

Runs of Homozygosity Revealed Reproductive Traits of Hu Sheep
 ,
,
Abstract
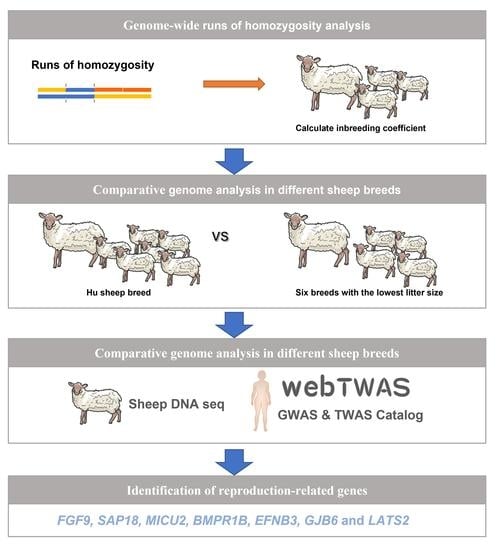
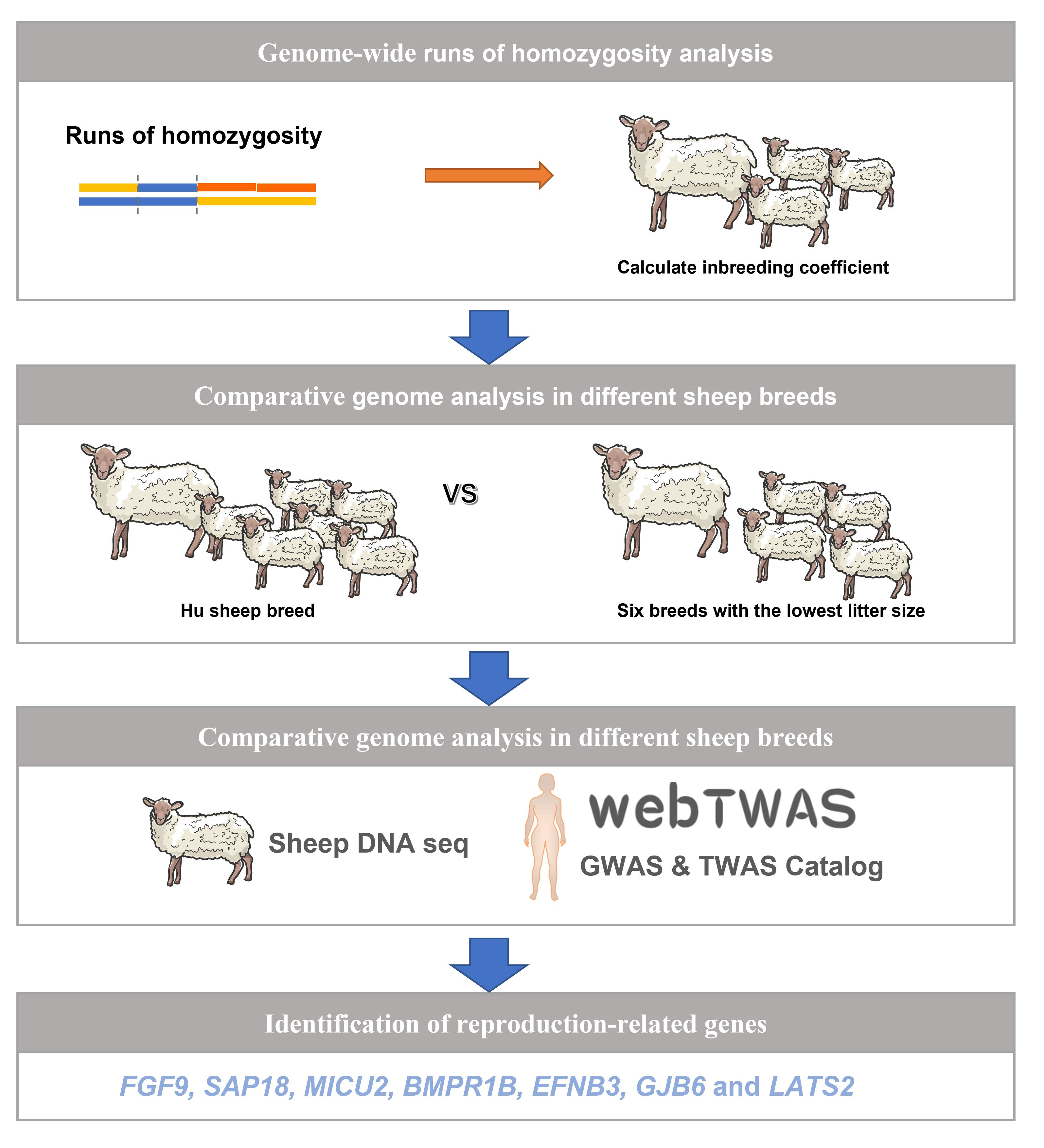
1. Introduction
2. Materials and Methods
2.1. Data
2.2. Definition of ROHs
2.3. Calculation of Inbreeding Coefficients
2.4. Candidate Gene Annotation within ROH Islands
3. Results
3.1. DNA Sequencing and Genetic Diversity
3.2. Statistics of Inbreeding Coefficients
3.3. Distribution of ROHs
3.4. Gene Annotation
4. Discussion
4.1. Pattern of ROHs
4.2. Inbreeding Level within the Population
4.3. Functional Enrichment Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.; Yang, J.; Shen, M.; Xie, X.-L.; Liu, G.-J.; Xu, Y.-X.; Lv, F.-H.; Yang, H.; Yang, Y.-L.; Liu, C.-B.; et al. Whole-Genome Resequencing of Wild and Domestic Sheep Identifies Genes Associated with Morphological and Agronomic Traits. Nat. Commun. 2020, 11, 2815. [Google Scholar] [CrossRef] [PubMed]
- EEr, H.; Ma, L.; Xie, X.; Ma, J.; Ma, X.; Yue, C.; Ma, Q.; Liang, X.; Ding, W.; Li, Y. Genetic Polymorphism Association Analysis of SNPs on the Species Conservation Genes of Tan Sheep and Hu Sheep. Trop. Anim. Health Prod. 2020, 52, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, S.; Li, F.; Pan, X.; Li, C.; Zhang, X.; Ma, Y.; La, Y.; Xi, R.; Li, T. Polymorphisms of the Ovine BMPR-IB, BMP-15 and FSHR and Their Associations with Litter Size in Two Chinese Indigenous Sheep Breeds. Int. J. Mol. Sci. 2015, 16, 11385–11397. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, L.; Li, Q.; Long, Y.; Lin, Y.; Yin, J.; Zeng, Y.; Huang, L.; Yao, T.; Abbasi, M.N.; et al. Functional Probiotics of Lactic Acid Bacteria from Hu Sheep Milk. BMC Microbiol. 2020, 20, 228. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Ciani, E.; Sardina, M.T.; Sottile, G.; Pilla, F.; Portolano, B.; Consortium, B.O.I. Runs of Homozygosity Reveal Genome-Wide Autozygosity in Italian Sheep Breeds. Anim. Genet. 2018, 49, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Peripolli, E.; Munari, D.P.; Silva, M.V.G.B.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of Homozygosity: Current Knowledge and Applications in Livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef]
