
Balanced Polymorphism at the Pgm-1 Locus of the Pompeii Worm Alvinella pompejana and Its Variant Adaptability Is Only Governed by Two QE Mutations at Linked Sites

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
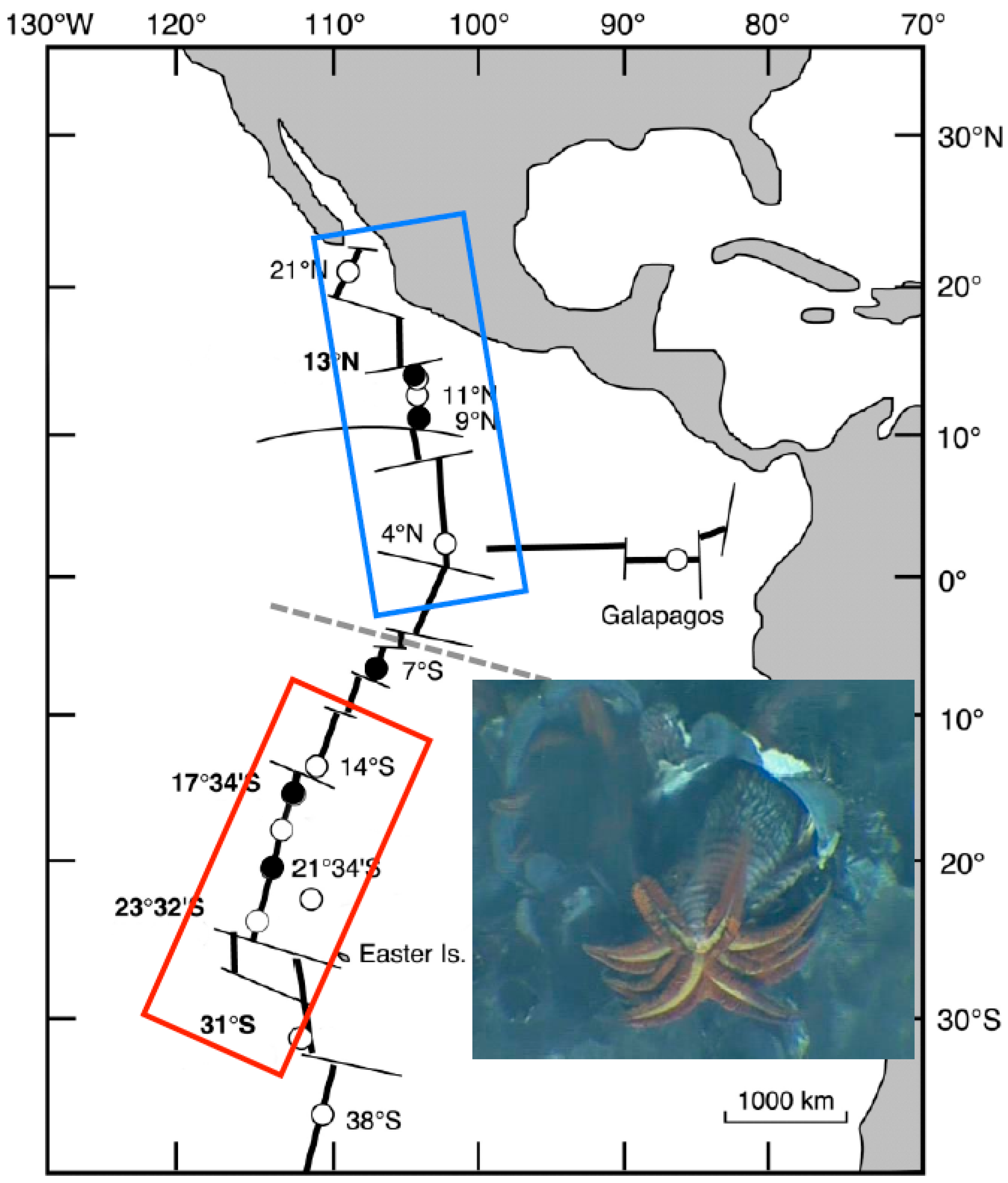
2.1. Animal Sampling
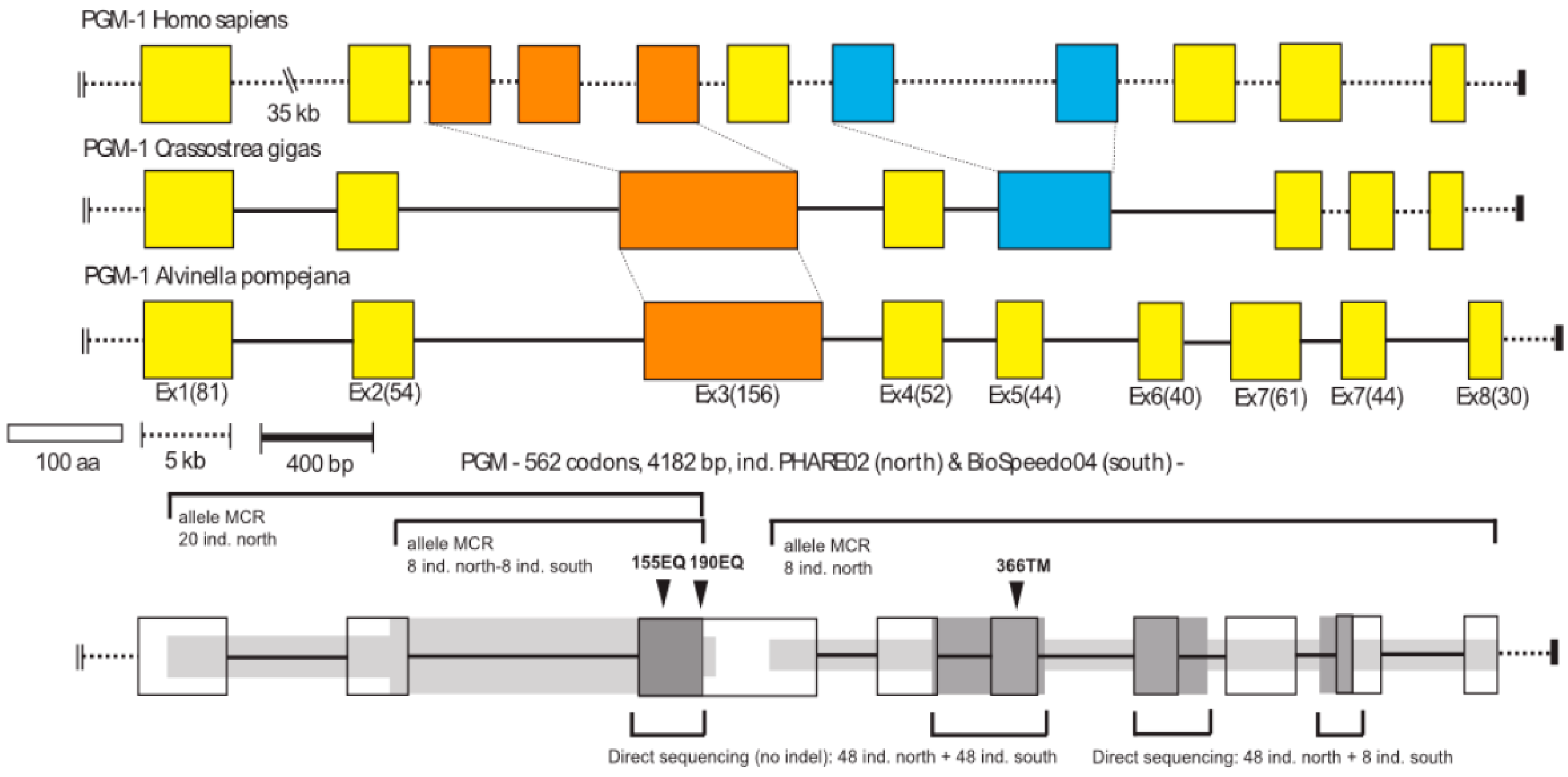
2.2. Identification and Characterization of the AP-Pgm-1 Gene
2.2.1. Sequencing of AP-Pgm-1 cDNA Using Homozygous Individuals
2.2.2. Sequencing the AP-Pgm-1 Gene with a Series of Specific Exonic Primers
2.2.3. Correspondence between Allozymes and Non-Synonymous Mutations of AP-Pgm-1
2.2.4. Examining the Synonymous and Non-Synonymous Changes along AP-Pgm-1
2.3. Coalescent Simulations Using Models of Selection
2.4. Functional and Structural Analysis of PGM-1 Recombinant Isoforms
2.4.1. Plasmid Construction for Enzyme Expression
2.4.2. Directed Mutagenesis
2.4.3. Protein Expression and Purification
2.4.4. Enzyme Activity Assay
2.4.5. Thermal Inactivation
2.5. Guanidinium Chloride-Induced Unfolding of PGM Isoforms
2.6. 3D-PGM Structure Modelling
3. Results
3.1. Sequencing AP-Pgm-1 cDNA from Homozygous Genotypes
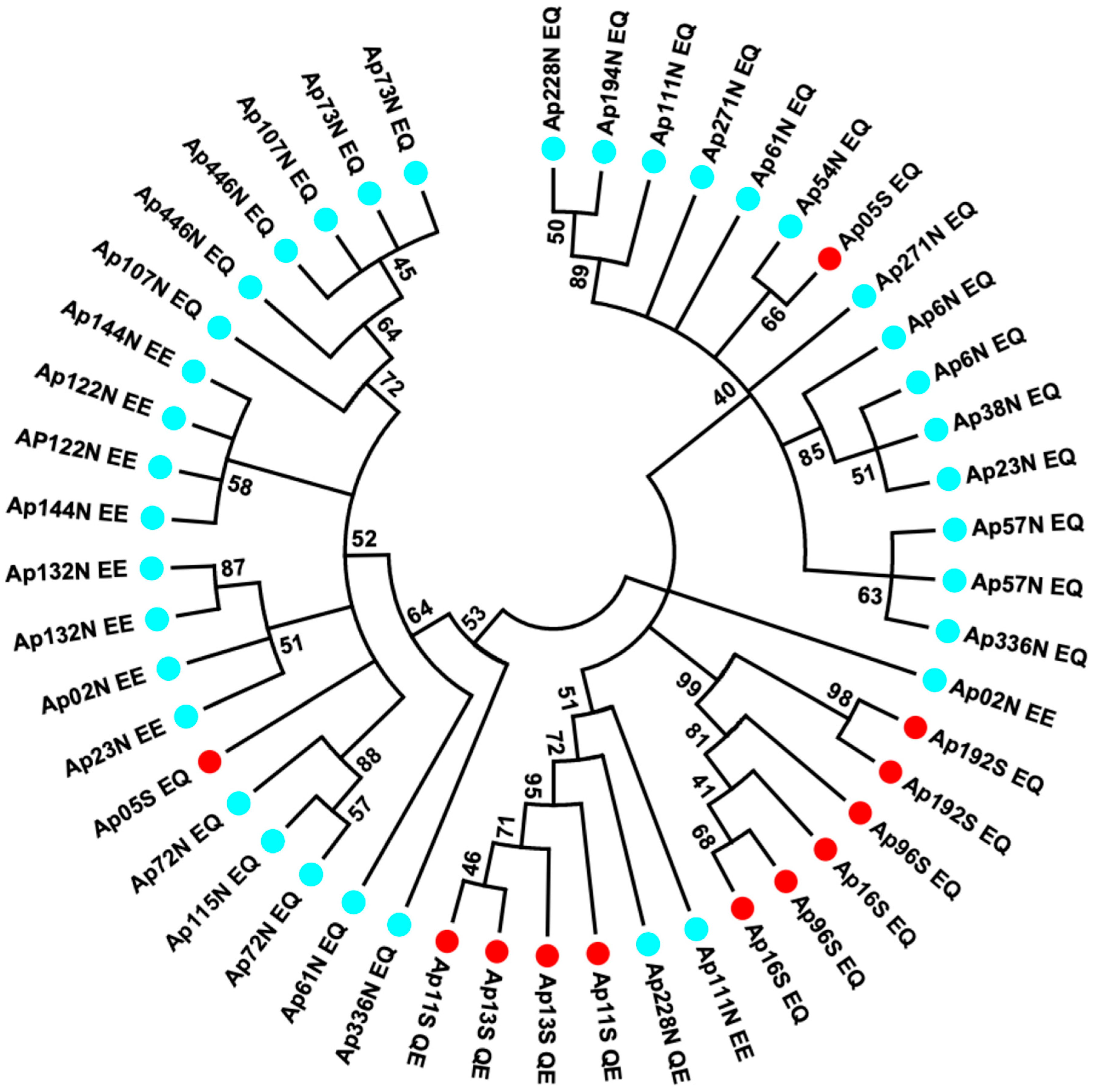
3.2. Assignment of the Two EQ Amino Acid Replacements to Allozymes in Natural Populations
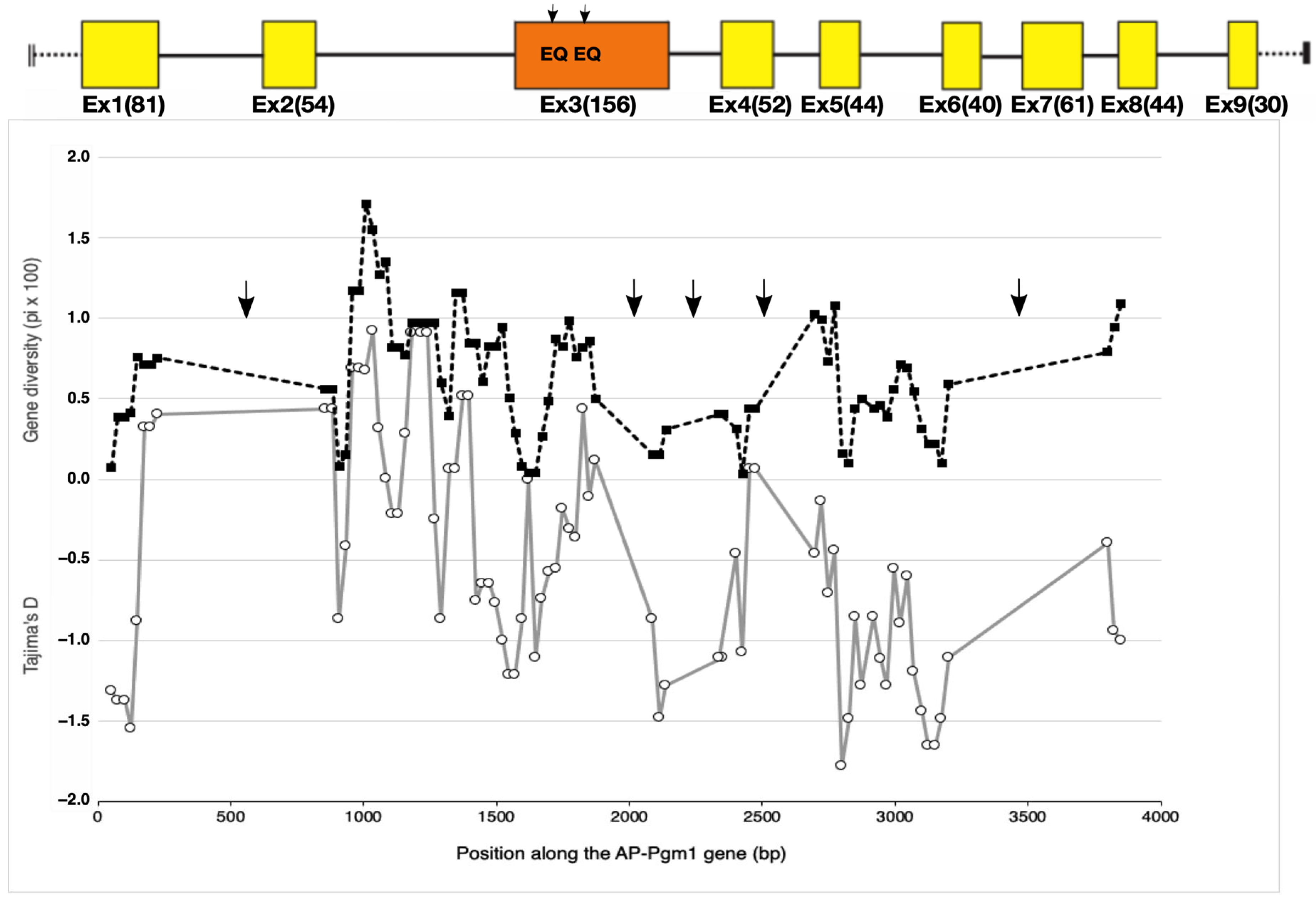
3.3. Cryptic Amino Acid Variation along the AP-Pgm-1 Gene
3.4. Estimating Allele Divergence and Linkage Disequilibria between Segregating Sites
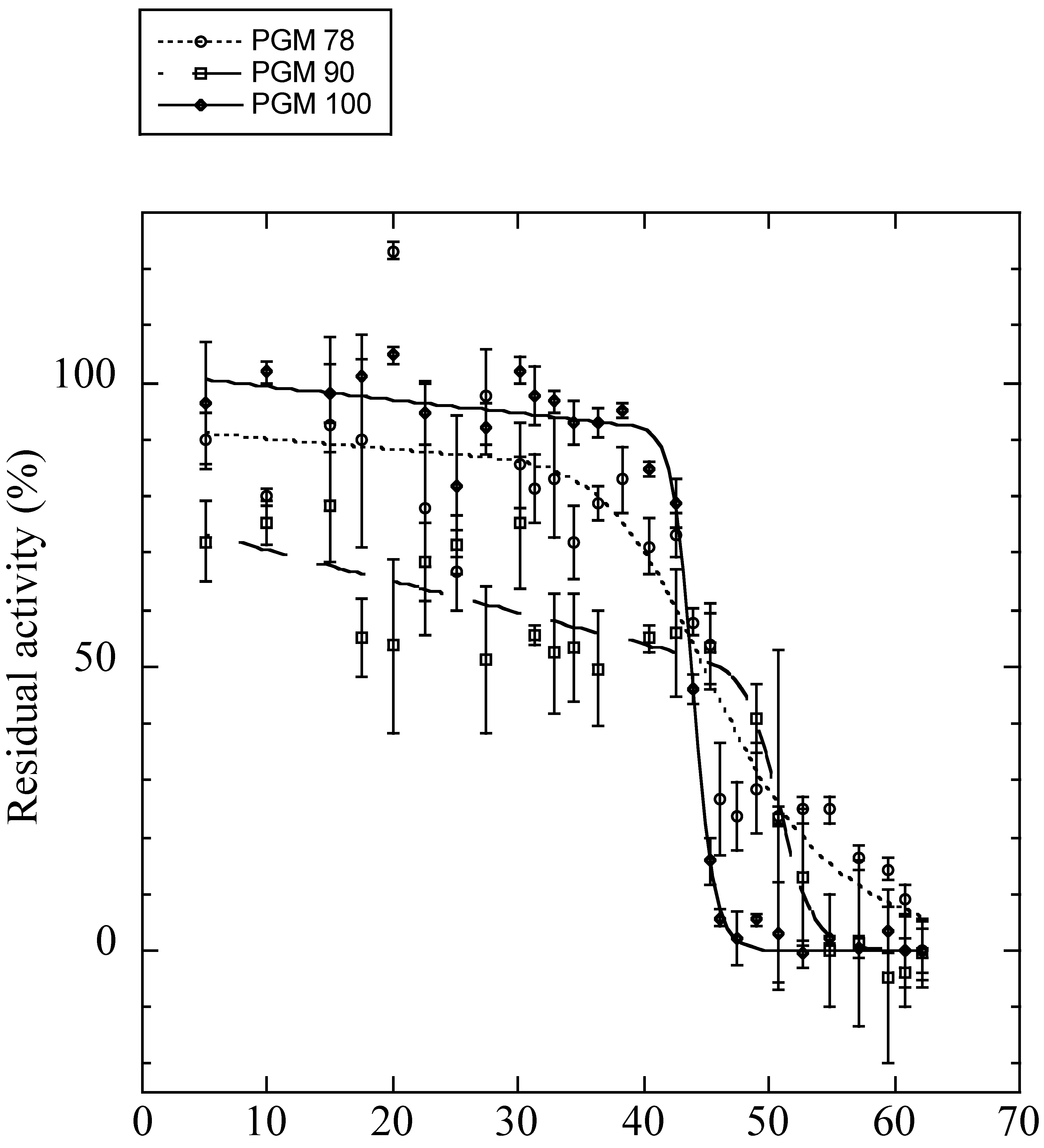
3.5. Conformational Stability, Thermal Inactivation, and Kinetics of the Mutated Isoforms
3.6. Fitness Cost of Individuals Carrying the Thermostable Allele in Terms of Female Fecundity
4. Discussion
4.1. Two-Allele Polymorphism at the Pgm Locus: A Long Story of Balancing Selection?
4.2. Selective Modalities for the Maintenance of a Balanced Polymorphism
4.3. Adaptive Polymorphism: A Trade-Off between Enzyme Thermostability and Catalysis
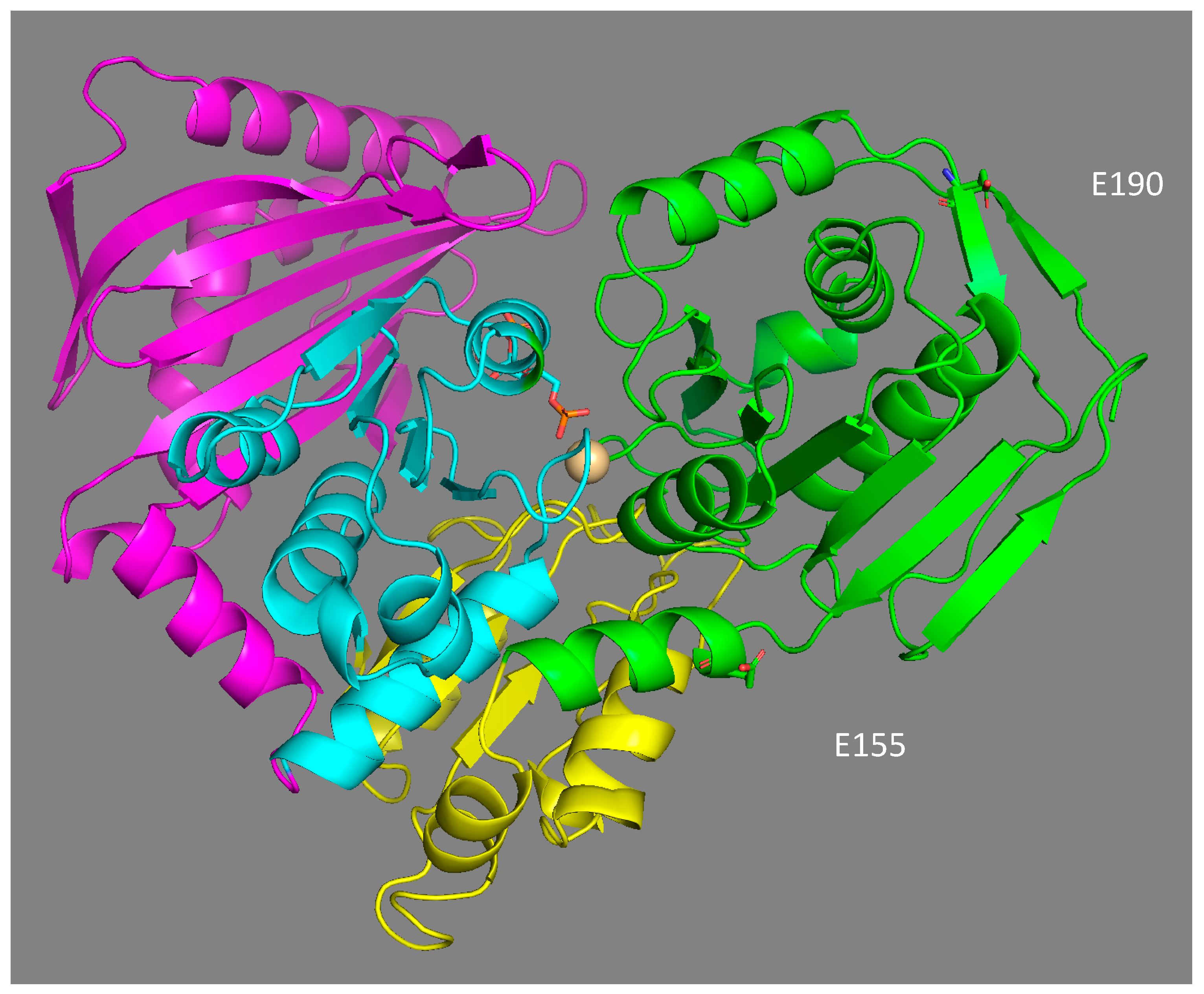
4.4. Structural Effect of Mutations E155Q and E190Q
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holt, R.D.; Gaines, M.S. Analysis of adaptation in heterogeneous landscapes: Implications for the evolution of fundamental niches. Evolut. Ecol. 1992, 6, 433–447. [Google Scholar] [CrossRef]
- Schmidt, P.S.; Serrão, E.A.; Pearson, G.A.; Riginos, C.; Rawson, P.D.; Hilbish, T.J.; Brawley, S.H.; Trussel, G.C.; Carrington, E.; Wethey, D.S.; et al. Ecological genetics in the North Atlantic: Environmental gradients and adaptation at specific loci. Ecology 2008, 89, 91–107. [Google Scholar] [CrossRef]
- Nevo, E.; Shimony, T.; Libni, M. Thermal selection of allozyme polymorphisms in barnacles. Nature 1977, 267, 99. [Google Scholar] [CrossRef]
- Nevo, E.; Lavie, E.; Ben-Shlomo, R. Selection of allelic isozyme polymorphisms in marine organisms: Pattern, theory, and application. Isozymes 1983, 10, 69–92. [Google Scholar] [PubMed]
- Nevo, E.; Noy, R.; Lavie, B.; Beiles, A.; Muchtar, S. Genetic diversity and resistance to marine pollution. Biol. J. Linn. Soc. 1986, 29, 39–144. [Google Scholar] [CrossRef]
