
Identification of Genetic Risk Factors of Severe COVID-19 Using Extensive Phenotypic Data: A Proof-of-Concept Study in a Cohort of Russian Patients

 , , , ,
, , , ,  ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Inclusion Criteria
2.2. Characteristics of Groups of Patients
2.3. Clinical and Biochemical Surveillances
2.4. Therapy for Patients with COVID-19 Infection
2.5. Library Preparation and Exome Sequencing
2.6. Variant Calling in Patient Exomes
2.7. Phenotype Processing
2.8. Common Variant Association Analysis (CVAS)
2.9. Rare Variants Associations Studies
2.10. Replication of the Identified Associations and Functional Evidence Mining
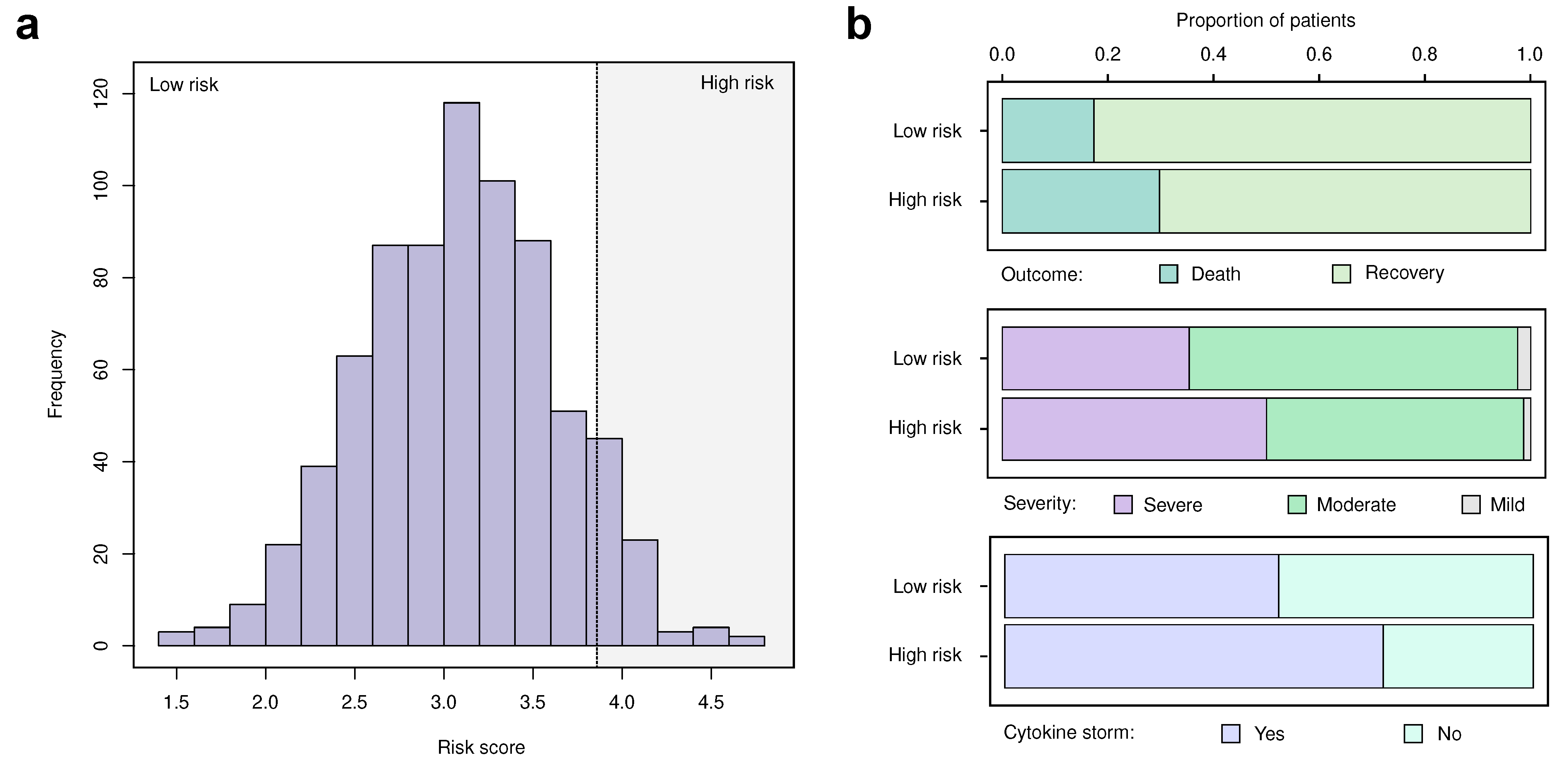
2.11. Construction of the Risk Score
3. Results
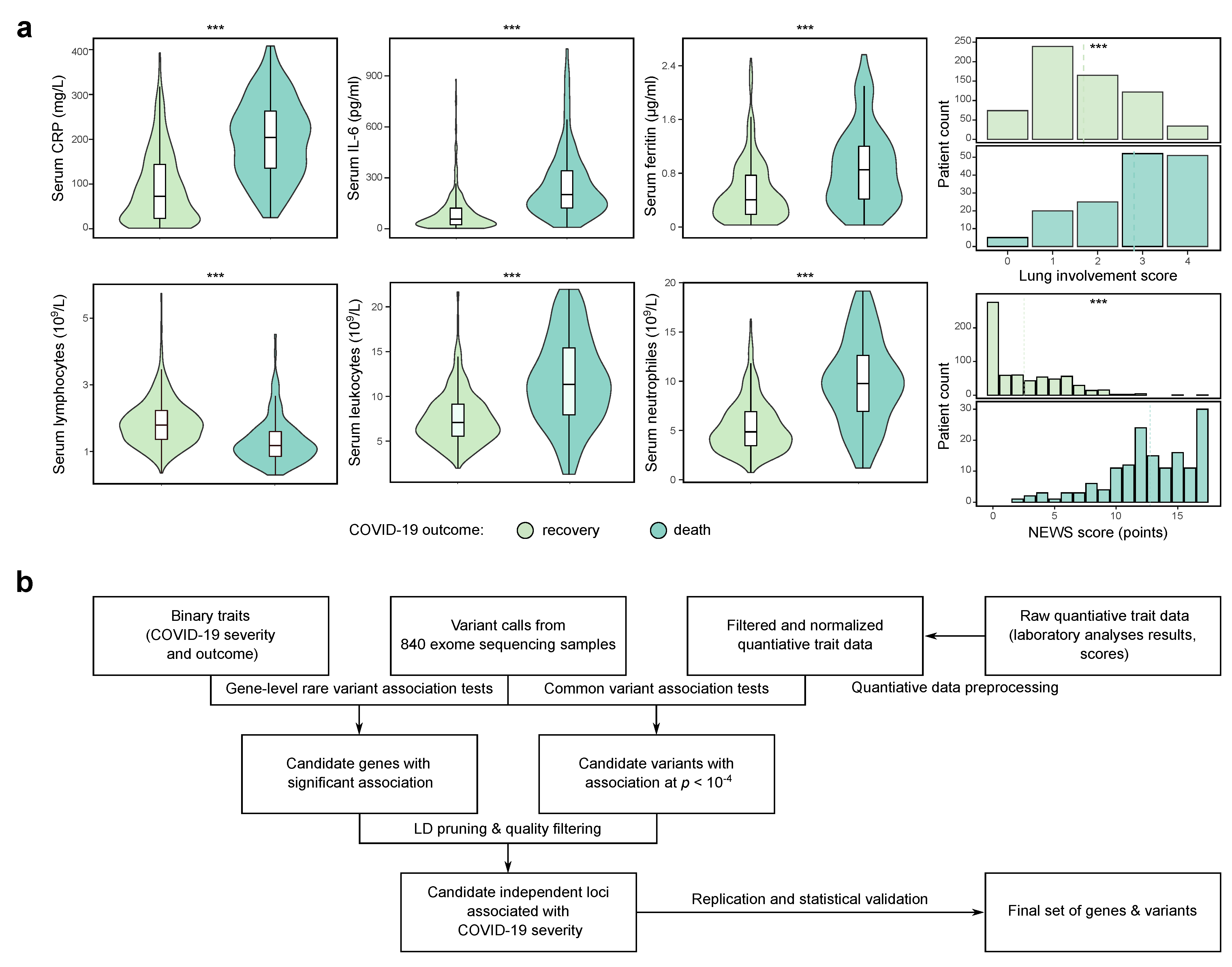
3.1. Study Design and Data Preprocessing
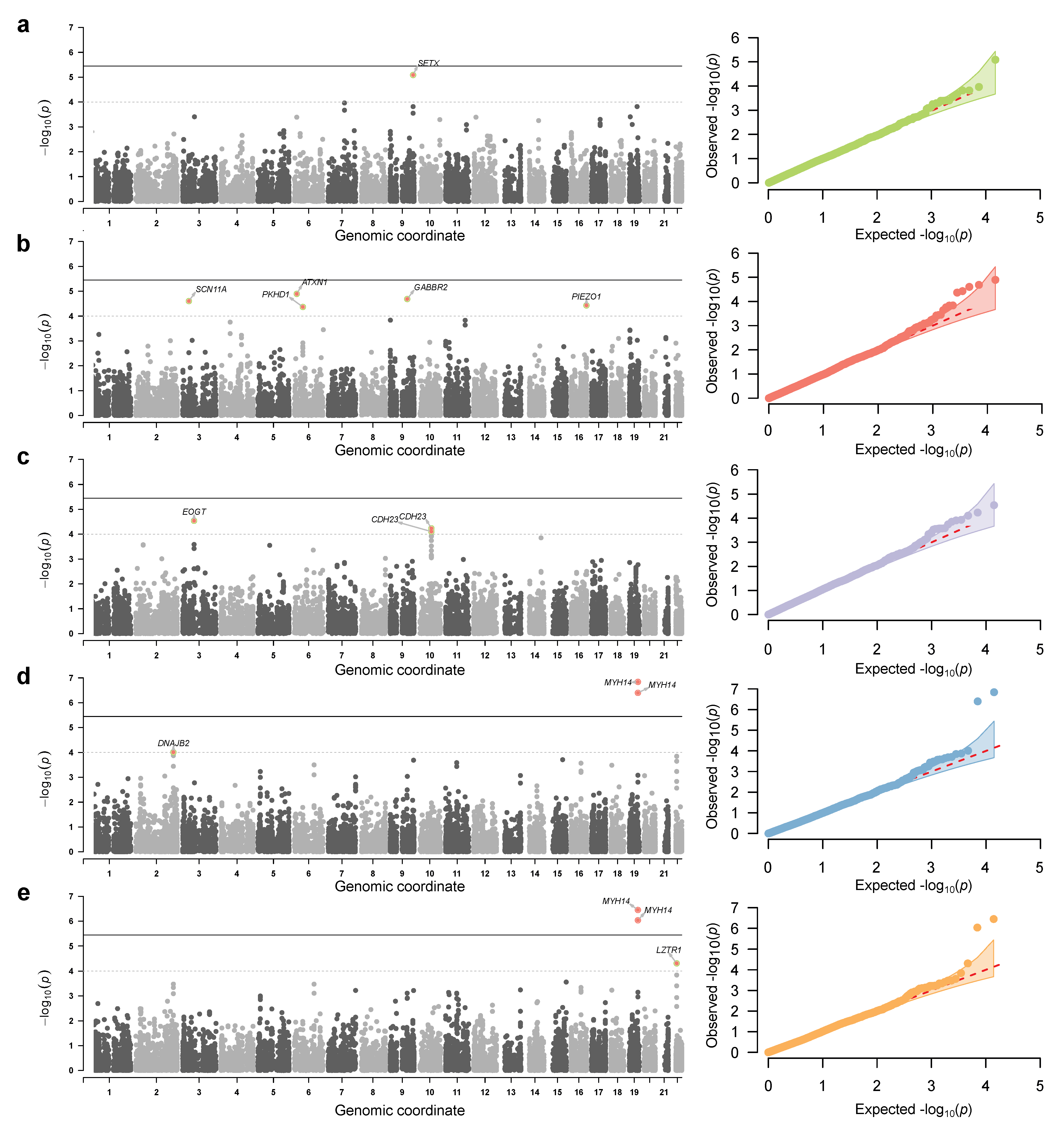
3.2. Genome-Wide Association Analysis Using a Deeply Phenotype Cohort
3.3. Replication and Validation of the Identified Markers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| COVID-19 | Coronavirus disease 2019 |
| eQTL | expression quantiative trait locus |
| GWAS | Genome-wide association study |
| SNP | single-nucleotide polymorphism |
| WES | Whole exome sequencing |
References
- Zhu, H.; Wei, L.; Niu, P. The novel coronavirus outbreak in Wuhan, China. Glob. Health Res. Policy 2020, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Prakrithi, P.; Lakra, P.; Sundar, D.; Kapoor, M.; Mukerji, M.; Gupta, I.; The Indian Genome Variation Consortium. Genetic Risk Prediction of COVID-19 Susceptibility and Severity in the Indian Population. Front. Genet. 2021, 12, 714185. [Google Scholar] [CrossRef]
- Suh, S.; Lee, S.; Gym, H.; Yoon, S.; Park, S.; Cha, J.; Kwon, D.H.; Yang, Y.; Jee, S.H. A systematic review on papers that study on Single Nucleotide Polymorphism that affects coronavirus 2019 severity. BMC Infect. Dis. 2022, 22, 47. [Google Scholar] [CrossRef] [PubMed]
- Mousa, M.; Vurivi, H.; Kannout, H.; Uddin, M.; Alkaabi, N.; Mahboub, B.; Tay, G.K.; Alsafar, H.S. Genome-wide association study of hospitalized COVID-19 patients in the United Arab Emirates. eBioMedicine 2021, 74, 103695. [Google Scholar] [CrossRef] [PubMed]