- Yang, R.; Guo, X.; Zhu, D.; Tan, C.; Bian, C.; Ren, J.; Huang, Z.; Zhao, Y.; Cai, G.; Liu, D.; et al. Accelerated Deciphering of the Genetic Architecture of Agricultural Economic Traits in Pigs Using a Low-Coverage Whole-Genome Sequencing Strategy. GigaScience 2021, 10, giab048. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Salavati, M.; Caulton, A.; Clark, R.; Gazova, I.; Smith, T.P.L.; Worley, K.C.; Cockett, N.E.; Archibald, A.L.; Clarke, S.M.; Murdoch, B.M.; et al. Global Analysis of Transcription Start Sites in the New Ovine Reference Genome (Oar Rambouillet v1.0). Front. Genet. 2020, 11, 580580. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; McCarthy, S.A. BCFtools/csq: Haplotype-aware variant consequences. Bioinformatics 2017, 33, 2037–2039. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; do Carmo Panetto, J.C.; Ventura, R.V.; Baldi, F.; da Silva, M.V.G.B. Assessment of Runs of Homozygosity Islands and Estimates of Genomic Inbreeding in Gyr (Bos Indicus) Dairy Cattle. BMC Genom. 2018, 19, 34. [Google Scholar] [CrossRef]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of Homozygosity Reveal Highly Penetrant Recessive Loci in Schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Wright, S. Coefficients of Inbreeding and Relationship. Am. Nat. 1922, 56, 330–338. [Google Scholar] [CrossRef]
- Fonseca, P.A.S.; Suárez-Vega, A.; Marras, G.; Cánovas, Á. GALLO: An R Package for Genomic Annotation and Integration of Multiple Data Sources in Livestock for Positional Candidate Loci. GigaScience 2020, 9, giaa149. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the Future: Meeting New Challenges and Providing Updated Services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef]
- Cao, C.; Wang, J.; Kwok, D.; Cui, F.; Zhang, Z.; Zhao, D.; Li, M.J.; Zou, Q. WebTWAS: A Resource for Disease Candidate Susceptibility Genes Identified by Transcriptome-Wide Association Study. Nucleic Acids Res. 2022, 50, D1123–D1130. [Google Scholar] [CrossRef]
- Fang, Y.; Hao, X.; Xu, Z.; Sun, H.; Zhao, Q.; Cao, R.; Zhang, Z.; Ma, P.; Sun, Y.; Qi, Z.; et al. Genome-Wide Detection of Runs of Homozygosity in Laiwu Pigs Revealed by Sequencing Data. Front. Genet. 2021, 12, 629966. [Google Scholar] [CrossRef] [PubMed]