- Pogson, G.H. Biochemical characterization of genotypes at the phosphoglucomutase-2 locus in the Pacific oyster, Crassostrea gigas. Biochem. Genet. 1989, 27, 571–589. [Google Scholar] [CrossRef]
- Riddoch, B. The adaptive significance of electrophoretic mobility in phosphoglucose isomerase (PGI). Biol. J. Linn. Soc. 1993, 50, 1–17. [Google Scholar] [CrossRef]
- Schmidt, P.S.; Rand, D.M. Adaptive maintenance of genetic polymorphism in an intertidal barnacle: Habitat-and life-stage-specific survivorship of MPI genotypes. Evolution 2001, 55, 1336–1344. [Google Scholar] [CrossRef]
- Boutet, I.; Tanguy, A.; le Guen, D.; Piccino, P.; Hourdez, S.; Ravaux, J.; Shillito, B.; Legendre, P.; Jollivet, D. Global depression in gene expression as a response to rapid thermal changes in vent mussels. Proc. R. Soc. Lond. B 2009, 276, 3071–3079. [Google Scholar] [CrossRef] [PubMed]
- Bougerol, M.; Boutet, I.; le Guen, D.; Jollivet, D.; Tanguy, A. Transcriptomic response of the hydrothermal mussel Bathymodiolus azoricus mediated by heavy metals is modulated by Pgm genotypes and symbiont content. Mar. Genom. 2015. [Google Scholar] [CrossRef]
- Eanes, W.F. Analysis of selection on enzyme polymorphisms. Ann. Rev. Ecol. Syst. 1999, 30, 301–326. [Google Scholar] [CrossRef]
- Piccino, P.; Viard, F.; Sarradin, P.-M.; le Bris, N.; le Guen, D.; Jollivet, D. Thermal selection of PGM allozymes in newly founded populations of the thermotolerant vent polychaete Alvinella pompejana. Proc. R. Soc. Lond. B 2004, 271, 2351–2359. [Google Scholar] [CrossRef] [PubMed]
- Pogson, G.H. Expression of overdominance for specific activity at the phosphoglucomutase-2 locus in the Pacific oyster, Crassostrea gigas. Genetics 1991, 128, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Verrelli, B.C.; Eanes, W.F. Clinal variation for amino acid polymorphisms at the Pgm locus in Drosophila melanogaster. Genetics 2001, 157, 1649–1663. [Google Scholar] [CrossRef]
- Vrijenhoek, R.C. Gene flow and genetic diversity in naturally fragmented metapopulations of deep-sea hydrothermal vent animals. J. Hered. 1997, 88, 285–293. [Google Scholar] [CrossRef]
- Jollivet, D.; Chevaldonné, P.; Planque, B. Hydrothermal-vent alvinellid polychaete dispersal in the Eastern Pacific. 2. A metapopulation model based on habitat shifts. Evolution 1999, 53, 1128–1142. [Google Scholar] [CrossRef]
- Vrijenhoek, R.C. Genetic diversity and connectivity of deep-sea hydrothermal vent metapopulations. Mol. Ecol. 2010, 19, 4391–4411. [Google Scholar] [CrossRef]
- Watremez, P.; Kervevan, C. 1990 Origine des variations de l'activité hydrothermale: Premiers éléments de réponse d'un modèle numérique simple. C. R. Acad. Sci Paris 2010, 311, 153–158. [Google Scholar]
- Johnson, K.S.; Childress, J.J.; Hessler, R.R.; Sakamoto-Arnold, C.M.; Beehler, C.L. Chemical and biological interactions in the Rose Garden hydrothermal vent field, Galapagos spreading center. Deep Sea Res. A 1988, 35, 1723–1744. [Google Scholar] [CrossRef]
