
Elucidating Hexanucleotide Repeat Number and Methylation within the X-Linked Dystonia-Parkinsonism (XDP)-Related SVA Retrotransposon in TAF1 with Nanopore Sequencing

 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Demographics
2.2. Single-Nucleotide Variants and Repeat Detection
2.3. Methylation Detection
2.4. Data Analysis
2.5. Statistical Analysis
3. Results
3.1. Single-Nucleotide Polymorphisms within the SVA TAF1 Insertion
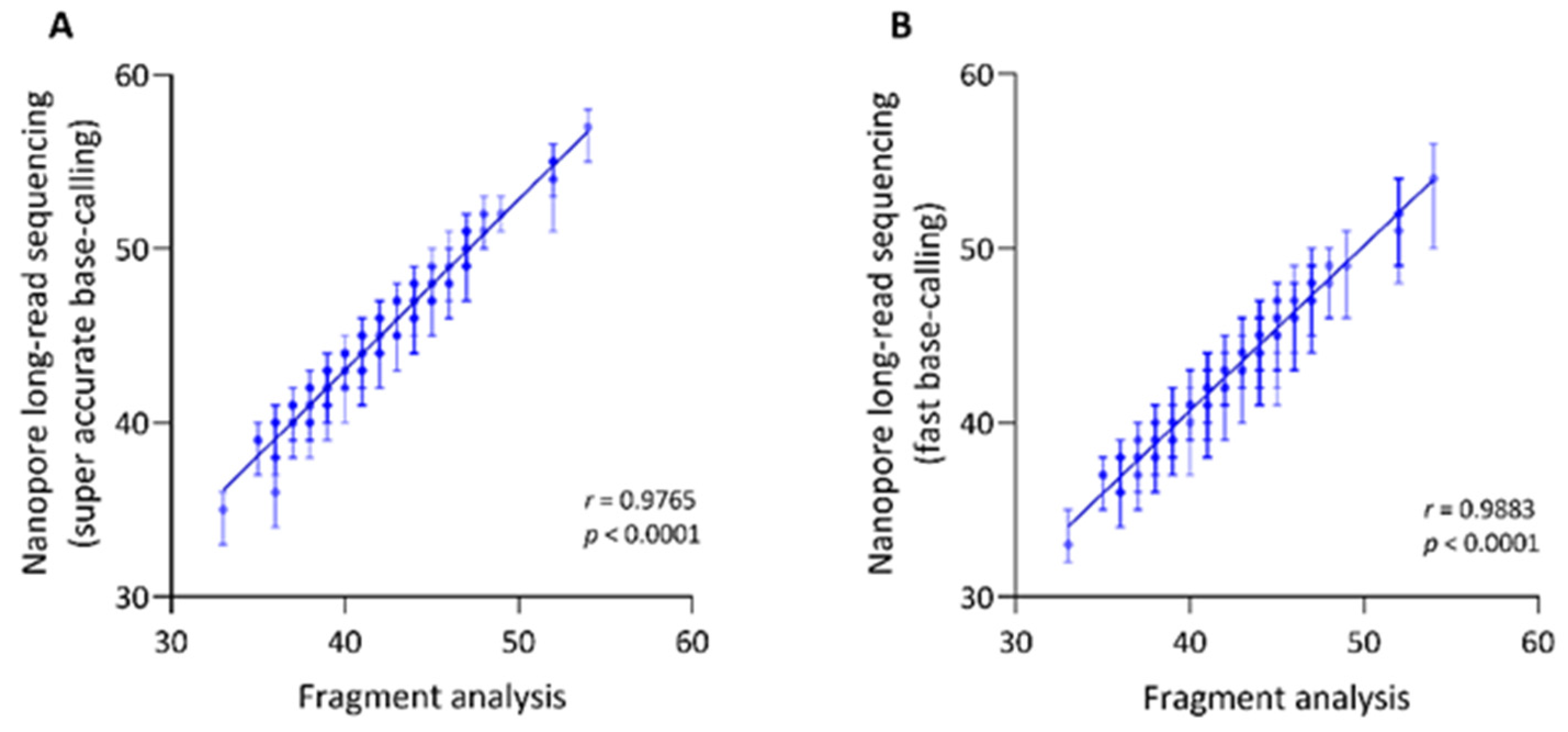
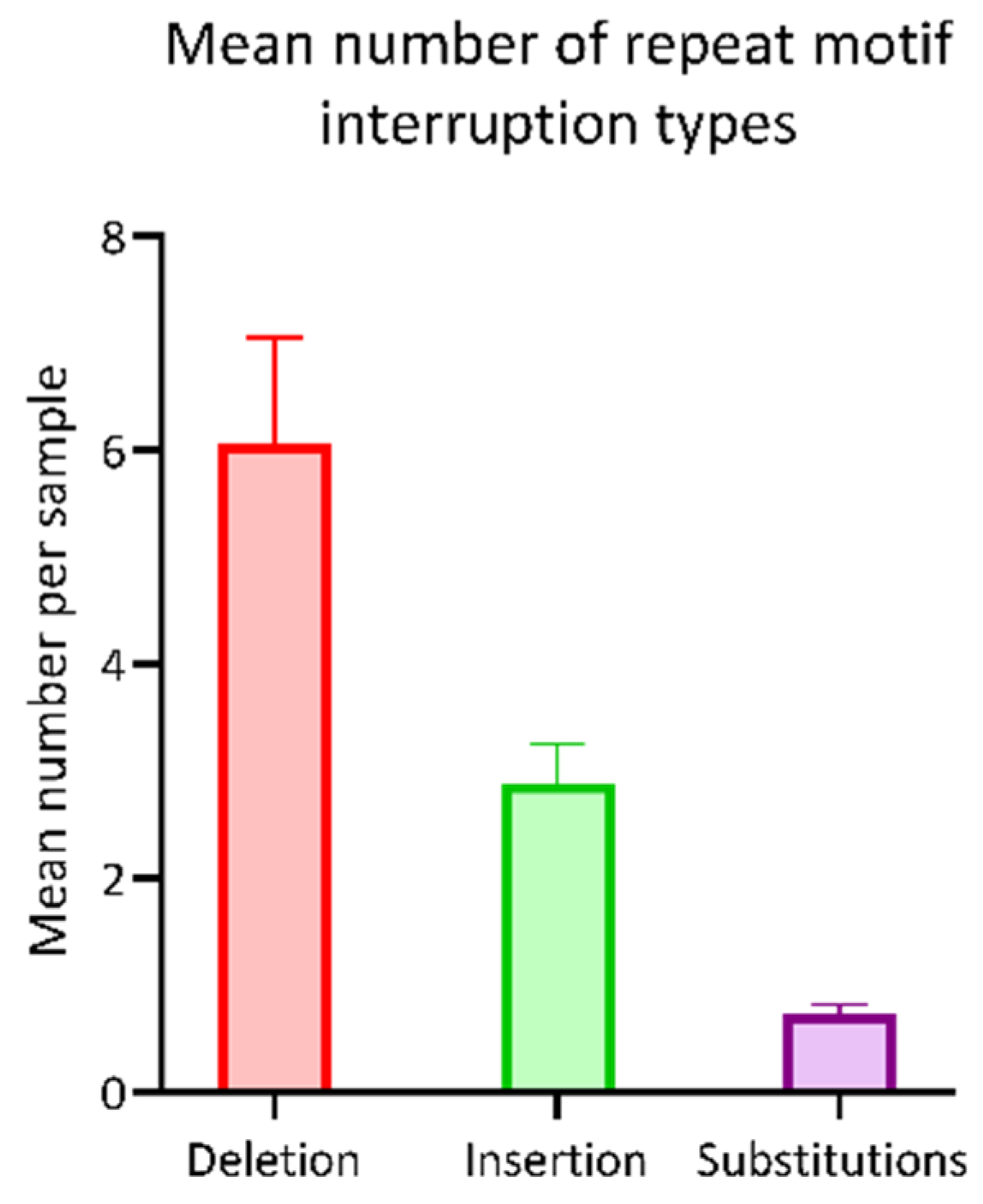
3.2. Assessment of the Hexanucleotide Repeat Length
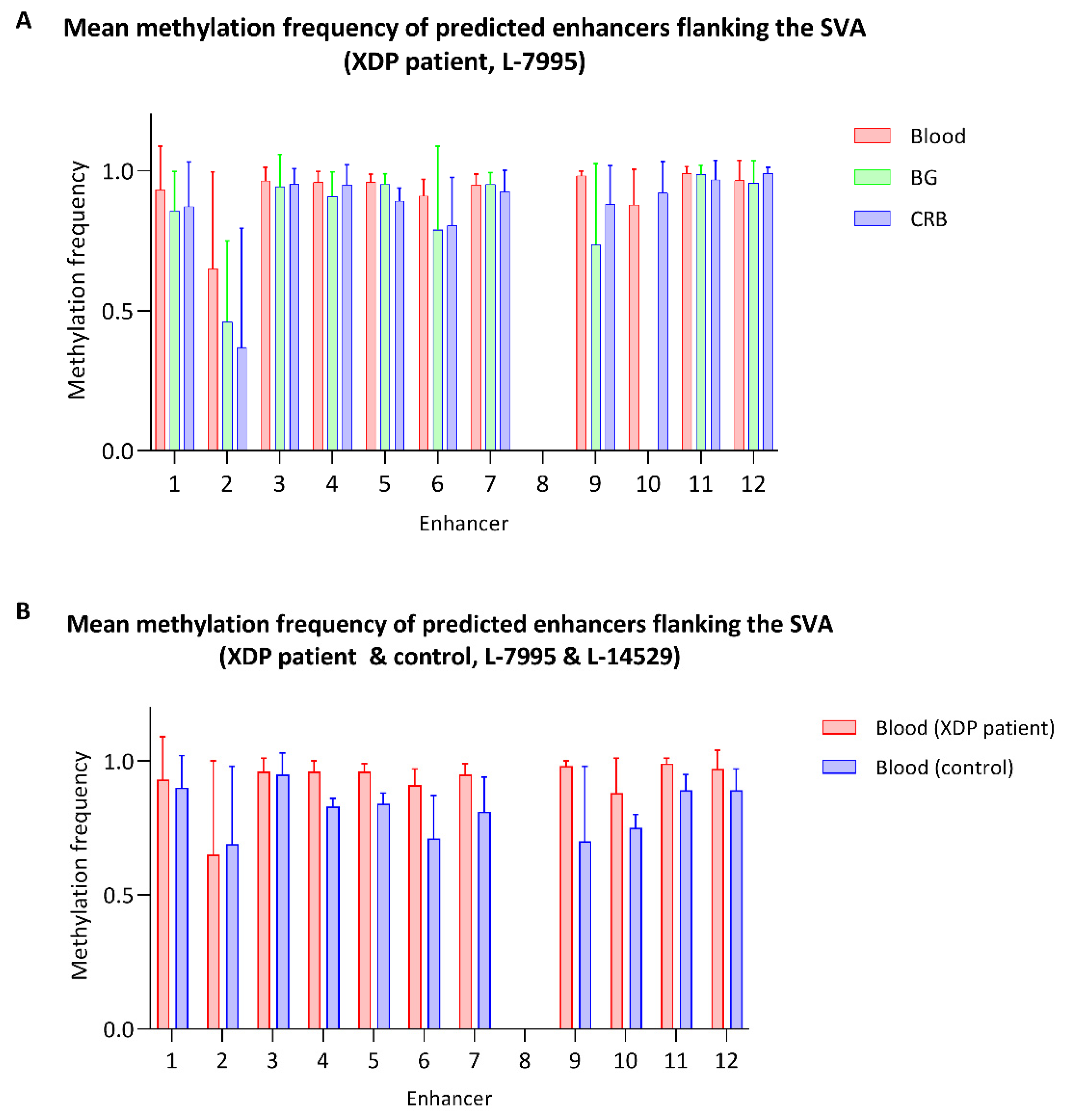
3.3. Methylation within the SVA and in the Flanking Regions
4. Discussion
4.1. Examination of the Hexanucleotide Repeat Domain
4.2. Methylation Status of the TAF1 SVA Insertion and Adjacent Enhancer Sites
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, L.V.; Pascasio, F.M.; Fuentes, F.D.; Viterbo, G.H. Torsion dystonia in Panay, Philippines. Adv. Neurol. 1976, 14, 137–151. [Google Scholar]
- Rosales, R.L. X-linked dystonia parkinsonism: Clinical phenotype, genetics and therapeutics. J. Mov. Disord. 2010, 3, 32–38. [Google Scholar] [CrossRef]
- Lee, L.V.; Munoz, E.L.; Tan, K.T.; Reyes, M.T. Sex linked recessive dystonia parkinsonism of Panay, Philippines (XDP). Mol. Pathol. 2001, 54, 362–368. [Google Scholar] [PubMed]
- Pauly, M.G.; Lopez, M.R.; Westenberger, A.; Saranza, G.; Bruggemann, N.; Weissbach, A.; Rosales, R.L.; Diesta, C.C.; Jamora, R.D.G.; Reyes, C.J.; et al. Expanding Data Collection for the MDSGene Database: X-linked Dystonia-Parkinsonism as Use Case Example. Mov. Disord. 2020, 35, 1933–1938. [Google Scholar] [CrossRef]
