
Network Meta-Analysis of Chicken Microarray Data following Avian Influenza Challenge—A Comparison of Highly and Lowly Pathogenic Strains



Abstract
:1. Introduction
2. Materials and Methods
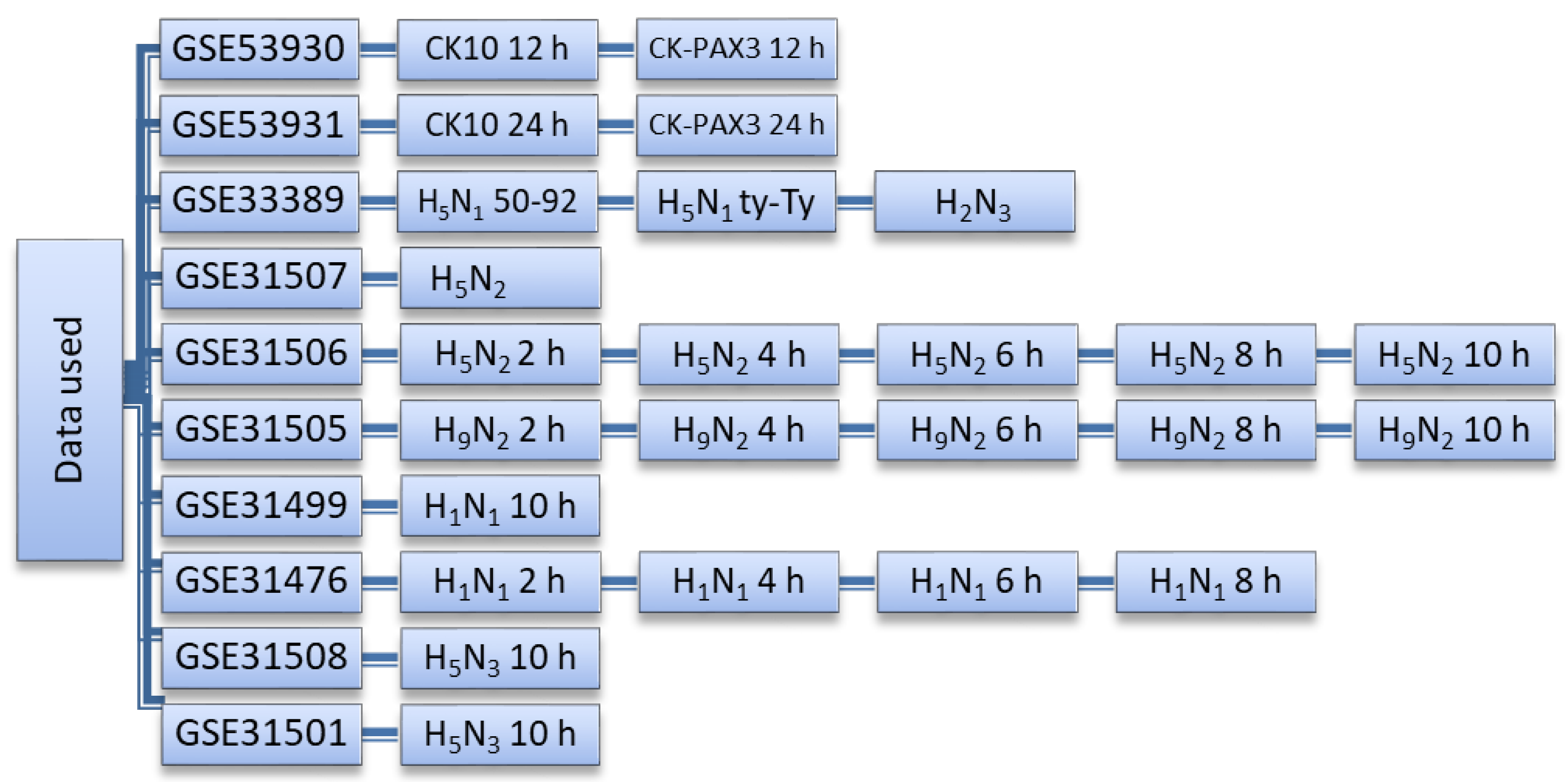
2.1. Datasets Used in This Study
2.2. Normalization of Microarray Data
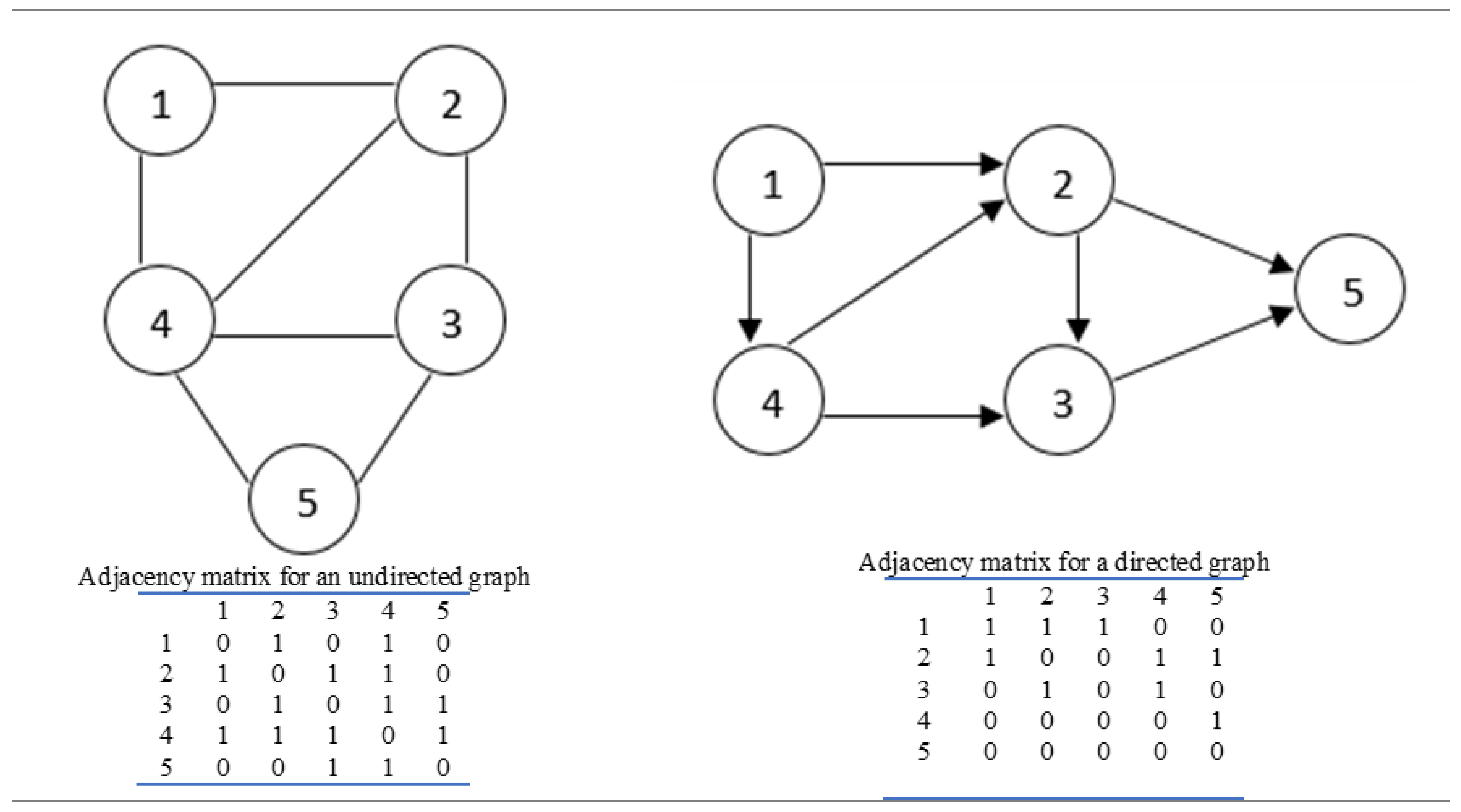
2.3. Network Construction
2.4. Derivation of Network Metrics
2.5. Downstream Analysis of Network Genes
3. Results
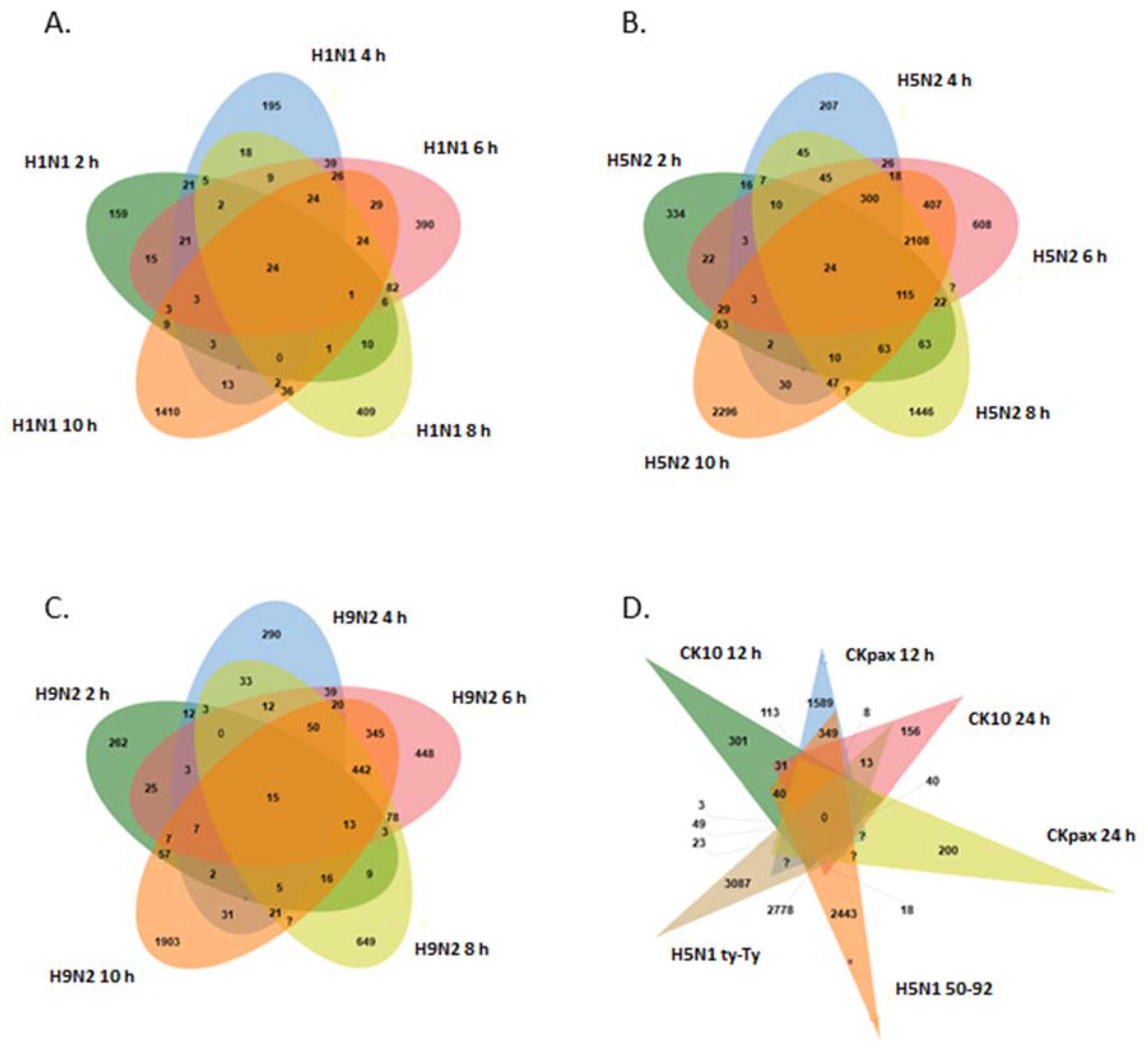
3.1. Analysis of LPAI Datasets
3.2. Analysis of HPAI Datasets
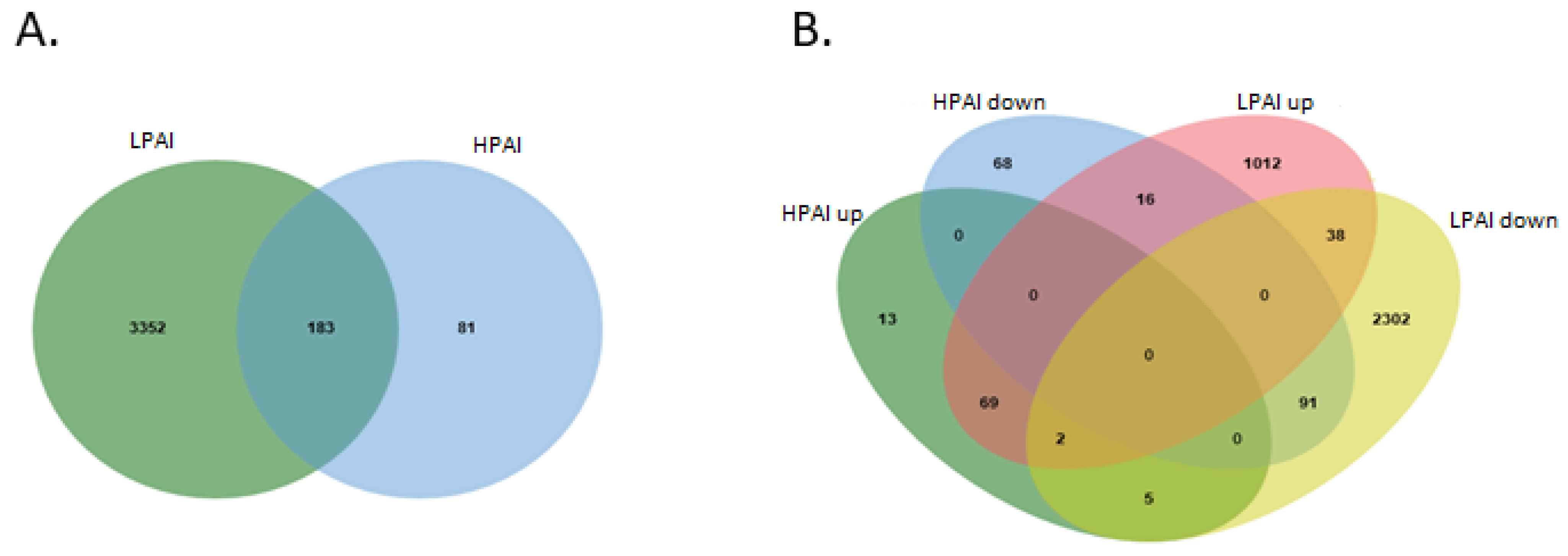
3.3. Comparison of LPAI and HPAI Datasets
3.4. Analysis of All Up- and All Down-Regulated Genes
3.5. Gene Ontology
3.6. Pathway Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reperant, L.A.; Kuiken, T.; Osterhaus, A.D. Adaptive pathways of zoonotic influenza viruses: From exposure to establishment in humans. Vaccine 2012, 30, 4419–4434. [Google Scholar] [CrossRef] [Green Version]
- Katz, J.M.; Veguilla, V.; Belser, J.A.; Maines, T.R.; Van Hoeven, N.; Pappas, C.; Hancock, K.; Tumpey, T.M. The public health impact of avian influenza viruses. Poult. Sci. 2009, 88, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, A.J.; Armstrong, J.S.; Downie, J.C. From where did the 2009 ‘swine-origin’ influenza A virus (H1N1) emerge? Virol. J. 2009, 6, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.; Chen, L.M.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taye, B.; Yeo, D.; Lee, R.T.C.; Tan, B.H.; Sugrue, R.J.; Maurer-Stroh, S. Inter-Species Host Gene Expression Differences in Response to Human and Avian Influenza A Virus Strains. Int. J. Mol. Sci. 2017, 18, 2295. [Google Scholar] [CrossRef] [Green Version]
- Zamperin, G.; Bianco, A.; Smith, J.; Bortolami, A.; Vervelde, L.; Schivo, A.; Fortin, A.; Marciano, S.; Panzarin, V.; Mazzetto, E.; et al. Heterogeneity of Early Host Response to Infection with Four Low-Pathogenic H7 Viruses with a Different Evolutionary History in the Field. Viruses 2021, 13, 2323. [Google Scholar] [CrossRef] [PubMed]
- Hancock, K.; Veguilla, V.; Lu, X.; Zhong, W.; Butler, E.N.; Sun, H.; Liu, F.; Dong, L.; DeVos, J.R.; Gargiullo, P.M.; et al. Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N. Engl. J. Med. 2009, 361, 1945–1952. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Shinya, K.; Kiso, M.; Watanabe, T.; Sakoda, Y.; Hatta, M.; Muramoto, Y.; Tamura, D.; Sakai-Tagawa, Y.; Noda, T.; et al. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 2009, 460, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Belshe, R.B. The origins of pandemic influenza-lessons from the 1918 virus. N. Engl. J. Med. 2005, 353, 2209–2211. [Google Scholar] [CrossRef] [PubMed]