- The Severe Covid-19 GWAS Group. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Shikov, A.E.; Barbitoff, Y.A.; Glotov, A.S.; Danilova, M.M.; Tonyan, Z.N.; Nasykhova, Y.A.; Mikhailova, A.A.; Bespalova, O.N.; Kalinin, R.S.; Mirzorustamova, A.M.; et al. Analysis of the Spectrum of ACE2 Variation Suggests a Possible Influence of Rare and Common Variants on Susceptibility to COVID-19 and Severity of Outcome. Front. Genet. 2020, 11, 551220. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, S. Statistical power in genome-wide association studies and quantitative trait locus mapping. Heredity 2019, 123, 287–306. [Google Scholar] [CrossRef]
- Belfiore, M.P.; Urraro, F.; Grassi, R.; Giacobbe, G.; Patelli, G.; Cappabianca, S.; Reginelli, A. Artificial intelligence to codify lung CT in Covid-19 patients. Radiol. Med. 2020, 125, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhang, H.; Xie, J.; Lin, M.; Ying, L.; Pang, P.; Ji, W. Sensitivity of Chest CT for COVID-19: Comparison to RT-PCR. Radiology 2020, 296, E115–E117. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Clinical Management of COVID-19: Interim Guidance, 27 May 2020; Technical Documents; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Smith, G.B.; Redfern, O.C.; Pimentel, M.A.; Gerry, S.; Collins, G.S.; Malycha, J.; Prytherch, D.; Schmidt, P.E.; Watkinson, P.J. The National Early Warning Score 2 (NEWS2). Clin. Med. 2019, 19, 260. [Google Scholar] [CrossRef] [PubMed]
- Carr, E.; Bendayan, R.; Bean, D.; Stammers, M.; Wang, W.; Zhang, H.; Searle, T.; Kraljevic, Z.; Shek, A.; Phan, H.T.T.; et al. Evaluation and improvement of the National Early Warning Score (NEWS2) for COVID-19: A multi-hospital study. BMC Med. 2021, 19, 23. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Goh, L.; Yap, V.B. Effects of normalization on quantitative traits in association test. BMC Bioinform. 2009, 10, 415. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- McInnes, G.; Tanigawa, Y.; DeBoever, C.; Lavertu, A.; Olivieri, J.E.; Aguirre, M.; Rivas, M.A. Global Biobank Engine: Enabling genotype-phenotype browsing for biobank summary statistics. Bioinformatics 2019, 35, 2495–2497. [Google Scholar] [CrossRef]