- Le Bris, N.; Gaill, F. How does the annelid Alvinella pompejana deal with an extreme hydrothermal environment? Rev. Environ. Sci. Biotechnol. 2007, 6, 197–221. [Google Scholar] [CrossRef]
- Hourdez, S.; Lallier, F.H. Adaptations to hypoxia in hydrothermal-vent and cold-seep invertebrates. In Life in Extreme Environments; Springer: Dordrecht, The Netherlands, 2006; pp. 297–313. [Google Scholar]
- Von Damm, K.L. Controls on the chemistry and temporal variability of seafloor hydrothermal fluids. In Seafloor Hydrothermal Systems: Physical, Chemical, Biological, and Geological Interactions; Washington DC American Geophysical Union Geophysical Monograph Series; Wiley: Hoboken, NJ, USA, 1995; Volume 91, pp. 222–247. [Google Scholar]
- Jollivet, D. Specific and genetic diversity at deep-sea hydrothermal vents: An overview. Biodiv. Cons. 1996, 5, 1619–1653. [Google Scholar] [CrossRef]
- Matabos, M.; le Bris, N.; Pendlebury, S.; Thiébaut, E. Role of physico-chemical environment on gastropod assemblages at hydrothermal vents on the East Pacific Rise (13 N/EPR). J. Mar. Biol. Assoc. UK 2008, 88, 995–1008. [Google Scholar] [CrossRef]
- Shank, T.M.; Fornari, D.J.; von Damm, K.L.; Lilley, M.D.; Haymon, R.M.; Lutz, R.A. Temporal and spatial patterns of biological community development at nascent deep-sea hydrothermal vents (9 50′ N. East Pacific Rise). Deep Sea Res. 1998, 45, 465–515. [Google Scholar] [CrossRef]
- Fontaine, F.J.; Cannat, M.; Escartin, J. Hydrothermal circulation at slow-spreading mid-ocean ridges: The role of along-axis variations in axial lithospheric thickness. Geology 2008, 36, 759–762. [Google Scholar] [CrossRef]
- Marcus, J.; Tunnicliffe, V.; Butterfield, D.A. Post-eruption succession of macrofaunal communities at diffuse flow hydrothermal vents on Axial Volcano, Juan de Fuca Ridge, Northeast Pacific. Deep Sea Res. 2009, 56, 1586–1598. [Google Scholar] [CrossRef]
- Chevaldonné, P.; Desbruyères, D.; Childress, J.J. ... and some even hotter. Nature 1992, 359, 593–594. [Google Scholar] [CrossRef]
- Cary, S.C.; Shank, T.; Stein, J. Worms bask in extreme temperatures. Nature 1998, 391, 545–546. [Google Scholar] [CrossRef]
- Ravaux, J.; Hamel, G.; Zbinden, M.; Tasiemski, A.A.; Boutet, I.; Léger, N.; Tanguy, A.; Jollivet, D.; Shillito, B. Thermal limit for metazoan life in question: In Vivo heat tolerance of the Pompeii worm. PLoS ONE 2013, 8, e64074. [Google Scholar]
- Jang, S.J.; Park, E.; Lee, W.K.; Johnson, S.B.; Vrijenhoek, R.C.; Won, Y.J. Population subdivision of hydrothermal vent polychaete Alvinella pompejana across equatorial and Easter Microplate boundaries. BMC Evolut. Biol. 2016, 16, 1–15. [Google Scholar] [CrossRef]
- Desbruyères, D.; Chevaldonné, P.; Alayse, A.-M.; Jollivet, D.; Lallier, F.H.; Jouin-Toulmond, C.; Zal, F.; Sarradin, P.-M.; Cosson, R.; Caprais, J.-C.; et al. Biology and ecology of the “Pompeii worm”(Alvinella pompejana Desbruyères and Laubier), a normal dweller of an extreme deep-sea environment: A synthesis of current knowledge and recent developments. Deep Sea Res. 1998, 45, 383–422. [Google Scholar] [CrossRef]