- Munster, V.J.; de Wit, E.; van den Brand, J.M.; Herfst, S.; Schrauwen, E.J.; Bestebroer, T.M.; van de Vijver, D.; Boucher, C.A.; Koopmans, M.; Rimmelzwaan, G.F.; et al. Pathogenesis and transmission of swine-origin 2009 A(H1N1) influenza virus in ferrets. Science 2009, 325, 481–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchipudi, S.V.; Tellabati, M.; Sebastian, S.; Londt, B.Z.; Jansen, C.; Vervelde, L.; Brookes, S.M.; Brown, I.H.; Dunham, S.P.; Chang, K.C. Highly pathogenic avian influenza virus infection in chickens but not ducks is associated with elevated host immune and pro-inflammatory responses. Vet. Res. 2014, 45, 118. [Google Scholar] [CrossRef] [Green Version]
- Thitithanyanont, A.; Engering, A.; Uiprasertkul, M.; Ekchariyawat, P.; Wiboon-Ut, S.; Kraivong, R.; Limsalakpetch, A.; Kum-Arb, U.; Yongvanitchit, K.; Sa-Ard-Iam, N.; et al. Antiviral immune responses in H5N1-infected human lung tissue and possible mechanisms underlying the hyperproduction of interferon-inducible protein IP-10. Biochem. Biophys. Res. Commun. 2010, 398, 752–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, G.B.; Kawaoka, Y.; Jones, D.J.; Bean, W.J.; Pryor, S.P.; Hinshaw, V.; Webster, R.G. Coinfection of wild ducks by influenza A viruses: Distribution patterns and biological significance. J. Virol. 1997, 71, 6128–6135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isoda, N.; Sakoda, Y.; Kishida, N.; Bai, G.R.; Matsuda, K.; Umemura, T.; Kida, H. Pathogenicity of a highly pathogenic avian influenza virus, A/chicken/Yamaguchi/7/04 (H5N1) in different species of birds and mammals. Arch. Virol. 2006, 151, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Perkins, L.E.; Swayne, D.E. Pathogenicity of a Hong Kong-origin H5N1 highly pathogenic avian influenza virus for emus, geese, ducks, and pigeons. Avian Dis. 2002, 46, 53–63. [Google Scholar] [CrossRef]
- Jeong, O.M.; Kim, M.C.; Kim, M.J.; Kang, H.M.; Kim, H.R.; Kim, Y.J.; Joh, S.J.; Kwon, J.H.; Lee, Y.J. Experimental infection of chickens, ducks and quails with the highly pathogenic H5N1 avian influenza virus. J. Vet. Sci. 2009, 10, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Watanabe, C.; Takemae, N.; Chaisingh, A.; Uchida, Y.; Buranathai, C.; Suzuki, H.; Okamatsu, M.; Imada, T.; Parchariyanon, S.; et al. Pathogenicity of highly pathogenic avian influenza viruses of H5N1 subtype isolated in Thailand for different poultry species. Vet. Microbiol. 2009, 133, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Shang, Y.H.; Yao, H.X.; Cui, B.A.; Zhang, H.Y.; Wang, Z.X.; Wang, Y.D.; Chao, A.J.; Duan, T.Y. Immune responses of chickens inoculated with a recombinant fowlpox vaccine coexpressing HA of H9N2 avain influenza virus and chicken IL-18. Antivir. Res. 2011, 91, 50–56. [Google Scholar] [CrossRef]