- Aguet, F.; Ardlie, K.G.; Cummings, B.B.; Gelfand, E.T.; Getz, G.; Hadley, K.; Handsaker, R.E.; Huang, K.H.; Kashin, S.; Karczewski, K.J.; et al. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Shcherbak, S.G.; Anisenkova, A.Y.; Mosenko, S.V.; Glotov, O.S.; Chernov, A.N.; Apalko, S.V.; Urazov, S.P.; Garbuzov, E.Y.; Khobotnikov, D.N.; Klitsenko, O.A.; et al. Basic Predictive Risk Factors for Cytokine Storms in COVID-19 Patients. Front. Immunol. 2021, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Povysil, G.; Butler-Laporte, G.; Shang, N.; Wang, C.; Khan, A.; Alaamery, M.; Nakanishi, T.; Zhou, S.; Forgetta, V.; Eveleigh, R.J.; et al. Rare loss-of-function variants in type I IFN immunity genes are not associated with severe COVID-19. J. Clin. Investig. 2021, 131, JCI147834. [Google Scholar] [CrossRef] [PubMed]
- Barbitoff, Y.A.; Skitchenko, R.K.; Poleshchuk, O.I.; Shikov, A.E.; Serebryakova, E.A.; Nasykhova, Y.A.; Polev, D.E.; Shuvalova, A.R.; Shcherbakova, I.V.; Fedyakov, M.A.; et al. Whole-exome sequencing provides insights into monogenic disease prevalence in Northwest Russia. Mol. Genet. Genom. Med. 2019, 7, e964. [Google Scholar] [CrossRef]
- Shikov, A.E.; Skitchenko, R.K.; Predeus, A.V.; Barbitoff, Y.A. Phenome-wide functional dissection of pleiotropic effects highlights key molecular pathways for human complex traits. Sci. Rep. 2020, 10, 1037. [Google Scholar] [CrossRef]
- Watanabe, K.; Taskesen, E.; van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ke, Y.; Xia, X.; Wang, Y.; Cheng, F.; Liu, X.; Jin, X.; Li, B.; Xie, C.; Liu, S.; et al. Genome-wide association study of COVID-19 severity among the Chinese population. Cell Discov. 2021, 7, 76. [Google Scholar] [CrossRef]
- Donaudy, F.; Snoeckx, R.; Pfister, M.; Zenner, H.P.; Blin, N.; Di Stazio, M.; Ferrara, A.; Lanzara, C.; Ficarella, R.; Declau, F.; et al. Nonmuscle Myosin Heavy-Chain Gene MYH14 Is Expressed in Cochlea and Mutated in Patients Affected by Autosomal Dominant Hearing Impairment (DFNA4). Am. J. Hum. Genet. 