
Stability and Oligomerization of Mutated SMN Protein Determine Clinical Severity of Spinal Muscular Atrophy
 , ,
, ,  , , , , , , and
, , , , , , and Highlights
- Spinal muscular atrophy (SMA) is caused by mutations in the SMN1 gene. The location and type of mutation in the SMN1 gene product, SMN, are predicted to determine the severity of SMA. However, the mechanism by which the location and type of mutation determine the severity of the disease remains unclear.
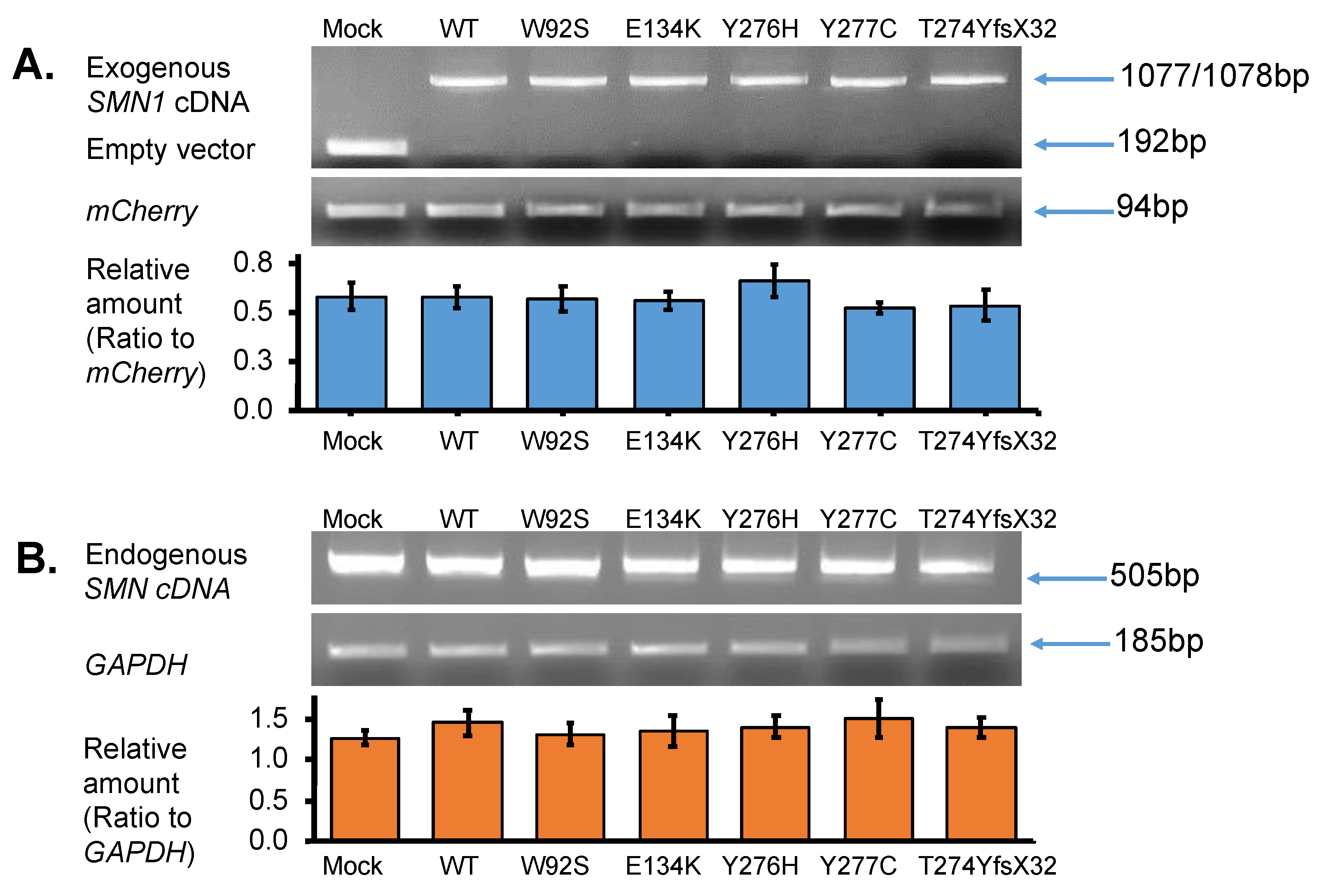
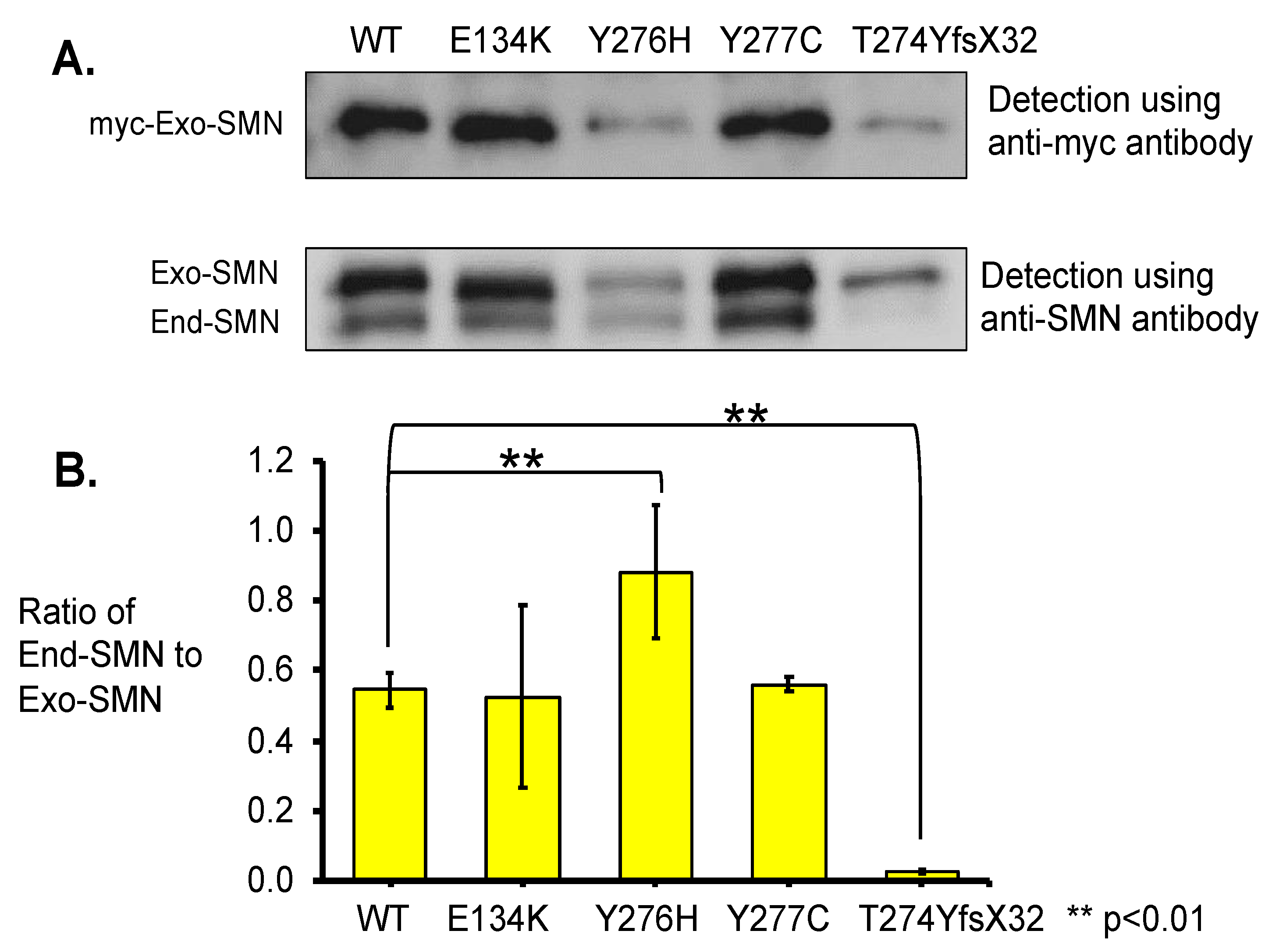
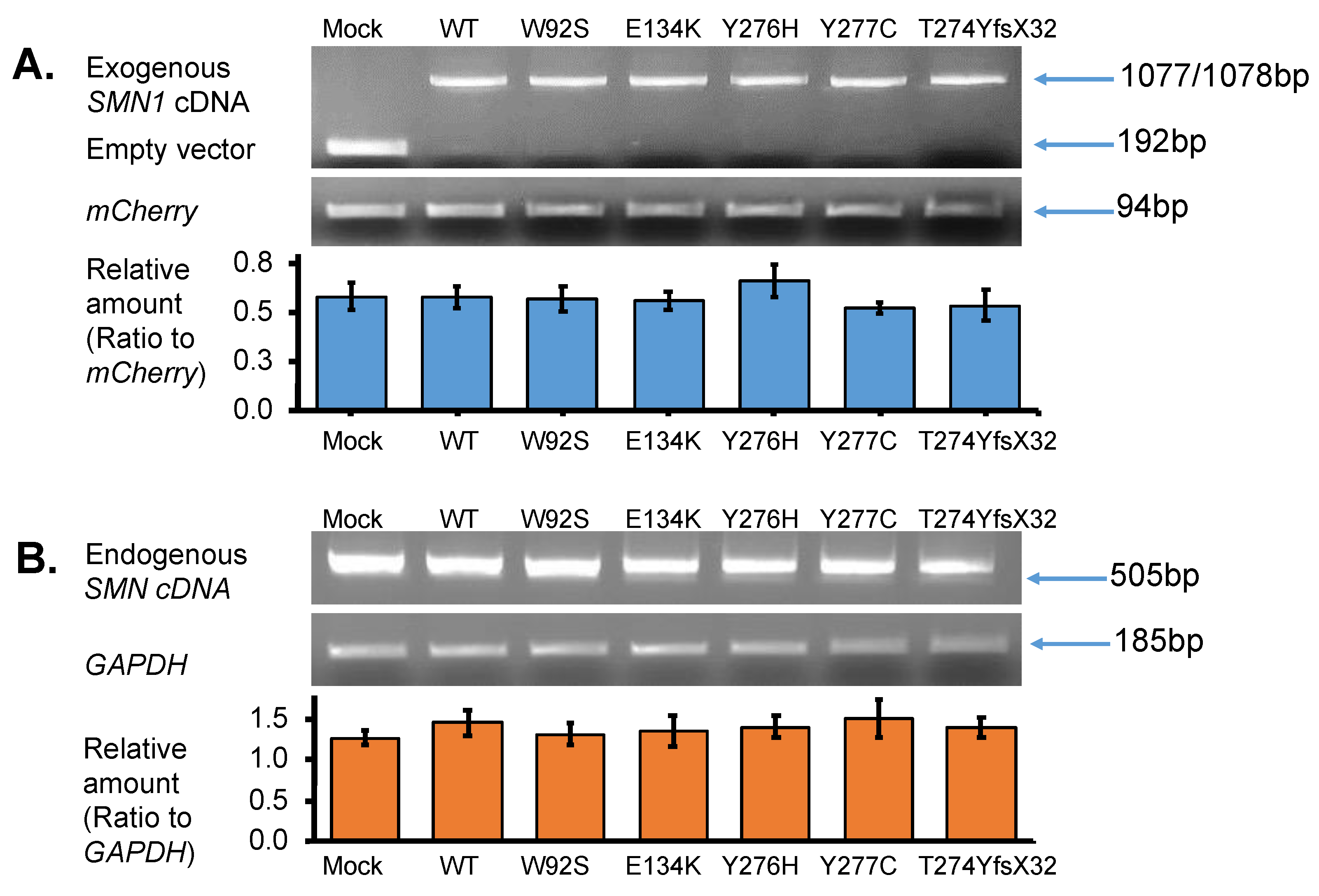
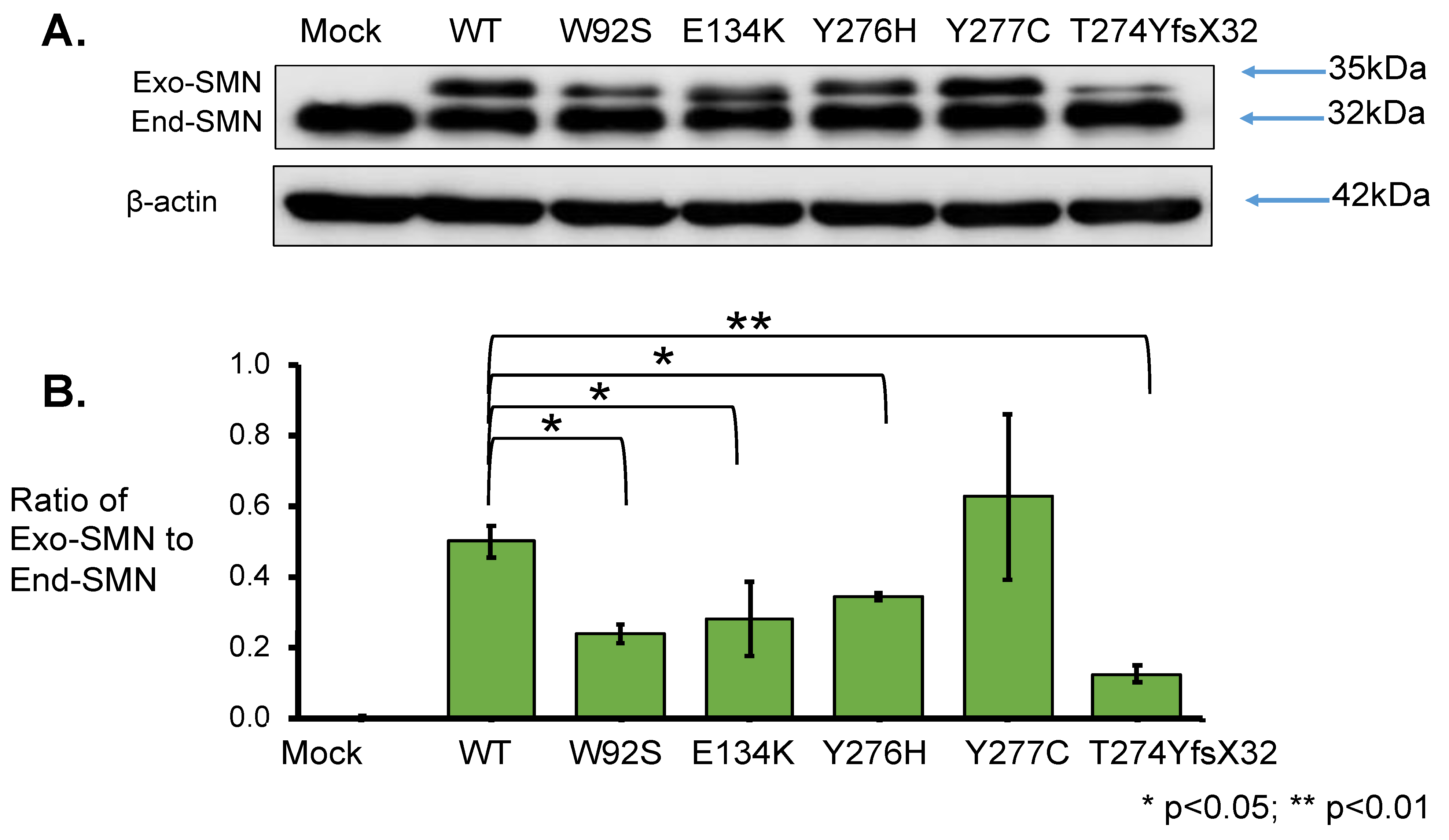
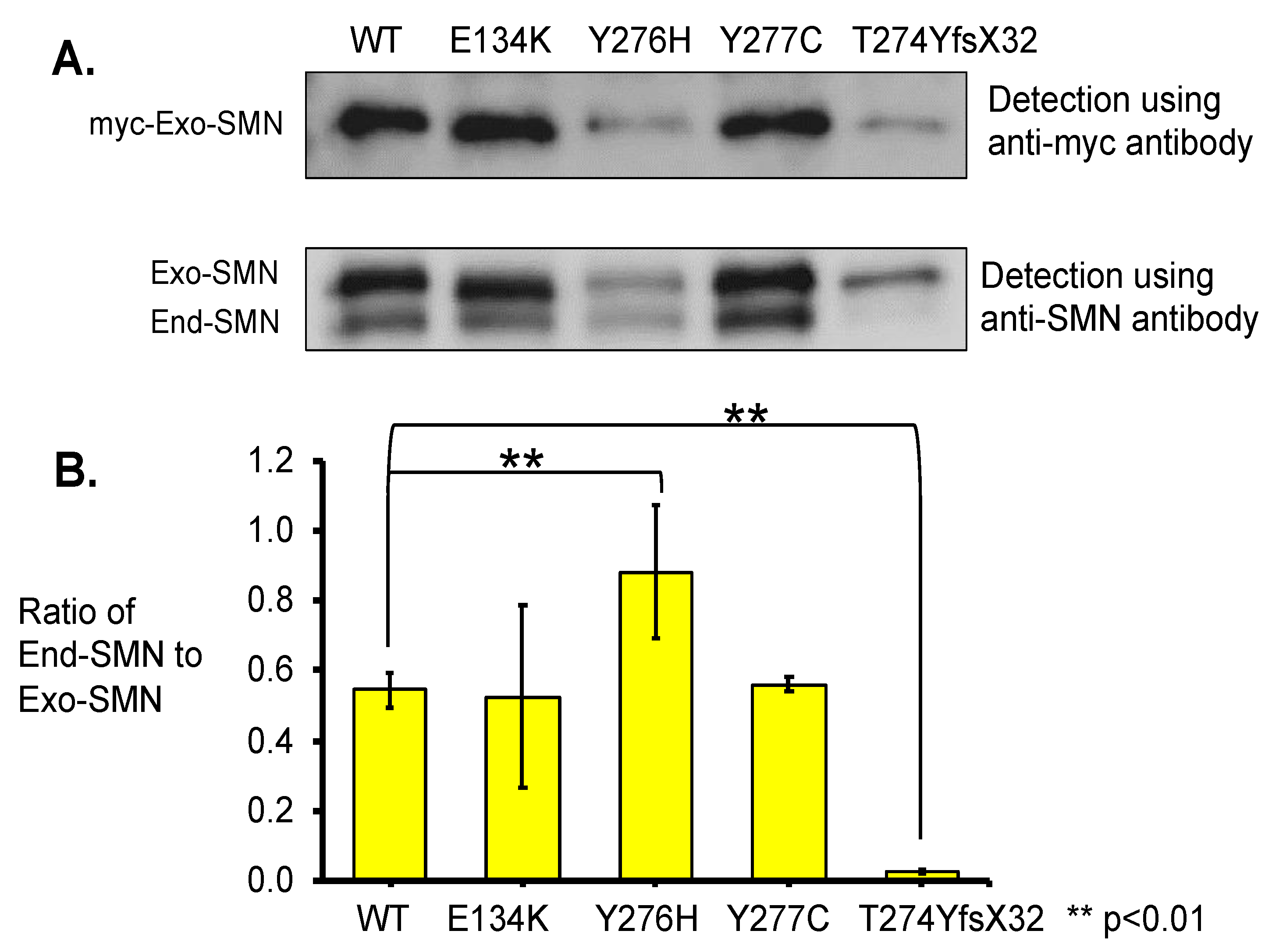
- In this study, we showed that the myc-SMN fusion protein (exogenous SMN protein) was expressed in HeLa cells transfected with plasmids carrying SMN1 cDNA with wild-type and mutant sequences (E134K, Y276H, Y277C, and T274YfsX32). In the experiment, the transcription levels of all plasmids were similar. However, the exogenous SMN protein containing T274YfsX32 showed much lower expression levels than other exogenous SMN1 proteins containing wild-type sequences, E134K, Y276H, and Y277C. This result suggests that protein stability is closely related to the position or type of mutation.