- Wibawa, H.; Henning, J.; Wong, F.; Selleck, P.; Junaidi, A.; Bingham, J.; Daniels, P.; Meers, J. A molecular and antigenic survey of H5N1 highly pathogenic avian influenza virus isolates from smallholder duck farms in Central Java, Indonesia during 2007–2008. Virol. J. 2011, 8, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; MacDonald, L.A.; Takimoto, T. Influenza A Virus Protein PA-X Contributes to Viral Growth and Suppression of the Host Antiviral and Immune Responses. J. Virol. 2015, 89, 6442–6452. [Google Scholar] [CrossRef] [Green Version]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.A.; Wills, N.M.; Xiao, Y.L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Mo, Y.; Wang, X.; Gu, M.; Hu, Z.; Zhong, L.; Wu, Q.; Hao, X.; Hu, S.; Liu, W.; et al. PA-X decreases the pathogenicity of highly pathogenic H5N1 influenza A virus in avian species by inhibiting virus replication and host response. J. Virol. 2015, 89, 4126–4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaucherand, L.; Porter, B.K.; Levene, R.E.; Price, E.L.; Schmaling, S.K.; Rycroft, C.H.; Kevorkian, Y.; McCormick, C.; Khaperskyy, D.A.; Gaglia, M.M. The Influenza A Virus Endoribonuclease PA-X Usurps Host mRNA Processing Machinery to Limit Host Gene Expression. Cell Rep. 2019, 27, 776–792.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, M.B.; Dawson, E.D.; Mehlmann, M.; Smagala, J.A.; Dankbar, D.M.; Moore, C.L.; Smith, C.B.; Cox, N.J.; Kuchta, R.D.; Rowlen, K.L. Experimental evaluation of the FluChip diagnostic microarray for influenza virus surveillance. J. Clin. Microbiol. 2006, 44, 2863–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, R.; Barabási, A.-L. Statistical mechanics of complex networks. Rev. Mod. Phys. 2002, 74, 47. [Google Scholar] [CrossRef] [Green Version]
- Schnitzler, A.; Gross, J. Normal and pathological oscillatory communication in the brain. Nat. Rev. Neurosci. 2005, 6, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitling, R.; Armengaud, P.; Amtmann, A.; Herzyk, P. Rank products: A simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004, 573, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Breitling, R.; McEntee, C.W.; Wittner, B.S.; Nemhauser, J.L.; Chory, J. RankProd: A bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 2006, 22, 2825–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.; Smith, N.; Yu, L.; Paton, I.R.; Gutowska, M.W.; Forrest, H.L.; Danner, A.F.; Seiler, J.P.; Digard, P.; Webster, R.G.; et al. A comparative analysis of host responses to avian influenza infection in ducks and chickens highlights a role for the interferon-induced transmembrane proteins in viral resistance. BMC Genom. 2015, 16, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujioka, Y.; Tsuda, M.; Nanbo, A.; Hattori, T.; Sasaki, J.; Sasaki, T.; Miyazaki, T.; Ohba, Y. A Ca2+-dependent signalling circuit regulates influenza A virus internalization and infection. Nat. Commun. 2013, 4, 2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, L.; Leymarie, O.; Chevalier, C.; Esnault, E.; Moroldo, M.; Da Costa, B.; Georgeault, S.; Roingeard, P.; Delmas, B.; Quéré, P.; et al. Transcriptomic profiling of a chicken lung epithelial cell line (CLEC213) reveals a mitochondrial respiratory chain activity boost during influenza virus infection. PLoS ONE 2017, 12, e0176355. [Google Scholar] [CrossRef] [PubMed]