2004, 74, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Almutawa, W.; Smith, C.; Sabouny, R.; Smit, R.B.; Zhao, T.; Wong, R.; Lee-Glover, L.; Desrochers-Goyette, J.; Ilamathi, H.S.; Suchowersky, O.; et al. The R941L mutation in MYH14 disrupts mitochondrial fission and associates with peripheral neuropathy. EBioMedicine 2019, 45, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.H.; Peng, S.; Hu, X.; Chen, C.; Rahman, M.R.; Uddin, S.; Quinn, J.M.; Moni, M.A. A Network-Based Bioinformatics Approach to Identify Molecular Biomarkers for Type 2 Diabetes that Are Linked to the Progression of Neurological Diseases. Int. J. Environ. Res. Public Health 2020, 17, 1035. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef]
- Craig, E.A.; Marszalek, J. How Do J-Proteins Get Hsp70 to Do So Many Different Things? Trends Biochem. Sci. 2017, 42, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cao, X.; Wang, J.; Li, Q.; Zhao, Y.; Jin, X. LZTR1: A promising adaptor of the CUL3 family (Review). Oncol. Lett. 2021, 22, 564. [Google Scholar] [CrossRef] [PubMed]
- Macfarlan, T.; Kutney, S.; Altman, B.; Montross, R.; Yu, J.; Chakravarti, D. Human THAP7 Is a Chromatin-associated, Histone Tail-binding Protein That Represses Transcription via Recruitment of HDAC3 and Nuclear Hormone Receptor Corepressor. J. Biol. Chem. 2005, 280, 7346–7358. [Google Scholar] [CrossRef] [PubMed]
- Dächert, C.; Gladilin, E.; Binder, M. Gene Expression Profiling of Different Huh7 Variants Reveals Novel Hepatitis C Virus Host Factors. Viruses 2019, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, J.V.; Martinac, B.; Cox, C.D. Loss-of-Function Piezo1 Mutations Display Altered Stability Driven by Ubiquitination and Proteasomal Degradation. Front. Pharmacol. 2021, 12, 766416. [Google Scholar] [CrossRef] [PubMed]
- Mustacich, D.J.; Lai, L.W.; Bernas, M.J.; Jones, J.A.; Myles, R.J.; Kuo, P.H.; Williams, W.H.; Witte, C.L.; Erickson, R.P.; Witte, M.H. Digenic Inheritance of a FOXC2 Mutation and Two PIEZO1 Mutations Underlies Congenital Lymphedema in a Multigeneration Family. Am. J. Med. 2021, 135, e31–e41. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Park, S.; Kim, H.Y.; Chae, J.H.; Ko, J.M. Extended phenotypes of PIEZO1-related lymphatic dysplasia caused by two novel compound heterozygous variants. Eur. J. Med. Genet. 2021, 64, 104295. [Google Scholar] [CrossRef]