- Bragg, D.C.; Mangkalaphiban, K.; Vaine, C.A.; Kulkarni, N.J.; Shin, D.; Yadav, R.; Dhakal, J.; Ton, M.L.; Cheng, A.; Russo, C.T.; et al. Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proc. Natl. Acad. Sci. USA 2017, 114, E11020–E11028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westenberger, A.; Reyes, C.J.; Saranza, G.; Dobricic, V.; Hanssen, H.; Domingo, A.; Laabs, B.H.; Schaake, S.; Pozojevic, J.; Rakovic, A.; et al. A hexanucleotide repeat modifies expressivity of X-linked dystonia parkinsonism. Ann. Neurol. 2019, 85, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Reyes, C.J.; Laabs, B.H.; Schaake, S.; Luth, T.; Ardicoglu, R.; Rakovic, A.; Grutz, K.; Alvarez-Fischer, D.; Jamora, R.D.; Rosales, R.L.; et al. Brain Regional Differences in Hexanucleotide Repeat Length in X-Linked Dystonia-Parkinsonism Using Nanopore Sequencing. Neurol. Genet. 2021, 7, e608. [Google Scholar] [CrossRef]
- Domingo, A.; Westenberger, A.; Lee, L.V.; Braenne, I.; Liu, T.; Vater, I.; Rosales, R.; Jamora, R.D.; Pasco, P.M.; Cutiongco-Dela Paz, E.M.; et al. New insights into the genetics of X-linked dystonia-parkinsonism (XDP, DYT3). Eur. J. Hum. Genet. 2015, 23, 1334–1340. [Google Scholar] [CrossRef]
- Makino, S.; Kaji, R.; Ando, S.; Tomizawa, M.; Yasuno, K.; Goto, S.; Matsumoto, S.; Tabuena, M.D.; Maranon, E.; Dantes, M.; et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am. J. Hum. Genet. 2007, 80, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakovic, A.; Ziegler, J.; Martensson, C.U.; Prasuhn, J.; Shurkewitsch, K.; Konig, P.; Paulson, H.L.; Klein, C. PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ. 2019, 26, 1428–1441. [Google Scholar] [CrossRef]