- Ozgür, A.; Vu, T.; Erkan, G.; Radev, D.R. Identifying gene-disease associations using centrality on a literature mined gene-interaction network. Bioinformatics 2008, 24, 277–285. [Google Scholar] [CrossRef]
- Zhuang, D.Y.; Jiang, L.; He, Q.Q.; Zhou, P.; Yue, T. Identification of hub subnetwork based on topological features of genes in breast cancer. Int. J. Mol. Med. 2015, 35, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Hamelin, J.I.; Dall’Asta, L.; Barrat, A.; Vespignani, A. K-core decomposition of internet graphs: Hierarchies, self-similarity and measurement biases. Netw. Heterog. Media 2008, 3, 371–393. [Google Scholar] [CrossRef]
- Tiku, V.; Tan, M.W.; Dikic, I. Mitochondrial Functions in Infection and Immunity. Trends Cell Biol. 2020, 30, 263–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Lee, J.R.; Hahn, M.J. Regulation of inflammatory gene expression in macrophages by epithelial-stromal interaction 1 (Epsti1). Biochem. Biophys. Res. Commun. 2018, 496, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Clohisey, S.M.; Chia, B.S.; Wang, B.; Cui, A.; Eisenhaure, T.; Schweitzer, L.D.; Hoover, P.; Parkinson, N.J.; Nachshon, A.; et al. Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nat. Commun. 2020, 11, 164. [Google Scholar] [CrossRef] [Green Version]
- Dahlin, A.; Denny, J.; Roden, D.M.; Brilliant, M.H.; Ingram, C.; Kitchner, T.E.; Linneman, J.G.; Shaffer, C.M.; Weeke, P.; Xu, H.; et al. CMTR1 is associated with increased asthma exacerbations in patients taking inhaled corticosteroids. Immun. Inflamm. Dis. 2015, 3, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Louer, E.M.M.; Günzel, D.; Rosenthal, R.; Carmone, C.; Yi, G.; Stunnenberg, H.G.; den Hollander, A.I.; Deen, P.M.T. Differential day-night expression of tight junction components in murine retinal pigment epithelium. Exp. Eye Res. 2020, 193, 107985. [Google Scholar] [CrossRef] [PubMed]
- Higgins, S.E.; Ellestad, L.E.; Trakooljul, N.; McCarthy, F.; Saliba, J.; Cogburn, L.A.; Porter, T.E. Transcriptional and pathway analysis in the hypothalamus of newly hatched chicks during fasting and delayed feeding. BMC Genom. 2010, 11, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, F.; Liu, X.; Han, Z.; Shao, Y.; Kong, X.; Liu, S. Transcriptome analysis of chicken kidney tissues following coronavirus avian infectious bronchitis virus infection. BMC Genom. 2013, 14, 743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.H.; Dinh, H.; Lillehoj, H.S.; Song, K.D.; Oh, J.D. Differential regulation of microRNA transcriptome in chicken lines resistant and susceptible to necrotic enteritis disease. Poult. Sci. 2014, 93, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Carrillo, J.A.; Menendez, K.R.; Tablante, N.L.; Song, J. Transcriptome analysis reveals an activation of major histocompatibility complex 1 and 2 pathways in chicken trachea immunized with infectious laryngotracheitis virus vaccine. Poult. Sci. 2014, 93, 848–855. [Google Scholar] [CrossRef]