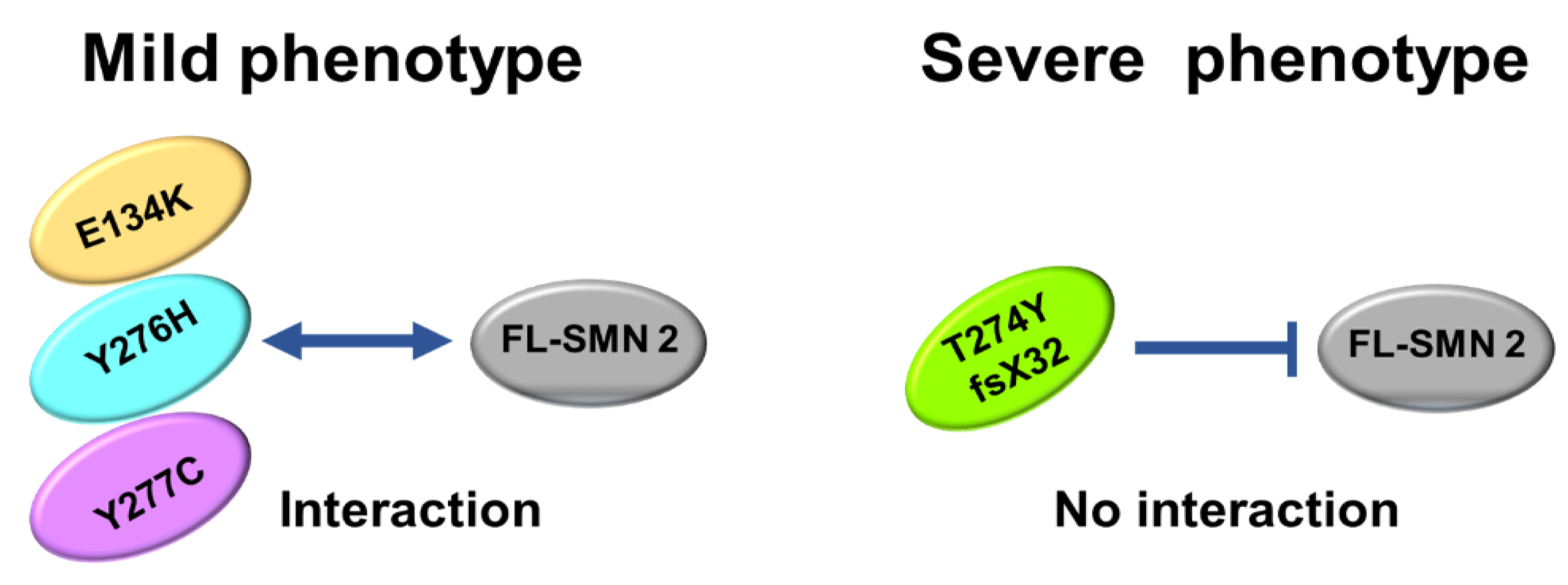
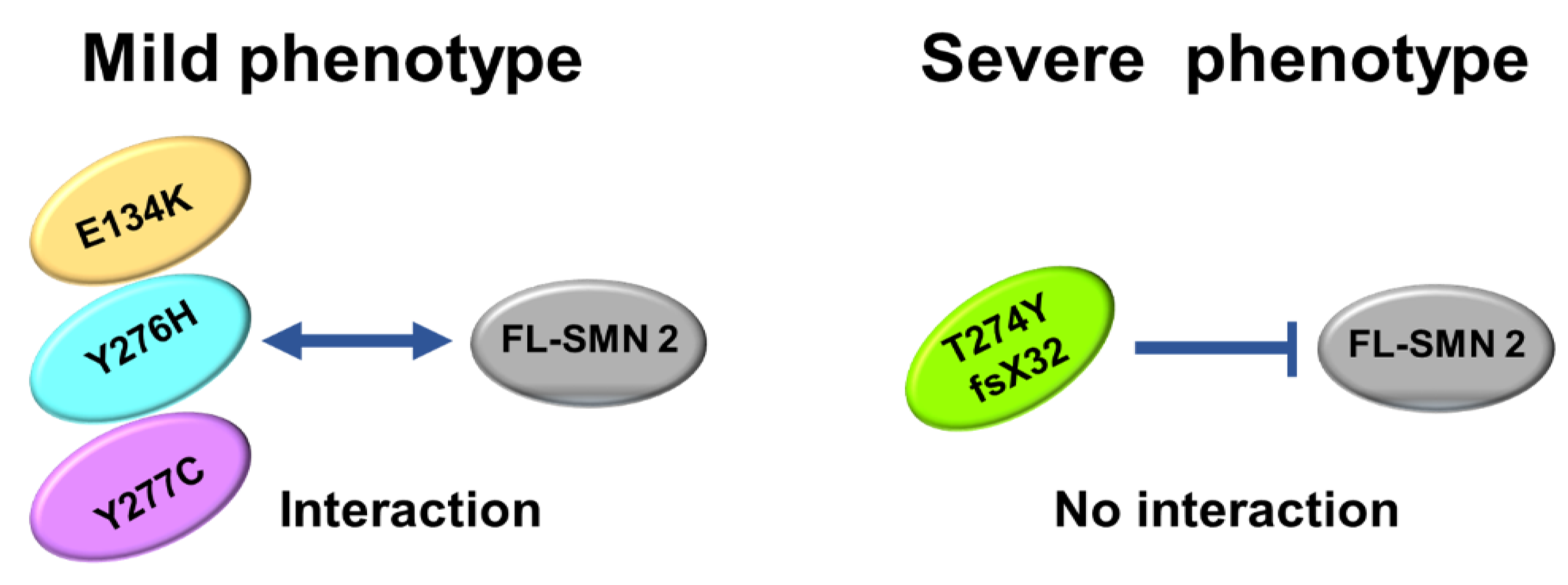
- Immunoprecipitation assay demonstrated that the exogenous SMN protein containing T274YfsX32 did not bind to the SMN protein already present in HeLa cells (endogenous SMN protein), indicating that this mutation prevented the formation of hetero-oligomers of exogenous and endogenous SMN proteins. However, exogenous SMN proteins containing E134K, Y276H, and Y277C bound to endogenous SMN protein to some extent, which means that they formed hetero-oligomers with endogenous SMN protein to some extent.