- Nandolo, W.; Utsunomiya, Y.T.; Mészáros, G.; Wurzinger, M.; Khayadzadeh, N.; Torrecilha, R.B.P.; Mulindwa, H.A.; Gondwe, T.N.; Waldmann, P.; Ferenčaković, M.; et al. Misidentification of Runs of Homozygosity Islands in Cattle Caused by Interference with Copy Number Variation or Large Intermarker Distances. Genet. Sel. Evol. 2018, 50, 43. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Medland, S.E.; Ferreira, M.A.R.; Morley, K.I.; Zhu, G.; Cornes, B.K.; Montgomery, G.W.; Martin, N.G. Assumption-Free Estimation of Heritability from Genome-Wide Identity-by-Descent Sharing between Full Siblings. PLoS Genet. 2006, 2, e41. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Bovo, S.; Bertolini, F.; Tinarelli, S.; Dall’Olio, S.; Nanni Costa, L.; Gallo, M.; Fontanesi, L. Comparative Evaluation of Genomic Inbreeding Parameters in Seven Commercial and Autochthonous Pig Breeds. Animal 2020, 14, 910–920. [Google Scholar] [CrossRef]
- Yang, R.; Hu, Z.; Kong, Q.; Li, W.; Zhang, L.; Du, X.; Huang, S.; Xia, X.; Sang, H. A known mutation in GJB6 in a large Chinese family with hidrotic ectodermal dysplasia. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1362–1365. [Google Scholar] [CrossRef]
- Gresakova, V.; Novosadova, V.; Prochazkova, M.; Prochazka, J.; Sedlacek, R. Dual Role of Fam208a during Zygotic Cleavage and Early Embryonic Development. Exp. Cell Res. 2021, 406, 112723. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Whelan, E.C.; Guan, X.; Deng, B.; Wang, S.; Sun, J.; Avarbock, M.R.; Wu, X.; Brinster, R.L. FGF9 Promotes Mouse Spermatogonial Stem Cell Proliferation Mediated by P38 MAPK Signalling. Cell Prolif. 2021, 54, e12933. [Google Scholar] [CrossRef]
- Šućurović, S.; Nikolić, T.; Brosens, J.J.; Mulac-Jeričević, B. Spatial and Temporal Analyses of FGF9 Expression during Early Pregnancy. Cell Physiol. Biochem. 2017, 42, 2318–2329. [Google Scholar] [CrossRef]
- Li, Y.-H.; Chen, T.-M.; Huang, B.-M.; Yang, S.-H.; Wu, C.-C.; Lin, Y.-M.; Chuang, J.-I.; Tsai, S.-J.; Sun, H.S. FGF9 Is a Downstream Target of SRY and Sufficient to Determine Male Sex Fate in Ex Vivo XX Gonad Culture. Biol. Reprod. 2020, 103, 1300–1313. [Google Scholar] [CrossRef]