- Coble, D.J.; Fleming, D.; Persia, M.E.; Ashwell, C.M.; Rothschild, M.F.; Schmidt, C.J.; Lamont, S.J. RNA-seq analysis of broiler liver transcriptome reveals novel responses to high ambient temperature. BMC Genom. 2014, 15, 1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.K.; Lillehoj, H.S.; Jang, S.I.; Lee, S.H.; Hong, Y.H.; Cheng, H.H. Transcriptional profiles of host-pathogen responses to necrotic enteritis and differential regulation of immune genes in two inbreed chicken lines showing disparate disease susceptibility. PLoS ONE 2014, 9, e114960. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Liu, P.; Nolan, L.K.; Lamont, S.J. Thymus transcriptome reveals novel pathways in response to avian pathogenic Escherichia coli infection. Poult. Sci. 2016, 95, 2803–2814. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Xu, J.; He, X.; Xu, H.; Li, G.; Du, H.; Nie, Q.; Zhang, X. A Genome-Wide mRNA Screen and Functional Analysis Reveal FOXO3 as a Candidate Gene for Chicken Growth. PLoS ONE 2015, 10, e0137087. [Google Scholar] [CrossRef]
- Tariq, M.; Chen, R.; Yuan, H.; Liu, Y.; Wu, Y.; Wang, J.; Xia, C. De novo transcriptomic analysis of peripheral blood lymphocytes from the Chinese goose: Gene discovery and immune system pathway description. PLoS ONE 2015, 10, e0121015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naraballobh, W.; Trakooljul, N.; Muráni, E.; Brunner, R.; Krischek, C.; Janisch, S.; Wicke, M.; Ponsuksili, S.; Wimmers, K. Immediate and long-term transcriptional response of hind muscle tissue to transient variation of incubation temperature in broilers. BMC Genom. 2016, 17, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, X.; Hsieh, J.C.; Schmidt, C.J.; Zhu, Q.; Lamont, S.J. Liver transcriptome response to hyperthermic stress in three distinct chicken lines. BMC Genom. 2016, 17, 955. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, X.; Zhang, Z.; Li, J.; Zou, W.; Zeng, F.; Wang, H. Comparative transcriptome analysis reveals induction of apoptosis in chicken kidney cells associated with the virulence of nephropathogenic infectious bronchitis virus. Microb. Pathog. 2017, 113, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Xu, L.; Wang, H.; Guo, X.; Li, Z.; Wan, F.; Chen, J.; Liu, L.; Liu, X.; Chang, G.; et al. Identification of the crucial genes in the elimination and survival process of Salmonella enterica ser. Pullorum in the chicken spleen. Anim. Genet. 2017, 48, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Q.; Liu, Z.; Guo, X.; Du, Y.; Yuan, Z.; Guo, M.; Kang, L.; Sun, Y.; Jiang, Y. Transcriptome Analysis on Single Small Yellow Follicles Reveals That Wnt4 Is Involved in Chicken Follicle Selection. Front. Endocrinol. 2017, 8, 317. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Yang, X.; Wang, D.; Zhu, F.; Yang, N.; Hou, Z.; Ning, Z. Thyroid transcriptome analysis reveals different adaptive responses to cold environmental conditions between two chicken breeds. PLoS ONE 2018, 13, e0191096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Dai, G.; Chen, F.; Chen, L.; Zhang, T.; Xie, K.; Wang, J.; Zhang, G. Transcriptome profile analysis of leg muscle tissues between slow- and fast-growing chickens. PLoS ONE 2018, 13, e0206131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, L.; Chang, G.; Li, Z.; Bi, Y.; Liu, X.; Chen, G. Comprehensive Transcriptome Analysis Reveals Competing Endogenous RNA Networks During Avian Leukosis Virus, Subgroup J-Induced Tumorigenesis in Chickens. Front. Physiol. 2018, 9, 996. [Google Scholar] [CrossRef]
- Freeman, L.C. A set of measures of centrality based on betweenness. Sociometry 1977, 40, 35–41. [Google Scholar] [CrossRef]
- Freeman, L.C. Centrality in social networks conceptual clarification. Soc. Netw. 1978, 1, 215–239. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.E. The structure and function of complex networks. SIAM Rev. 2003, 45, 167–256. [Google Scholar] [CrossRef] [Green Version]
- Rochat, Y. Closeness Centrality Extended to Unconnected Graphs: The Harmonic Centrality Index; ASNA: Zurich, Switzerland, 2009; pp. 26–28. [Google Scholar]
- Maccari, L.; Ghiro, L.; Guerrieri, A.; Montresor, A.; Cigno, R.L. On the Distributed Computation of Load Centrality and its Application to DV Routing. In Proceedings of the IEEE INFOCOM 2018—IEEE Conference on Computer Communications, Honolulu, HI, USA, 15–19 April 2018; pp. 2582–2590. [Google Scholar] [CrossRef] [Green Version]
- Estrada, E.; Rodríguez-Velázquez, J.A. Subgraph centrality in complex networks. Phys. Rev. E 2005, 71, 056103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saramäki, J.; Kivelä, M.; Onnela, J.P.; Kaski, K.; Kertész, J. Generalizations of the clustering coefficient to weighted complex networks. Phys. Rev. E 2007, 75, 027105. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Python Network for LPAI Microarray Data | ||
|---|---|---|
| Regulation | Number of Nodes | Number of Edges |
| up-regulation | 1842 | 60,606 |
| down-regulation | 4162 | 243,604 |
| Python Network for HPAI Microarray Data | ||
| Regulation | Number of Nodes | Number of Edges |
| up-regulation | 305 | 114 |
| down-regulation | 1813 | 276 |
| LPAI: Up-Regulated Genes | ||||||
|---|---|---|---|---|---|---|
| Gene | Number of Connections | Degree | Harmonic | Closeness | Eigenvector | Subgraph |
| RBM18 | 384 | 0.37 | 647.81 | 0.5 | 0.07 | 1.21 × 10103 |
| NDUFB1 | 375 | 0.36 | 639.53 | 0.49 | 0.07 | 1.17 × 10103 |
| DCTN3 | 372 | 0.36 | 641.19 | 0.5 | 0.07 | 1.16 × 10103 |
| COX7C | 370 | 0.36 | 639.55 | 0.49 | 0.06 | 1.03 × 10103 |
| IMMP2L | 368 | 0.36 | 638.38 | 0.49 | 0.07 | 1.16 × 10103 |
| ZDHHC9 | 368 | 0.36 | 639.43 | 0.5 | 0.07 | 1.11 × 10103 |
| LPAI: Down-Regulated Genes | ||||||
| Gene | Number of Connections | Degree | Harmonic | Closeness | Eigenvector | Subgraph |
| GTF3C5 | 849 | 0.36 | 1491.63 | 0.52 | 0.06 | 8.47 × 10184 |
| RFWD2 | 840 | 0.36 | 1493.43 | 0.52 | 0.05 | 8.04 × 10184 |