- Chevaldonné, P.; Fisher, C.R.; Childress, J.J.; Desbruyères, D.; Jollivet, D.; Zal, F.; Toulmond, A. Thermotolerance and the ‘Pompeii worms’. Mar. Ecol. Prog. Ser. 2000, 208, 293–295. [Google Scholar] [CrossRef]
- Jollivet, D.; Desbruyères, D.; Bonhomme, F.; Moraga, D. Genetic differentiation of deep-sea hydrothermal vent alvinellid populations (Annelida: Polychaeta) along the East Pacific Rise. Heredity 1995, 74, 376–391. [Google Scholar] [CrossRef]
- Plouviez, S.; le Guen, D.; Lecompte, O.; Lallier, F.H.; Jollivet, D. Determining gene flow and the influence of selection across the equatorial barrier of the East Pacific Rise in the tube-dwelling polychaete Alvinella pompejana. BMC Evol. Biol. 2010, 10, 220. [Google Scholar] [CrossRef] [PubMed]
- Jollivet, D.; Desbruyères, D.; Ladrat, C.; Laubier, L. Evidence for differences in the allozyme thermostability of deep-sea hydrothermal vent polychaetes (Alvinellidae): A possible selection by habitat. Mar. Ecol. Prog. Ser. 1995, 123, 125–136. [Google Scholar] [CrossRef]
- Verrelli, B.C.; Eanes, W.F. The functional impact of Pgm amino acid polymorphism on glycogen content in Drosophila melanogaster. Genetics 2001, 159, 201–210. [Google Scholar] [CrossRef]
- Charlesworth, D. Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet. 2006, 2, e64. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W. Balancing selection. Curr. Biol. 2007, 17, 230–231. [Google Scholar] [CrossRef]
- Plouviez, S.; Shank, T.M.; Faure, B.; Daguin-Thiébaut, C.; Viard, F.; Lallier, F.H.; Jollivet, D. Comparative phylogeography among hydrothermal vent species along the East Pacific Rise reveals vicariant processes and population expansion in the South. Mol. Ecol. 2009, 18, 3903–3917. [Google Scholar] [PubMed]
- Pradillon, F.; Shillito, B.; Young, C.M.; Gaill, F. Deep-sea ecology: Developmental arrest in vent worm embryos. Nature 2001, 413, 698. [Google Scholar] [CrossRef]
- Faure, B.; Chevaldonné, F.P.; Jollivet, D. Spatial and temporal dynamics of reproduction and settlement in the Pompeii worm Alvinella pompejana (Polychaeta: Alvinellidae). Mar. Ecol. Prog. Ser. 2007, 348, 197–211. [Google Scholar] [CrossRef]
- Mishra, R.N.; Singla-Pareek, S.L.; Nair, S.; Sopory, S.K.; Reddy, M.K. Directional genome walking using PCR. Biotechniques 2002, 33, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Bierne, N.; Tanguy, A.; Faure, M.; Faure, B.; David, E.; Boutet, I.; Boon, E.; Quéré, N.; Plouviez, S.; Kemppainen, P.; et al. Mark-recapture cloning: A straightforward and cost-effective cloning method for population genetics of single copy nuclear DNA sequences in diploids. Mol. Ecol. Notes 2007, 7, 562–566. [Google Scholar] [CrossRef]