- Zhang, Z.; Bai, H.; Blumenfeld, J.; Ramnauth, A.B.; Barash, I.; Prince, M.; Tan, A.Y.; Michaeel, A.; Liu, G.; Chicos, I.; et al. Detection of PKD1 and PKD2 Somatic Variants in Autosomal Dominant Polycystic Kidney Cyst Epithelial Cells by Whole-Genome Sequencing. J. Am. Soc. Nephrol. 2021, 32, 3114–3129. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.; Mei, H.; Tin, A.; Bressler, J.; Pruett, W.A.; Fornage, M.; Huang, J.; Boerwinkle, E.; Mosley, T.H.; Simino, J. Whole Exome Sequence Study of Mild Cognitive Impairment in African and European Americans; the Atherosclerosis Risk in Communities-Neurocognitive Study. Alzheimer’s Dement. 2021, 17, e058619. [Google Scholar] [CrossRef]
- Ohni, S.; Yamaguchi, H.; Hirotani, Y.; Nakanishi, Y.; Midorikawa, Y.; Sugitani, M.; Naruse, H.; Nakayama, T.; Makishima, M.; Esumi, M. Direct molecular evidence for both multicentric and monoclonal carcinogenesis followed by transdifferentiation from hepatocellular carcinoma to cholangiocarcinoma in a case of metachronous liver cancer. Oncol. Lett. 2021, 23, 22. [Google Scholar] [CrossRef]
- Kumaran, D.; Balagopal, K.; Tharmaraj, R.G.A.; Aaron, S.; George, K.; Muliyil, J.; Sivadasan, A.; Danda, S.; Alexander, M.; Hasan, G. Genetic characterization of Spinocerebellar ataxia 1 in a South Indian cohort. BMC Med. Genet. 2014, 15, 114. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.E.; Bird, T.D. Molecular genetic testing for hereditary ataxia. Neurol. Clin. Pract. 2018, 8, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, A.; Kanahara, N.; Kogure, M.; Otsuka, I.; Okazaki, S.; Watanabe, Y.; Yamasaki, F.; Nakata, Y.; Oda, Y.; Hishimoto, A.; et al. A preliminary genetic association study of GAD1 and GABAB receptor genes in patients with treatment-resistant schizophrenia. Mol. Biol. Rep. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hadjinicolaou, A.; Ngo, K.J.; Conway, D.Y.; Provias, J.P.; Baker, S.K.; Brady, L.I.; Bennett, C.L.; La Spada, A.R.; Fogel, B.L.; Yoon, G. De novo pathogenic variant in SETX causes a rapidly progressive neurodegenerative disorder of early childhood-onset with severe axonal polyneuropathy. Acta Neuropathol. Commun. 2021, 9, 194. [Google Scholar] [CrossRef]