- Aneichyk, T.; Hendriks, W.T.; Yadav, R.; Shin, D.; Gao, D.; Vaine, C.A.; Collins, R.L.; Domingo, A.; Currall, B.; Stortchevoi, A.; et al. Dissecting the Causal Mechanism of X-Linked Dystonia-Parkinsonism by Integrating Genome and Transcriptome Assembly. Cell 2018, 172, 897–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewing, A.D.; Smits, N.; Sanchez-Luque, F.J.; Faivre, J.; Brennan, P.M.; Richardson, S.R.; Cheetham, S.W.; Faulkner, G.J. Nanopore Sequencing Enables Comprehensive Transposable Element Epigenomic Profiling. Mol. Cell 2020, 80, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xing, J.; Grover, D.; Hedges, D.J.; Han, K.; Walker, J.A.; Batzer, M.A. SVA elements: A hominid-specific retroposon family. J. Mol. Biol. 2005, 354, 994–1007. [Google Scholar] [CrossRef]
- Watkins, W.S.; Feusier, J.E.; Thomas, J.; Goubert, C.; Mallick, S.; Jorde, L.B. The Simons Genome Diversity Project: A Global Analysis of Mobile Element Diversity. Genome Biol. Evol. 2020, 12, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, A.L.; Bubb, V.J.; Quinn, J.P.; Koks, S. Reference SVA insertion polymorphisms are associated with Parkinson’s Disease progression and differential gene expression. npj Parkinsons Dis. 2021, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Montague, T.G.; Cruz, J.M.; Gagnon, J.A.; Church, G.M.; Valen, E. CHOPCHOP: A CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014, 42, W401–W407. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.S.; Cechova, M.; Makova, K.D. Noise-cancelling repeat finder: Uncovering tandem repeats in error-prone long-read sequencing data. Bioinformatics 2019, 35, 4809–4811. [Google Scholar] [CrossRef] [Green Version]