- Black, W.C.; Krafsur, E.S. A FORTRAN program for the calculation and analysis of two-locus linkage disequilibrium coefficients. Theor. Appl. Genet. 1985, 70, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Belkhir, K.; Borsa, P.; Chikhi, L.; Raufaste, N.; Bonhomme, F. GENETIX 4.0. 5.2, Software under Windows™ for the Genetics of the Populations; University of Montpellier: Montpellier, France, 2004. [Google Scholar]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Kelly, J.K. A test of neutrality based on interlocus associations. Genetics 1997, 146, 1197–1206. [Google Scholar] [CrossRef]
- Hudson, R.R.; Kaplan, N.L. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 1985, 111, 147–164. [Google Scholar] [CrossRef]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef]
- Li, N.; Stephens, M. Modelling linkage disequilibrium and identifying recombination hotspots using snp data. Genetics 2003, 165, 2213–2233. [Google Scholar] [CrossRef]
- Hudson, R.R.; Slatkin, M.; Maddison, W.P. Estimation of levels of gene flow from DNA sequence data. Genetics 1992, 132, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Ewing, G.; Hermisson, J. MSMS: A coalescent simulation program including recombination, demographic structure and selection at a single locus. Bioinformatics 2010, 26, 2064–2065. [Google Scholar] [CrossRef] [PubMed]
- Thornton, K. Libsequence: A c++ class library for evolutionary genetic analysis. Bioinformatics 2003, 19, 2325–2327. [Google Scholar] [CrossRef]
- Reikofski, J.; Tao, B.Y. Polymerase chain reaction (PCR) techniques for site-directed mutagenesis. Biotechnol. Adv. 1992, 10, 535–547. [Google Scholar] [CrossRef]
- Pace, C.N.; Scholtz, J.M. Measuring the conformational stability of a protein. In Protein Structure: A Practical Approach; Oxford University Press: Oxford, UK, 1997; Volume 2, pp. 299–321. [Google Scholar]
- Pace, C.N. Determination and analysis of urea and guanidine hydrochloride denaturation curves. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1986; Volume 131, pp. 266–280. [Google Scholar]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Whitehouse, D.B.; Putt, W.; Lovegrove, J.U.; Morrison, K.; Hollyoake, M.; Fox, M.F.; Hopkinson, D.A.; Edwards, Y.H. Phosphoglucomutase 1: Complete human and rabbit mRNA sequences and direct mapping of this highly polymorphic marker on human chromosome 1. Proc. Nat. Acad. Sci. USA 1992, 89, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Tanguy, A.; Boutet, I.; Boudry, P.; Degremont, L.; Laroche, J.; Moraga, D. Molecular identification and expression of the phosphoglucomutase (PGM) gene from the Pacific oyster Crassostrea gigas. Gene 2006, 382, 20–27. [Google Scholar] [CrossRef]
- Matabos, M.; Plouviez, S.; Hourdez, S.; Desbruyères, D.; Legendre, P.; Warèn, A.; Jollivet, D.; Thiébaut, E. Faunal changes and geographic crypticism indicate the occurrence of a biogeographic transition zone along the Southern East-Pacific Rise. J. Biogeogr. 2011, 38, 575–594. [Google Scholar] [CrossRef]
- Matabos, M.; Jollivet, D. Revisiting the species’ complex of Lepetodrilus elevatus (Vetigastropod, Lepetodrilidae) using gastropod samples from the Galápagos and Guaymas hydrothermal vent systems. J. Moll. Stud. 2019, 85, 154–165. [Google Scholar] [CrossRef]