- Gao, Y.; Hao, Q.; Cang, M.; Wang, J.; Yu, H.; Liu, Y.; Zhang, W.; Tong, B. Association between Novel Variants in BMPR1B Gene and Litter Size in Mongolia and Ujimqin Sheep Breeds. Reprod. Domest. Anim. 2021, 56, 1562–1571. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, Z.; Yang, H.; Yao, X.; Yang, P.; Ren, C.; Wang, F.; Zhang, Y. Pituitary Transcriptomic Study Reveals the Differential Regulation of LncRNAs and MRNAs Related to Prolificacy in Different FecB Genotyping Sheep. Genes 2019, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Peng, X.; Li, S.; Gong, Y. Isolation and Characterization of Sexual Dimorphism Genes Expressed in Chicken Embryonic Gonads. Acta Biochim. Biophys. Sin. 2009, 41, 285–294. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wei, Q.; Zhong, L.; Zhang, S.; Mu, H.; Xiang, J.; Yue, L.; Dai, Y.; Han, J. Bovine Lineage Specification Revealed by Single-Cell Gene Expression Analysis from Zygote to Blastocyst. Biol. Reprod. 2017, 97, 5–17. [Google Scholar] [CrossRef]
- Wu, Z.-H.; Tang, Y.; Yu, H.; Li, H.-D. The Role of Ferroptosis in Breast Cancer Patients: A Comprehensive Analysis. Cell Death Discov. 2021, 7, 93. [Google Scholar] [CrossRef]
- Li, X.P.; Lan, J.Y.; Liu, D.Q.; Zhou, H.; Qian, M.M.; Wang, W.W.; Yang, M. OCA2 rs4778137 polymorphism predicts survival of breast cancer patients receiving neoadjuvant chemotherapy. Gene 2018, 651, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wu, W.; Togashi, Y.; Liang, W.; Ohta, T. HERC2 Inactivation Abrogates Nucleolar Localization of RecQ Helicases BLM and WRN. Sci. Rep. 2021, 11, 360. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inbreeding Coefficient | Mean | Min | Max | SD |
|---|---|---|---|---|
| FROH1–5Mb | 0.034 | 0.001 | 0.113 | 0.028 |
| FROH5–10Mb | 0.014 | 0.002 | 0.065 | 0.014 |
| FROH>10Mb | 0.010 | 0.004 | 0.026 | 0.006 |
| FROH-all | 0.041 | 0.001 | 0.176 | 0.040 |
| FSNP1 | 0.921 | 0.685 | 1.109 | 0.086 |
| FSNP2 | 0.921 | 0.882 | 0.964 | 0.019 |
| FSNP3 | 0.921 | 0.817 | 1.002 | 0.038 |
| Correlation | FROH1–5Mb | FROH5–10Mb | FROH>10Mb | FROH-all | FSNP1 | FSNP2 | FSNP3 |
|---|---|---|---|---|---|---|---|
| FROH1–5Mb | 1 | ||||||
| FROH5–10Mb | 0.649 | 1 | |||||
| FROH>10Mb | 0.654 | 0.674 | 1 | ||||
| FROH-all | 0.965 | 0.817 | 0.768 | 1 | |||
| FSNP1 | −0.423 | −0.067 | −0.327 | −0.356 | 1 | ||