- Here, we analyzed four SMN1 mutations from different patients. T274YfsX32 was found in patients with the most severe phenotype, while the other mutations, E134K, Y276H, and Y277C, were found in patients with less severe phenotypes. The results suggest that we have found that stability and oligomerization of mutated SMN proteins impacts disease phenotype.
- When considering treatment for SMA, increasing the stability or promoting oligomerization of SMN protein will be used as an alternative goal to develop new drugs or re-position existing drugs for SMA in the laboratories.
Abstract
:1. Introduction
2. Materials and Methods
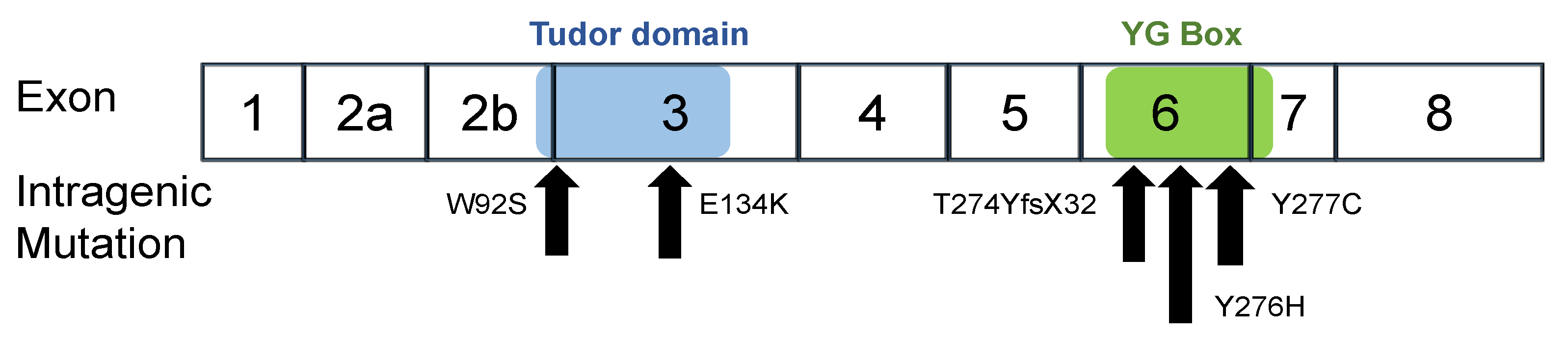
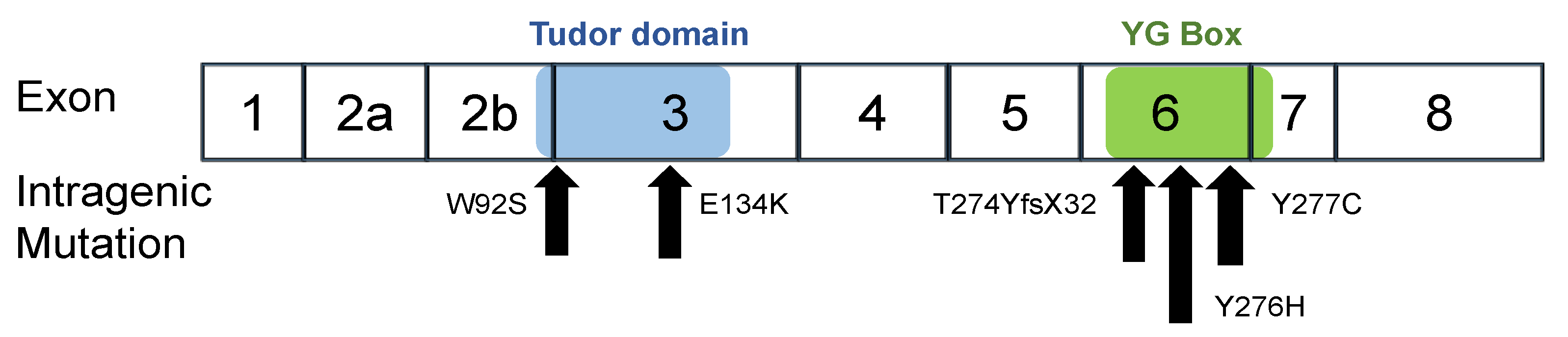
2.1. Patients and Their Intragenic SMN1 Mutations
2.2. Outline of the Experiments
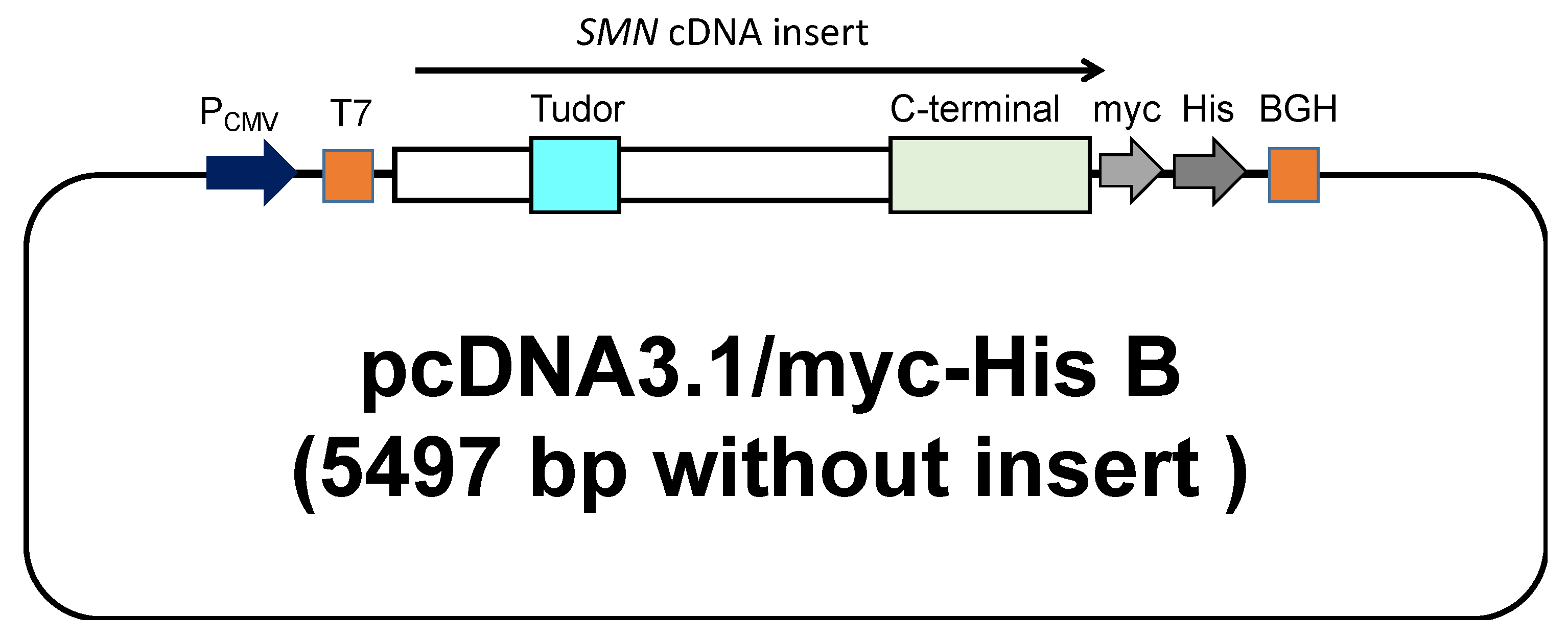
2.3. Plasmid Construction
2.4. Transfection into HeLa Cells
2.5. RNA Analysis by RT-PCR
2.6. SMN1 Protein Quantification by Western Blotting
2.7. Pull-Down Assay with Immunoprecipitation
2.8. Statistics
3. Results
3.1. Plasmid Construction and Transfection
3.1.1. Plasmid Construct Carrying Exogenous SMN cDNA
3.1.2. Transfection of the Plasmid Containing SMN1 cDNA
3.2. SMN Transcript Analysis
3.2.1. Expression of Exogenous SMN Transcript in HeLa Cells
3.2.2. Expression of Endogenous SMN Transcript in HeLa Cells
3.2.3. Quantification of Exogenous and Endogenous SMN Transcripts by RT-PCR
3.3. SMN1 Protein Analysis
3.3.1. Western Blotting Analysis with Anti-SMN Antibody
3.3.2. Pull-Down Assay with Immunoprecipitation
4. Discussion
4.1. SMA Patients with an Intragenic Mutation in the Retaining SMN1 Allele
4.2. Stability of the Mutated SMN1 Proteins
4.3. Oligomerization of SMN Proteins
4.4. Clinical Phenotype of the Patients with an Intragenic Mutation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and Management of Spinal Muscular Atrophy: Part 1: Recommendations for Diagnosis, Rehabilitation, Orthopedic and Nutritional Care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurputra, D.K.; Lai, P.S.; Harahap, N.I.F.; Morikawa, S.; Yamamoto, T.; Nishimura, N.; Kubo, Y.; Takeuchi, A.; Saito, T.; Takeshima, Y.; et al. Spinal Muscular Atrophy: From Gene Discovery to Clinical Trials: SMA Gene Discovery to Clinical Trials. Ann. Hum. Genet. 2013, 77, 435–463. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmüller, H. Prevalence, Incidence and Carrier Frequency of 5q–Linked Spinal Muscular Atrophy—A Literature Review. Orphanet. J. Rare Dis. 2017, 12, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouzier, C.; Chaussenot, A.; Paquis-Flucklinger, V. Molecular Diagnosis and Genetic Counseling for Spinal Muscular Atrophy (SMA). Arch. Pédiatr. 2020, 27, 7S9–7S14. [Google Scholar] [CrossRef]
- Kimizu, T.; Ida, S.; Okamoto, K.; Awano, H.; Niba, E.T.E.; Wijaya, Y.O.S.; Okazaki, S.; Shimomura, H.; Lee, T.; Tominaga, K.; et al. Spinal Muscular Atrophy: Diagnosis, Incidence, and Newborn Screening in Japan. IJNS 2021, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.D.; Kassar, D.; Kissel, J.T. Spinal Muscular Atrophy: Diagnosis and Management in a New Therapeutic Era: Spinal Muscular Atrophy. Muscle Nerve 2015, 51, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Melki, J.; Abdelhak, S.; Sheth, P.; Bachelot, M.F.; Burlet, P.; Marcadet, A.; Aicardi, J.; Barois, A.; Carriere, J.P.; Fardeau, M.; et al. Gene for Chronic Proximal Spinal Muscular Atrophies Maps to Chromosome 5q. Nature 1990, 344, 767–768. [Google Scholar] [CrossRef]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A Single Nucleotide in the SMN Gene Regulates Splicing and Is Responsible for Spinal Muscular Atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorson, C.L. An Exonic Enhancer Is Required for Inclusion of an Essential Exon in the SMA-Determining Gene SMN. Hum. Mol. Genet. 