- Eanes, W.F. Molecular population genetics and selection in the glycolytic pathway. J. Exp. Biol. 2011, 214, 165–171. [Google Scholar] [CrossRef][Green Version]
- Verrelli, B.C.; Eanes, W.F. Extensive amino acid polymorphism at the Pgm locus is consistent with adaptive protein evolution in Drosoph. Melanogaster. Genet. 2000, 156, 1737–1752. [Google Scholar] [CrossRef] [PubMed]
- Wheat, C.W.; Watt, W.B.; Pollock, D.D.; Schulte, P.M. From DNA to fitness differences: Sequences and structures of adaptive variants of Colias phosphoglucose isomerase (PGI). Mol. Biol. Evol. 2005, 23, 499–512. [Google Scholar] [CrossRef]
- McDonald, J.H.; Kreitman, M. Adaptive protein evolution at the Adh locus in Drosophila. Nature 1991, 351, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Begun, D.J.; Betancourt, A.J.; Langley, C.H.; Stephan, W. Is the fast/slow allozyme variation at the Adh locus of Drosophila melanogaster an ancient balanced polymorphism? Mol. Biol. Evol. 1999, 16, 1816–1819. [Google Scholar] [CrossRef]
- Fontanillas, E.; Galzitskaya, O.V.; Lecompte, O.; Lobanov, M.Y.; Tanguy, A.; Mary, J.; Girguis, P.R.; Hourdez, S.; Jollivet, D. Proteome evolution of deep-sea hydrothermal vent alvinellid polychaetes supports the ancestry of thermophily and subsequent adaptation to the cold for some lineages. Genome Biol. Evol. 2017, 9, 279–296. [Google Scholar] [CrossRef]
- Bioy, A. Histoire Évolutive et Influence de la Sélection sur la Diversité génétique des Annélides Polychètes D’environnements Extrêmes. Thèse de Doctorat, Sorbonne Université, Paris, France, 2018; pp. 248p. [Google Scholar]
- Hoekstra, R.F.; Bijlsma, R.; Dolman, A.J. Polymorphism from environmental heterogeneity: Models are only robust if the heterozygote is close in fitness to the favoured homozygote in each environment. Genet. Res. 1985, 45, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.P.A.; Lobkov, I. A test for overdominance at the phosphoglucomutase-2 locus in Pacific oysters (Crassostrea gigas) from B-New Zealand. Aquaculture 2005, 244, 29–39. [Google Scholar] [CrossRef]
- Levene, H. Genetic equilibrium when more than one ecological niche is available. Am. Nat. 1953, 87, 331–333. [Google Scholar] [CrossRef]
- Gillespie, J.H. The interaction of genetic drift and mutation with selection in a fluctuating environment. Theor. Pop. Biol. 1985, 27, 222–237. [Google Scholar] [CrossRef]
- Hedrick, P.W.; Ginevan, M.E.; Ewing, E.P. Genetic polymorphism in heterogeneous environments. Ann. Rev. Ecol. Syst. 1976, 7, 1–32. [Google Scholar] [CrossRef]
- Hedrick, P.W. Genetic polymorphism in heterogeneous environments: A decade later. Ann. Rev. Ecol. Syst. 1986, 17, 535–566. [Google Scholar] [CrossRef]
- Watt, W.B. Adaptation at specific loci. I. Natural selection on phosphoglucose isomerase of Colias butterflies: Biochemical and population aspects. Genetics 1977, 87, 177–194. [Google Scholar] [CrossRef]