- Saleem, I.B.; Masoud, M.S.; Qasim, M.; Ali, M.; Ahmed, Z.M. Identification and Computational Analysis of Rare Variants of Known Hearing Loss Genes Present in Five Deaf Members of a Pakistani Kindred. Genes 2021, 12, 1940. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Locus | rsID | Substitution | AF * | Trait(s) | Gene | Consequence | β ** | p-Value | GTEx eQTLs *** |
|---|---|---|---|---|---|---|---|---|---|
| 2:219280564 | rs2276638 | 6247C>G | Leukocytes | DNAJB2 | intron variant | Multiple genes and tissues | |||
| 3:38894643 | rs33985936 | c.2725G>T (p.Val909Phe) | CT score | SCN11A | missense variant | Multiple genes and tissues | |||
| 3:68997990 | rs4855544 | g.20905C>A | Lymphocytes | EOGT | intron variant | Multiple genes and tissues | |||
| 6:16306520 | rs16885 | c.2257C>T (p.Pro753Ala) | CT score | ATXN1 | missense variant | none | |||
| 6:51830849 | rs1571084 | g.261777T>A | CT score | PKHD1 | intron variant | PKHD1 (skin) | |||
| 9:98299383 | rs41273925 | g.414815C>G | CT score | GABBR2 | intron variant | TBC1D2 (thyroid) | |||
| 9:132278286 | rs11243705 | g.81700A>G | CRP | SETX | intron variant | SETX (multi-tissue) | |||
| 10:71799129 | rs4747194 | c.7073G>T (p.Arg2358Gln) | Lymphocytes | CDH23 | missense variant | CDH23 (colon, testis), PSAP (multi-tissue) | |||
| 16:88738516 | rs34600315 | c.*648_*649del | CT score | PIEZO1 | non coding transcript exon variant | PIEZO1 (whole blood) | |||
| 19:50259161 | rs1651553 | c.2127A>G | Leukocytes, neutrophiles | MYH14 | synonymous variant | , | , | none | |
| 22:20992196 | rs112544 | g.14928T>G | Neutrophiles | LZTR1 | intron variant | Multiple genes and tissues |
| Variant | Gene | p-Value (This Work) | A2 †,* | B1 †,** | B2 †,*** | C2 †,**** | The Severe COVID-19 GWAS Group †† | UK Biobank PheWAS Traits ††† |
|---|---|---|---|---|---|---|---|---|
| rs2276638 | DNAJB2 | none | ||||||