2000, 9, 259–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, B.G.; Muñoz, E.; Tandon, A.; Kwon, D.Y.; Sumner, C.J.; Fischbeck, K.H. Regulation of SMN Protein Stability. Mol. Cell Biol. 2009, 29, 1107–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Dreyfuss, G. A Degron Created by SMN2 Exon 7 Skipping Is a Principal Contributor to Spinal Muscular Atrophy Severity. Genes Dev. 2010, 24, 438–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, B. An Update of the Mutation Spectrum of the Survival Motor Neuron Gene (SMN1) in Autosomal Recessive Spinal Muscular Atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- Sun, Y.; Grimmler, M.; Schwarzer, V.; Schoenen, F.; Fischer, U.; Wirth, B. Molecular and Functional Analysis of Intragenic SMN1 Mutations in Patients with Spinal Muscular Atrophy. Hum. Mutat. 2005, 25, 64–71. [Google Scholar] [CrossRef]
- Wijaya, Y.O.S.; Ar Rohmah, M.; Niba, E.T.E.; Morisada, N.; Noguchi, Y.; Hidaka, Y.; Ozasa, S.; Inoue, T.; Shimazu, T.; Takahashi, Y.; et al. Phenotypes of SMA Patients Retaining SMN1 with Intragenic Mutation. Brain Dev. 2021, 43, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Kirwin, S.M.; Vinette, K.M.B.; Gonzalez, I.L.; Abdulwahed, H.A.; Al-Sannaa, N.; Funanage, V.L. A Homozygous Double Mutation in SMN 1: A Complicated Genetic Diagnosis of SMA. Mol. Genet. Genom. Med. 2013, 1, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Cuscó, I.; López, E.; Soler-Botija, C.; Jesús Barceló, M.; Baiget, M.; Tizzano, E.F. A Genetic and Phenotypic Analysis in Spanish Spinal Muscular Atrophy Patients with c.399_402del AGAG, the Most Frequently Found Subtle Mutation in the SMN1 Gene: Genotype-phenotype analysis in SMN1. Hum. Mutat. 2003, 22, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Fraidakis, M.J.; Drunat, S.; Maisonobe, T.; Gerard, B.; Pradat, P.F.; Meininger, V.; Salachas, F. Genotype-Phenotype Relationship in 2 SMA III Patients with Novel Mutations in the Tudor Domain. Neurology 2012, 78, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Sato, H.; Lai, P.S.; Nurputra, D.K.; Harahap, N.I.F.; Morikawa, S.; Nishimura, N.; Kurashige, T.; Ohshita, T.; Nakajima, H.; et al. Intragenic Mutations in SMN1 May Contribute More Significantly to Clinical Severity than SMN2 Copy Numbers in Some Spinal Muscular Atrophy (SMA) Patients. Brain Dev. 2014, 36, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Kotani, T.; Sutomo, R.; Sasongko, T.H.; Sadewa, A.H.; Gunadi; Minato, T.; Fujii, E.; Endo, S.; Lee, M.J.; Ayaki, H.; et al. A Novel Mutation at the N-Terminal of SMN Tudor Domain Inhibits Its Interaction with Target Proteins. J. Neurol. 2007, 254, 624–630. [Google Scholar] [CrossRef]
- de Holanda Mendonça, R.; Matsui, C.; Polido, G.J.; Silva, A.M.S.; Kulikowski, L.; Torchio Dias, A.; Zanardo, E.A.; Solla, D.J.F.; Gurgel-Giannetti, J.; de Moura, A.C.M.L.; et al. Intragenic Variants in the SMN1 Gene Determine the Clinical Phenotype in 5q Spinal Muscular Atrophy. Neurol. Genet. 2020, 6, e505. [Google Scholar] [CrossRef]