- Watt, W.B.; Cassin, R.C.; Swan, M.S. Adaptation at specific loci. III. Field behavior and survivorship differences among Colias PGI genotypes are predictable from In Vitro biochemistry. Genetics 1983, 103, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.A.; Watt, W.B. Adaptation at specific loci. V. Metabolically adjacent enzyme loci may have very distinct experiences of selective pressures. Genetics 1988, 119, 913–924. [Google Scholar] [CrossRef]
- Hartl, D.L.; Dykhuizen, D.E.; Dean, A.M. Limits of adaptation: The evolution of selective neutrality. Genetics 1985, 111, 655–674. [Google Scholar] [CrossRef]
- Somero, G.N. Temperature adaptation of enzymes: Biological optimization through structure-function compromises. Ann. Rev. Ecol. Syst. 1978, 9, 1–29. [Google Scholar] [CrossRef]
- Somero, G.N. Proteins and temperature. Ann. Rev. Physiol. 1995, 57, 43–68. [Google Scholar] [CrossRef]
- Vogt, G.; Woell, S.; Argos, P. Protein thermal stability, hydrogen bonds, and ion pairs. J. Mol. Biol. 1997, 269, 631–643. [Google Scholar] [CrossRef]
- Shin, D.S.; DiDonato, M.; Barondeau, D.P.; Hura, G.L.; Hitomi, C.; Berglund, J.A.; Getzoff, E.D.; Cary, S.C.; Tainer, J.A. Superoxide dismutase from the eukaryotic thermophile Alvinella pompejana: Structures, stability, mechanism, and insights into amyotrophic lateral sclerosis. J. Mol. Biol. 2009, 385, 1534–1555. [Google Scholar] [CrossRef] [PubMed]
- Bruneaux, M.; Mary, J.; Verheye, M.; Lecompte, O.; Poch, O.; Jollivet, D.; Tanguy, A. Detection and characterisation of mutations responsible for allele-specific protein thermostabilities at the Mn-superoxide dismutase gene in the deep-sea hydrothermal vent polychaete Alvinella pompejana. J. Mol. Evol. 2013, 76, 295–310. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Dij | Rij | Chi2 | p-Value |
|---|---|---|---|---|
| EE-100 | 0.274 | 0.908 | 87.2 | 0.0001 *** |
| EQ-90 | 0.115 | 0.725 | 55.7 | 0.0001 *** |
| QE-78 | 0.357 | 0.907 | 87.2 | 0.0001 *** |
| EE-112 | 0.013 | 0.230 | 5.6 | 0.0178 * |
| Statistics | E1 North | E1 South | E2-I2 North | E2-I2 South | E3 North | E3 South | E4-E5 North | E4-E5 South | E7-E9 North | E7-E9 South |
|---|---|---|---|---|---|---|---|---|---|---|
| Fragment length (bp) | 273 | 273 | 1110 | 1110 | 278 | 278 | 803 | 803 | 576 | 576 |
| N | 38 | 40 | 36 | 12 | 156 | 218 | 20 | 45 | 62 | 12 |
| Hd | 0.77 ± 0.06 | 0.39 ± 0.01 | 0.95 ± 0.03 | 0.85 ± 0.02 | 0.72 ± 0.03 | 0.76 ± 0.04 | 0.68 ± 0.10 | 0.88 ± 0.04 | 0.91 ± 0.03 | 0.98 ± 0.04 |
| Overall π | 0.0059 ± 0.0006 | 0.0017 ± 0.0005 | 0.0056 ± 0.0003 | 0.0087 ± 0.0005 | 0.0045 ± 0.0003 | 0.0070 ± 0.0006 | 0.0023 ± 0.0005 | 0.0045 ± 0.0005 | 0.0044 ± 0.0005 | 0.0068 ± 0.0010 |
| π s | 0.0016 (62.6) | 0.0008 (62.6) | 0.0126 (58.7) | 0.0404 (58.7) | 0.0034 (48.7) | 0.0167 (48.7) | 0.0094 (88.1) | 0.0164 (88.1) | 0.0000 (27.8) | 0.0000 (27.8) |
| π n | 0.0029 (210.4) | 0.0020 (210.4) | 0.0027 (205.3) | 0.0056 (205.3) | 0.0030 (170.3) | 0.0028 (170.3) | 0.0003 (307.9) | 0.0012 (307.9) | 0.0018 (110.2) | 0.0028 (110.2) |
| S | 8 | 5 | 26 | 26 | 16 | 18 | 5 | 10 | 24 | 13 |