| FSNP2 | 0.675 | 0.277 | 0.474 | 0.610 | −0.652 | 1 | |
| FSNP3 | −0.318 | −0.009 | −0.257 | −0.257 | 0.982 | −0.499 | 1 |
| ROH Length (Mb) | ROH Number | Percent (%) | Mean Length (Mb) | Standard Deviation |
|---|---|---|---|---|
| 1–5 | 5638 | 95.49 | 1.55 | 0.74 |
| 5–10 | 223 | 3.78 | 6.62 | 1.21 |
| >10 | 43 | 0.73 | 12.86 | 3.25 |
| Total (>1) | 5904 | 100 | 1.82 | 1.58 |
| Chr | Start (MB) | End (MB) | No. SNPs | No. Genes | Candidate Genes | Gene Functions |
|---|---|---|---|---|---|---|
| 1 | 25.96 | 26.05 | 91 | 1 | - | - |
| 2 | 116.88 | 119.76 | 6054 | 8 | OCA2, LGSN | Meat and milk |
| 2 | 127.24 | 128.51 | 4739 | 0 | - | - |
| 3 | 107.12 | 107.77 | 851 | 5 | MERTK, ZC3H8 | Meat and heath |
| 3 | 167.83 | 168.54 | 791 | 0 | - | - |
| 5 | 43.42 | 43.83 | 302 | 1 | - | - |
| 5 | 49.58 | 50.67 | 1518 | 20 | GFRA3 | Health |
| 6 | 32.26 | 33.36 | 4024 | 4 | BMPR1B | Reproduction |
| 6 | 82.18 | 85.35 | 3911 | 1 | EPHA5 | Health |
| 10 | 35.46 | 39.69 | 12137 | 17 | FGF9, MICU2 | Reproduction |
| 10 | 42.23 | 45.97 | 11643 | 0 | - | - |
| 10 | 64.55 | 65.82 | 2732 | 0 | - | - |
| 11 | 34.06 | 34.77 | 517 | 36 | ALOX15B | Reproduction |
| 13 | 49.23 | 50.55 | 2321 | 2 | HAO1 | Meat |
| 13 | 53.31 | 54.35 | 1383 | 30 | GINS1 | Health |
| 15 | 3.23 | 3.45 | 968 | 0 | - | - |
| 15 | 61.59 | 62.62 | 562 | 0 | - | - |
| 17 | 38.73 | 38.89 | 332 | 17 | SPATA5 | Meat |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Chen, Z.; Fang, Y.; Cao, C.; Zhang, Z.; Pan, Y.; Wang, Q. Runs of Homozygosity Revealed Reproductive Traits of Hu Sheep. Genes 2022, 13, 1848. https://doi.org/10.3390/genes13101848
Li Y, Chen Z, Fang Y, Cao C, Zhang Z, Pan Y, Wang Q. Runs of Homozygosity Revealed Reproductive Traits of Hu Sheep. Genes. 2022; 13(10):1848. https://doi.org/10.3390/genes13101848
Chicago/Turabian StyleLi, Yuzhe, Zitao Chen, Yifei Fang, Caiyun Cao, Zhe Zhang, Yuchun Pan, and Qishan Wang. 2022. "Runs of Homozygosity Revealed Reproductive Traits of Hu Sheep" Genes 13, no. 10: 1848. https://doi.org/10.3390/genes13101848
APA StyleLi, Y., Chen, Z., Fang, Y., Cao, C., Zhang, Z., Pan, Y., & Wang, Q. (2022). Runs of Homozygosity Revealed Reproductive Traits of Hu Sheep. Genes, 13(10), 1848. https://doi.org/10.3390/genes13101848