatellite Instability—A Distinct Entity or Blurred Overlap between Stable and MSI Tumors" Genes 14, no. 7: 1474. https://doi.org/10.3390/genes14071474
APA StyleVuković Đerfi, K., Salar, A., Cacev, T., & Kapitanović, S. (2023). EMAST Type of Microsatellite Instability—A Distinct Entity or Blurred Overlap between Stable and MSI Tumors. Genes, 14(7), 1474. https://doi.org/10.3390/genes14071474