| MED23 | 828 | 0.35 | 1486.05 | 0.52 | 0.05 | 7.78 × 10184 |
| SEC23B | 798 | 0.34 | 1469.68 | 0.52 | 0.05 | 7.52 × 10184 |
| ATRX | 796 | 0.34 | 1450.85 | 0.5 | 0.05 | 6.77 × 10184 |
| DROSHA | 794 | 0.34 | 1456.03 | 0.5 | 0.05 | 7.31 × 10184 |
| HPAI: Up-Regulated Genes | ||||||
| Gene | Number of Connections | Degree | Harmonic | Closeness | Eigenvector | Subgraph |
| CMTR1 | 27 | 0.52 | 37.08 | 0.61 | 0.26 | 2.23 × 106 |
| HERC4L | 24 | 0.47 | 35.58 | 0.58 | 0.24 | 1.91 × 106 |
| IFIT5 | 23 | 0.45 | 34.11 | 0.53 | 0.24 | 1.84 × 106 |
| LY96 | 23 | 0.45 | 34.03 | 0.52 | 0.21 | 1.45 × 106 |
| RNF213 | 22 | 0.43 | 33.61 | 0.52 | 0.24 | 1.78 × 106 |
| EPSTI1 | 22 | 0.43 | 33.53 | 0.52 | 0.22 | 1.56 × 106 |
| HPAI: Down-Regulated Genes | ||||||
| Gene | Number of Connections | Degree | Harmonic | Closeness | Eigenvector | Subgraph |
| PEX14 | 60 | 0.45 | 81.7 | 0.5 | 0.16 | 5.62 × 1017 |
| HMG20A | 59 | 0.44 | 81.2 | 0.5 | 0.16 | 5.53 × 1017 |
| TLK1L | 59 | 0.44 | 81.2 | 0.5 | 0.16 | 5.52 × 1017 |
| RNF151 | 59 | 0.44 | 81.78 | 0.51 | 0.16 | 5.43 × 1017 |
| PARD6G | 58 | 0.43 | 80.78 | 0.5 | 0.15 | 5.01 × 1017 |
| CAV2 | 55 | 0.41 | 79.2 | 0.49 | 0.15 | 4.88 × 1017 |
| Gene Symbol | Gene Name | Entrez Gene ID | HPAI | PAI | ||
|---|---|---|---|---|---|---|
| Log2 Fold Change | Regulation | Log2 Fold Change | Expression | |||
| CMTR1 | cap methyltransferase 1 | 14306 | 2.10 | UP | ||
| HERC4L | hect domain and RLD 4-like | 4297 | 2.05 | UP | ||
| IFIT5 | interferon induced protein with tetratricopeptide repeats 5 | 33635 | 2.64 | UP | ||
| LY96 | lymphocyte antigen 96 | 5508 | 1.91 | UP | ||
| RNF213 | ring finger protein 213 | 10972 | 2.06 | UP | ||
| EPSTI1 | epithelial stromal interaction 1 | 11241 | 2.01 | UP | ||
| RBM18 | RNA binding motif protein 18 | 7150 | 1.30 | UP | ||
| NDUFB1 | NADH:ubiquinone oxidoreductase subunit B1 | 4970 | 1.27 | UP | ||
| DCTN3 | dynactin subunit 3 | 4824 | 1.29 | UP | ||
| COX7C | cytochrome c oxidase subunit 7C | 4726 | 1.32 | UP | ||
| IMMP2L | inner mitochondrial membrane peptidase subunit 2 | 9006 | 1.27 | UP | ||
| ZDHHC9 | zinc finger DHHC-type containing 9 | 2532 | 1.19 | UP | ||
| PEX14 | peroxisomal biogenesis factor 14 | 6768 | −0.34 | down | ||
| HMG20A | high mobility group 20A | 20936 | −0.34 | down | ||
| TLK1L | tousled like kinase 1 like | 6751 | −0.27 | down | ||
| RNF151 | ring finger protein 151 | 9782 | −0.36 | down | ||
| PARD6G | par-6 family cell polarity regulator gamma | 9912 | −0.42 | down | ||
| CAV2 | caveolin 2 | 9078 | −0.36 | down | ||
| GTF3C5 | general transcription factor IIIC subunit 5 | 21008 | −0.70 | down | ||
| RFWD2 | ring finger and WD repeat domain 2 | 37706 | −0.64 | down | ||
| MED23 | mediator complex subunit 23 | 8401 | −0.68 | down | ||
| SEC23B | Sec23 homolog B, coat complex II component | 21262 | −0.76 | down | ||
| ATRX | alpha thalassemia/mental retardation syndrome X-linked | 7476 | −0.68 | down | ||
| DROSHA | drosha ribonuclease III | 20908 | −0.70 | down | ||