- De Coster, W.; De Rijk, P.; De Roeck, A.; De Pooter, T.; D’Hert, S.; Strazisar, M.; Sleegers, K.; Van Broeckhoven, C. Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res. 2019, 29, 1178–1187. [Google Scholar] [CrossRef] [Green Version]
- Bragg, D.C.; Sharma, N.; Ozelius, L.J. X-Linked Dystonia-Parkinsonism: Recent advances. Curr. Opin. Neurol. 2019, 32, 604–609. [Google Scholar] [CrossRef]
- Nethisinghe, S.; Kesavan, M.; Ging, H.; Labrum, R.; Polke, J.M.; Islam, S.; Garcia-Moreno, H.; Callaghan, M.F.; Cavalcanti, F.; Pook, M.A.; et al. Interruptions of the FXN GAA Repeat Tract Delay the Age at Onset of Friedreich’s Ataxia in a Location Dependent Manner. Int. J. Mol. Sci. 2021, 22, 7507. [Google Scholar] [CrossRef]
- Findlay Black, H.; Wright, G.E.B.; Collins, J.A.; Caron, N.; Kay, C.; Xia, Q.; Arning, L.; Bijlsma, E.K.; Squitieri, F.; Nguyen, H.P.; et al. Frequency of the loss of CAA interruption in the HTT CAG tract and implications for Huntington disease in the reduced penetrance range. Genet. Med. 2020, 22, 2108–2113. [Google Scholar] [CrossRef]
- Giesselmann, P.; Brandl, B.; Raimondeau, E.; Bowen, R.; Rohrandt, C.; Tandon, R.; Kretzmer, H.; Assum, G.; Galonska, C.; Siebert, R.; et al. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat. Biotechnol. 2019, 37, 1478–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 2020, 38, 433–438. [Google Scholar] [CrossRef]
- Capponi, S.; Stöffler, N.; Penney, E.B.; Grütz, K.; Nizamuddin, S.; Vermunt, M.W.; Castelijns, B.; Fernandez-Cerado, C.; Legarda, G.P.; Velasco-Andrada, M.S.; et al. Dissection of TAF1 neuronal splicing and implications for neurodegeneration in X-linked dystonia-parkinsonism. Brain Commun. 2021, 3, fcab253. [Google Scholar] [CrossRef]