| θW (S) | 0.0070 ± 0.0031 | 0.0043 ± 0.0022 | 0.0057 ± 0.0033 | 0.0078 ± 0.0042 | 0.0102 ± 0.0026 | 0.0102 ± 0.0026 | 0.0036 ± 0.0022 | 0.0058 ± 0.0024 | 0.0094 ± 0.0030 | 0.0076 ± 0.0035 |
| ZnS | 0.054 (1/15/1 B) | 0.008 (0/1/0 B) | 0.110 (43/325/19 B) | 0.466 (46/153/0 B) | 0.0093 (2/36/1 B) | 0.0540 (11/66/7 B) | 0.0226 (0/3/0 B) | 0.0324 (3/21/0 B) | 0.0261 (4/120/1 B) | 0.1641 (5/36/0 B) |
| Rm | 1 (RDP n.d.) | 0 (RDP n.d.) | 4 (RDP = 1) | 0 (RDP n.d.) | 3 (RDP n.d.) | 6 (RDP n.d.) | 0 (RDP n.d.) | 3 (RDP n.d.) | 5 (RDP n.d.) | 2 (RDP n.d.) |
| Fst | 0.256 *** | 0.262 *** | 0.510 *** | 0.291 ** | 0.015 * | |||||
| Dxy | 0.0051 | 0.0098 | 0.0117 | 0.0059 | 0.0059 | |||||
| Tajima’s D | −0.46 NS | −1.52 NS | −0.07 NS | +0.46 NS | −1.57 NS | −1.12 NS | −1.07 NS | −0.66 NS | −1.73 NS | −0.41 NS |
| Fu & Li’s F | −0.19 NS | −1.89 NS | +0.07 NS | +0.34 NS | −2.31 * | −1.15 NS | −0.69 NS | −0.57 NS | −1.40 NS | +0.07 NS |
| PGM 78 | PGM 90 | PGM 100 | ||||
|---|---|---|---|---|---|---|
| 1st Transition | 2nd Transition | 1st Transition | 2nd Transition | 1st Transition | 2nd Transition | |
| Cm (M) | 0.50 ± 0.01 | 2.32 ± 0.02 | 0.53 ± 0.02 | 2.42 ± 0.05 | 0.41 ± 0.02 | 2.31 ± 0.03 |
| m (kJ.mol−1.M−1) | 16.00 ± 0.88 | 6.65 ± 0.39 | 21.62 ± 3.73 | 7.48 ± 1.13 | 10.45 ± 1.28 | 6.55 ± 0.42 |
| ΔG0H2O (kJ.mol−1) | 8.00 ± 0.46 | 15.43 ± 0.93 | 11.46 ± 2.03 | 18.10 ± 2.76 | 6.06 ± 0.27 | 15.13 ± 0.98 |
| Tm (°C) | 46.5 ± 1.7 | 50.9 ± 0.7 | 44 ± 0.1 | |||
| Kappm (mM) | 0.76 ± 0.07 | 6.25 ± 0.35 | 5.22 ± 0.74 | |||
| Kcat (sec−1) | 192 ± 3.3 | 12.7 ± 0.5 | 18.3 ± 0.2 | |||
| Kcat/Kappm (sec−1.M−1) | 252.63 ± 17.06 | 2.0 ± 0.1 | 3.51 ± 0.49 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bioy, A.; Le Port, A.-S.; Sabourin, E.; Verheye, M.; Piccino, P.; Faure, B.; Hourdez, S.; Mary, J.; Jollivet, D. Balanced Polymorphism at the Pgm-1 Locus of the Pompeii Worm Alvinella pompejana and Its Variant Adaptability Is Only Governed by Two QE Mutations at Linked Sites. Genes 2022, 13, 206. https://doi.org/10.3390/genes13020206
Bioy A, Le Port A-S, Sabourin E, Verheye M, Piccino P, Faure B, Hourdez S, Mary J, Jollivet D. Balanced Polymorphism at the Pgm-1 Locus of the Pompeii Worm Alvinella pompejana and Its Variant Adaptability Is Only Governed by Two QE Mutations at Linked Sites. Genes. 2022; 13(2):206. https://doi.org/10.3390/genes13020206
Chicago/Turabian StyleBioy, Alexis, Anne-Sophie Le Port, Emeline Sabourin, Marie Verheye, Patrice Piccino, Baptiste Faure, Stéphane Hourdez, Jean Mary, and Didier Jollivet. 2022. "Balanced Polymorphism at the Pgm-1 Locus of the Pompeii Worm Alvinella pompejana and Its Variant Adaptability Is Only Governed by Two QE Mutations at Linked Sites" Genes 13, no. 2: 206. https://doi.org/10.3390/genes13020206
APA StyleBioy, A., Le Port, A.-S., Sabourin, E., Verheye, M., Piccino, P., Faure, B., Hourdez, S., Mary, J., & Jollivet, D. (2022). Balanced Polymorphism at the Pgm-1 Locus of the Pompeii Worm Alvinella pompejana and Its Variant Adaptability Is Only Governed by Two QE Mutations at Linked Sites. Genes, 13(2), 206. https://doi.org/10.3390/genes13020206