| Gene Symbol | Gene Name | Probe ID | Number of Connections |
|---|---|---|---|
| SELENOK | selenoprotein K | Gga.1058.1.S1_at | 209 |
| NDUFA1 | NADH:ubiquinone oxidoreductase subunit A1 | Gga.5918.1.A1_a_at | 207 |
| PPP1R7 | protein phosphatase 1 regulatory subunit 7 | Gga.5583.1.S1_a_at | 202 |
| SMDT1 | single-pass membrane protein with aspartate rich tail 1 | Gga.9946.1.S1_at | 200 |
| COX7C | cytochrome c oxidase subunit 7C | Gga.6171.1.S1_a_at | 200 |
| PRELID3B | PRELI domain containing 3B | Gga.9900.2.S1_at | 199 |
| CIB1 | calcium and integrin binding 1 | Gga.5965.2.S1_a_at | 198 |
| OST4 | oligosaccharyltransferase complex subunit 4, non-catalytic | Gga.6184.1.S1_at | 196 |
| NDUFB2 | NADH:ubiquinone oxidoreductase subunit B2 | Gga.17299.1.S1_a_at | 196 |
| Gene Symbol | Gene Name | Probe ID | Number of Connections |
|---|---|---|---|
| PUS10 | pseudouridylate synthase 10 | GgaAffx.4897.1.S1_at | 509 |
| ERBIN | erbb2 interacting protein | Gga.17560.1.S1_at | 496 |
| SYDE2 | synapse defective Rho GTPase homolog 2 | Gga.11842.1.S1_s_at | 492 |
| PCGF6 | polycomb group ring finger 6 | Gga.16959.1.S1_at | 490 |
| FZD6 | frizzled class receptor 6 | Gga.2690.1.S1_at | 489 |
| ROR1 | receptor tyrosine kinase like orphan receptor 1 | Gga.9476.1.S1_at | 485 |
| LRIG2 | leucine rich repeats and immunoglobulin like domains 2 | Gga.17165.1.S1_at | 479 |
| SUPT7L | SPT7 like, STAGA complex gamma subunit | Gga.16763.1.S1_at | 475 |
| EXOC8 | exocyst complex component 8 | Gga.14199.1.S1_at | 470 |
| KIF1C | kinesin family member 1C | Gga.15878.1.S1_s_at | 469 |
| PCM1 | pericentriolar material 1 | Gga.3449.1.S1_at | 468 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirbaluty, A.M.; Mehrban, H.; Kadkhodaei, S.; Ravash, R.; Oryan, A.; Ghaderi-Zefrehei, M.; Smith, J. Network Meta-Analysis of Chicken Microarray Data following Avian Influenza Challenge—A Comparison of Highly and Lowly Pathogenic Strains. Genes 2022, 13, 435. https://doi.org/10.3390/genes13030435
Pirbaluty AM, Mehrban H, Kadkhodaei S, Ravash R, Oryan A, Ghaderi-Zefrehei M, Smith J. Network Meta-Analysis of Chicken Microarray Data following Avian Influenza Challenge—A Comparison of Highly and Lowly Pathogenic Strains. Genes. 2022; 13(3):435. https://doi.org/10.3390/genes13030435
Chicago/Turabian StylePirbaluty, Azadeh Moradi, Hossein Mehrban, Saeid Kadkhodaei, Rudabeh Ravash, Ahmad Oryan, Mostafa Ghaderi-Zefrehei, and Jacqueline Smith. 2022. "Network Meta-Analysis of Chicken Microarray Data following Avian Influenza Challenge—A Comparison of Highly and Lowly Pathogenic Strains" Genes 13, no. 3: 435. https://doi.org/10.3390/genes13030435
APA StylePirbaluty, A. M., Mehrban, H., Kadkhodaei, S., Ravash, R., Oryan, A., Ghaderi-Zefrehei, M., & Smith, J. (2022). Network Meta-Analysis of Chicken Microarray Data following Avian Influenza Challenge—A Comparison of Highly and Lowly Pathogenic Strains. Genes, 13(3), 435. https://doi.org/10.3390/genes13030435