- Takarada, T.; Ar Rochmah, M.; Harahap, N.I.F.; Shinohara, M.; Saito, T.; Saito, K.; Lai, P.S.; Bouike, Y.; Takeshima, Y.; Awano, H.; et al. SMA Mutations in SMN Tudor and C-Terminal Domains Destabilize the Protein. Brain Dev. 2017, 39, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Strasswimmer, J.; Yao, J.-M.; Baleja, J.D.; Hahnen, E.; Wirth, B.; Le, T.; Burghes, A.H.M.; Androphy, E.J. SMN Oligomerization Defect Correlates with Spinal Muscular Atrophy Severity. Nat. Genet. 1998, 19, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Pellizzoni, L.; Charroux, B.; Dreyfuss, G. SMN Mutants of Spinal Muscular Atrophy Patients Are Defective in Binding to SnRNP Proteins. Proc. Nat. Acad. Sci. USA 1999, 96, 11167–11172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, K.; Martin, R.; Sharp, R.; Sarachan, K.L.; Ninan, N.S.; Van Duyne, G.D. Oligomeric Properties of Survival Motor Neuron·Gemin2 Complexes. J. Biol. Chem. 2015, 290, 20185–20199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, L.; Battle, D.J.; Yong, J.; Gubitz, A.K.; Kolb, S.J.; Wang, J.; Dreyfuss, G. The Survival of Motor Neurons Protein Determines the Capacity for SnRNP Assembly: Biochemical Deficiency in Spinal Muscular Atrophy. Mol. Cell Biol. 2005, 25, 5543–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burghes, A.H.M.; Beattie, C.E. Spinal Muscular Atrophy: Why Do Low Levels of Survival Motor Neuron Protein Make Motor Neurons Sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ar Rochmah, M.; Awano, H.; Awaya, T.; Harahap, N.I.F.; Morisada, N.; Bouike, Y.; Saito, T.; Kubo, Y.; Saito, K.; Lai, P.S.; et al. Spinal Muscular Atrophy Carriers with Two SMN1 Copies. Brain Dev. 2017, 39, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Nishida, Y.; Maihara, T.; Sa’adah, N.; Harahap, N.I.F.; Nurputra, D.K.; Ar Rochmah, M.; Nishimura, N.; Saito, T.; Kubo, Y.; et al. Two Japanese Patients With SMA Type 1 Suggest That Axonal-SMN May Not Modify the Disease Severity. Pediatr. Neurol. 2015, 52, 638–641. [Google Scholar] [CrossRef]
- Shpargel, K.B.; Matera, A.G. Gemin Proteins Are Required for Efficient Assembly of Sm-Class Ribonucleoproteins. Proc. Nat. Acad. Sci. USA 2005, 102, 17372–17377. [Google Scholar] [CrossRef] [Green Version]
- Harahap, N.I.F.; Niba, E.T.E.; Ar Rochmah, M.; Wijaya, Y.O.S.; Saito, T.; Saito, K.; Awano, H.; Morioka, I.; Iijima, K.; Lai, P.S.; et al. Intron-Retained Transcripts of the Spinal Muscular Atrophy Genes, SMN1 and SMN2. Brain Dev. 2018, 40, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Bürglen, L.; Lefebvre, S.; Clermont, O.; Burlet, P.; Viollet, L.; Cruaud, C.; Munnich, A.; Melki, J. Structure and Organization of the Human Survival Motor Neurone (SMN) Gene. Genomics 1996, 32, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Rochmah, M.A.; Wijaya, Y.O.S.; Harahap, N.I.F.; Tode, C.; Takeuchi, A.; Ohuchi, K.; Shimazawa, M.; Hara, H.; Funato, M.; Saito, T.; et al. Phosphoethanolamine Elevation in Plasma of Spinal Muscular Atrophy Type 1 Patients. Kobe J. Med. Sci. 2020, 66, E1–E11. [Google Scholar] [PubMed]
- Martin, R.; Gupta, K.; Ninan, N.S.; Perry, K.; Van Duyne, G.D. The Survival Motor Neuron Protein Forms Soluble Glycine Zipper Oligomers. Structure 2012, 20, 1929–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, K.; Wen, Y.; Ninan, N.S.; Raimer, A.C.; Sharp, R.; Spring, A.M.; Sarachan, K.L.; Johnson, M.C.; Van Duyne, G.D.; Matera, A.G. Assembly of Higher-Order SMN Oligomers Is Essential for Metazoan Viability and Requires an Exposed Structural Motif Present in the YG Zipper Dimer. Nucleic Acids Res. 2021, 49, 7644–7664. [Google Scholar] [CrossRef]
- Coady, T.H.; Lorson, C.L. SMN in Spinal Muscular Atrophy and SnRNP Biogenesis: SMN in Spinal Muscular Atrophy and SnRNP Biogenesis. WIREs RNA 2011, 2, 546–564. [Google Scholar] [CrossRef]
- Lee, M.-S.; Lin, Y.-S.; Deng, Y.-F.; Hsu, W.-T.; Shen, C.-C.; Cheng, Y.-H.; Huang, Y.-T.; Li, C. Modulation of Alternative Splicing by Expression of Small Nuclear Ribonucleoprotein Polypeptide N. FEBS J. 2014, 281, 5194–5207. [Google Scholar] [CrossRef] [Green Version]