| rs33985936 | SCN11A | Platelet count, platelet crit | ||||||
| rs4855544 | EOGT | none | ||||||
| rs16885 | ATXN1 | Mean corpuscular hemoglobin | ||||||
| rs1571084 | PKHD1 | none | ||||||
| rs41273925 | GABBR2 | none | ||||||
| rs11243705 | SETX | none | ||||||
| rs4747194 | CDH23 | Monocyte % | ||||||
| rs34600315 | PIEZO1 | n.a. | none | |||||
| rs1651553 | MYH14 | none | ||||||
| rs112544 | LZTR1 | none |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shcherbak, S.G.; Changalidi, A.I.; Barbitoff, Y.A.; Anisenkova, A.Y.; Mosenko, S.V.; Asaulenko, Z.P.; Tsay, V.V.; Polev, D.E.; Kalinin, R.S.; Eismont, Y.A.; et al. Identification of Genetic Risk Factors of Severe COVID-19 Using Extensive Phenotypic Data: A Proof-of-Concept Study in a Cohort of Russian Patients. Genes 2022, 13, 534. https://doi.org/10.3390/genes13030534
Shcherbak SG, Changalidi AI, Barbitoff YA, Anisenkova AY, Mosenko SV, Asaulenko ZP, Tsay VV, Polev DE, Kalinin RS, Eismont YA, et al. Identification of Genetic Risk Factors of Severe COVID-19 Using Extensive Phenotypic Data: A Proof-of-Concept Study in a Cohort of Russian Patients. Genes. 2022; 13(3):534. https://doi.org/10.3390/genes13030534
Chicago/Turabian StyleShcherbak, Sergey G., Anton I. Changalidi, Yury A. Barbitoff, Anna Yu. Anisenkova, Sergei V. Mosenko, Zakhar P. Asaulenko, Victoria V. Tsay, Dmitrii E. Polev, Roman S. Kalinin, Yuri A. Eismont, and et al. 2022. "Identification of Genetic Risk Factors of Severe COVID-19 Using Extensive Phenotypic Data: A Proof-of-Concept Study in a Cohort of Russian Patients" Genes 13, no. 3: 534. https://doi.org/10.3390/genes13030534
APA StyleShcherbak, S. G., Changalidi, A. I., Barbitoff, Y. A., Anisenkova, A. Y., Mosenko, S. V., Asaulenko, Z. P., Tsay, V. V., Polev, D. E., Kalinin, R. S., Eismont, Y. A., Glotov, A. S., Garbuzov, E. Y., Chernov, A. N., Klitsenko, O. A., Ushakov, M. O., Shikov, A. E., Urazov, S. P., Baranov, V. S., & Glotov, O. S. (2022). Identification of Genetic Risk Factors of Severe COVID-19 Using Extensive Phenotypic Data: A Proof-of-Concept Study in a Cohort of Russian Patients. Genes, 13(3), 534. https://doi.org/10.3390/genes13030534