- Valente, E.M.; Bhatia, K.P. Solving Mendelian Mysteries: The Non-coding Genome May Hold the Key. Cell 2018, 172, 889–891. [Google Scholar] [CrossRef] [Green Version]
- Yates, P.A.; Burman, R.W.; Mummaneni, P.; Krussel, S.; Turker, M.S. Tandem B1 elements located in a mouse methylation center provide a target for de novo DNA methylation. J. Biol. Chem. 1999, 274, 36357–36361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.; Razaghi, R.; Gilpatrick, T.; Molnar, M.; Gershman, A.; Sadowski, N.; Sedlazeck, F.J.; Hansen, K.D.; Simpson, J.T.; Timp, W. Simultaneous profiling of chromatin accessibility and methylation on human cell lines with nanopore sequencing. Nat. Methods 2020, 17, 1191–1199. [Google Scholar] [CrossRef]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.K.; Kilaru, V.; Klengel, T.; Mercer, K.B.; Bradley, B.; Conneely, K.N.; Ressler, K.J.; Binder, E.B. DNA extracted from saliva for methylation studies of psychiatric traits: Evidence tissue specificity and relatedness to brain. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2015, 168B, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, P.R.; Han, S.; Hing, B.; Nagahama, Y.; Gaul, L.N.; Heinzman, J.T.; Grossbach, A.J.; Close, L.; Dlouhy, B.J.; Howard, M.A., 3rd; et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl. Psychiatry 2019, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Krause, C.; Schaake, S.; Grutz, K.; Sievert, H.; Reyes, C.J.; Konig, I.R.; Laabs, B.H.; Jamora, R.D.; Rosales, R.L.; Diesta, C.C.E.; et al. DNA Methylation as a Potential Molecular Mechanism in X-linked Dystonia-Parkinsonism. Mov. Disord. 2020, 35, 2220–2229. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lüth, T.; Laβ, J.; Schaake, S.; Wohlers, I.; Pozojevic, J.; Jamora, R.D.G.; Rosales, R.L.; Brüggemann, N.; Saranza, G.; Diesta, C.C.E.; et al. Elucidating Hexanucleotide Repeat Number and Methylation within the X-Linked Dystonia-Parkinsonism (XDP)-Related SVA Retrotransposon in TAF1 with Nanopore Sequencing. Genes 2022, 13, 126. https://doi.org/10.3390/genes13010126
Lüth T, Laβ J, Schaake S, Wohlers I, Pozojevic J, Jamora RDG, Rosales RL, Brüggemann N, Saranza G, Diesta CCE, et al. Elucidating Hexanucleotide Repeat Number and Methylation within the X-Linked Dystonia-Parkinsonism (XDP)-Related SVA Retrotransposon in TAF1 with Nanopore Sequencing. Genes. 2022; 13(1):126. https://doi.org/10.3390/genes13010126
Chicago/Turabian StyleLüth, Theresa, Joshua Laβ, Susen Schaake, Inken Wohlers, Jelena Pozojevic, Roland Dominic G. Jamora, Raymond L. Rosales, Norbert Brüggemann, Gerard Saranza, Cid Czarina E. Diesta, and et al. 2022. "Elucidating Hexanucleotide Repeat Number and Methylation within the X-Linked Dystonia-Parkinsonism (XDP)-Related SVA Retrotransposon in TAF1 with Nanopore Sequencing" Genes 13, no. 1: 126. https://doi.org/10.3390/genes13010126
APA StyleLüth, T., Laβ, J., Schaake, S., Wohlers, I., Pozojevic, J., Jamora, R. D. G., Rosales, R. L., Brüggemann, N., Saranza, G., Diesta, C. C. E., Schlüter, K., Tse, R., Reyes, C. J., Brand, M., Busch, H., Klein, C., Westenberger, A., & Trinh, J. (2022). Elucidating Hexanucleotide Repeat Number and Methylation within the X-Linked Dystonia-Parkinsonism (XDP)-Related SVA Retrotransposon in TAF1 with Nanopore Sequencing. Genes, 13(1), 126. https://doi.org/10.3390/genes13010126