- Akten, B.; Kye, M.J.; Hao, L.T.; Wertz, M.H.; Singh, S.; Nie, D.; Huang, J.; Merianda, T.T.; Twiss, J.L.; Beattie, C.E.; et al. Interaction of Survival of Motor Neuron (SMN) and HuD Proteins with MRNA Cpg15 Rescues Motor Neuron Axonal Deficits. Proc. Nat. Acad. Sci. USA 2011, 108, 10337–10342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallini, C.; Zhang, H.; Su, Y.; Silani, V.; Singer, R.H.; Rossoll, W.; Bassell, G.J. The Survival of Motor Neuron (SMN) Protein Interacts with the MRNA-Binding Protein HuD and Regulates Localization of Poly(A) MRNA in Primary Motor Neuron Axons. J. Neurosci. 2011, 31, 3914–3925. [Google Scholar] [CrossRef] [PubMed]
- Hubers, L.; Valderrama-Carvajal, H.; Laframboise, J.; Timbers, J.; Sanchez, G.; Côté, J. HuD Interacts with Survival Motor Neuron Protein and Can Rescue Spinal Muscular Atrophy-like Neuronal Defects. Hum. Mol. Genet. 2011, 20, 553–579. [Google Scholar] [CrossRef]
- Hao le, T.; Duy, P.Q.; An, M.; Talbot, J.; Iyer, C.C.; Wolman, M.; Beattie, C.E. HuD and the Survival Motor Neuron Protein Interact in Motoneurons and Are Essential for Motoneuron Development, Function, and MRNA Regulation. J. Neurosci. 2017, 37, 11559–11571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.K.; Tisdale, S.; Lotti, F.; Pellizzoni, L. SMN Control of RNP Assembly: From Post-Transcriptional Gene Regulation to Motor Neuron Disease. Semin. Cell Dev. Biol. 2014, 32, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, C.C.; Corlett, K.M.; Massoni-Laporte, A.; Duque, S.I.; Madabusi, N.; Tisdale, S.; McGovern, V.L.; Le, T.T.; Zaworski, P.G.; Arnold, W.D.; et al. Mild SMN Missense Alleles Are Only Functional in the Presence of SMN2 in Mammals. Hum. Mol. Genet. 2018, 27, 3404–3416. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Mutation Name | Clinical Subtype | SMN1 Copy Number | SMN2 Copy Number | Mutation Type | Nucleotide Change | Amino Acid Change | Exon Domain | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 01 02 | W92S | 1 | 1 | 3 | Missense | c.275 G > C | p.Trp92Ser | Ex3 Tudor | [20,22] |
| 03 | E134K | 2 | 1 | 2 | Missense | c.400 G > A | p.Glu134Lys | Ex3 Tudor | [27,30] |
| 04 | Y276H | 2 | 1 | 2 | Missense | c.826 T > C | p.Tyr276His | Ex6 C-term. * | [28] |
| 05 | Y277C | 2/3 | 1 | 1 | Missense | c.830 A > G | p.Tyr277Cys | Ex6 C-term. * | [19] |
| 06 | T274Y fsX32 | 1 | 1 | 2 | Frame shift | c.819_820 insT | p.Thr274TyrfsX32 | Ex6 C-term. * | [19,22,29] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niba, E.T.E.; Nishio, H.; Wijaya, Y.O.S.; Ar Rochmah, M.; Takarada, T.; Takeuchi, A.; Kimizu, T.; Okamoto, K.; Saito, T.; Awano, H.; et al. Stability and Oligomerization of Mutated SMN Protein Determine Clinical Severity of Spinal Muscular Atrophy. Genes 2022, 13, 205. https://doi.org/10.3390/genes13020205
Niba ETE, Nishio H, Wijaya YOS, Ar Rochmah M, Takarada T, Takeuchi A, Kimizu T, Okamoto K, Saito T, Awano H, et al. Stability and Oligomerization of Mutated SMN Protein Determine Clinical Severity of Spinal Muscular Atrophy. Genes. 2022; 13(2):205. https://doi.org/10.3390/genes13020205
Chicago/Turabian StyleNiba, Emma Tabe Eko, Hisahide Nishio, Yogik Onky Silvana Wijaya, Mawaddah Ar Rochmah, Toru Takarada, Atsuko Takeuchi, Tomokazu Kimizu, Kentaro Okamoto, Toshio Saito, Hiroyuki Awano, and et al. 2022. "Stability and Oligomerization of Mutated SMN Protein Determine Clinical Severity of Spinal Muscular Atrophy" Genes 13, no. 2: 205. https://doi.org/10.3390/genes13020205
APA StyleNiba, E. T. E., Nishio, H., Wijaya, Y. O. S., Ar Rochmah, M., Takarada, T., Takeuchi, A., Kimizu, T., Okamoto, K., Saito, T., Awano, H., Takeshima, Y., & Shinohara, M. (2022). Stability and Oligomerization of Mutated SMN Protein Determine Clinical Severity of Spinal Muscular Atrophy. Genes, 13(2), 205. https://doi.org/10.3390/genes13020205