
Ancient Mitogenomes Suggest Stable Mitochondrial Clades of the Siberian Roe Deer

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. DNA Extraction
2.3. Library Construction
2.4. Data Processing and Data Sets
2.5. Bioinformatics Analyses
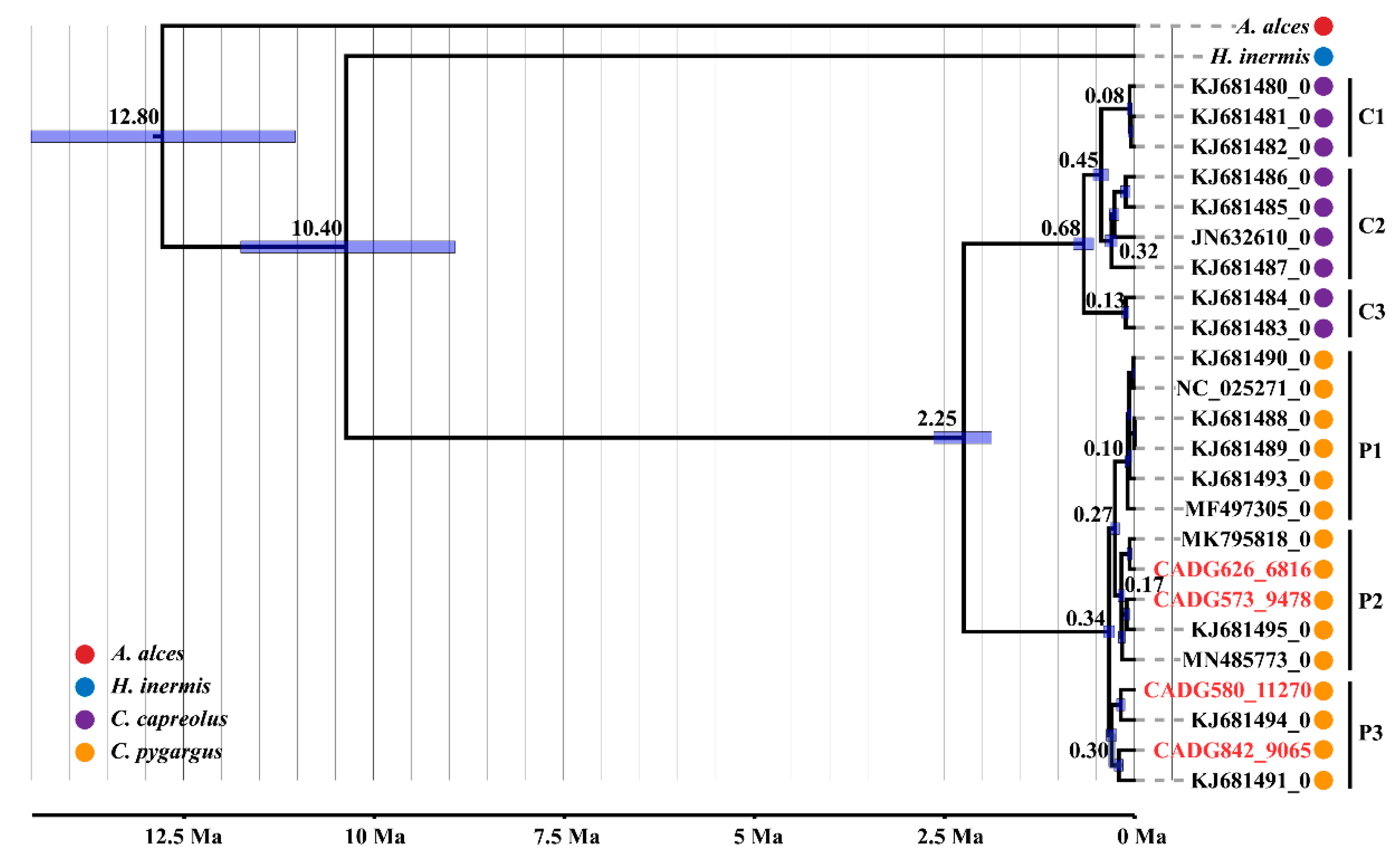
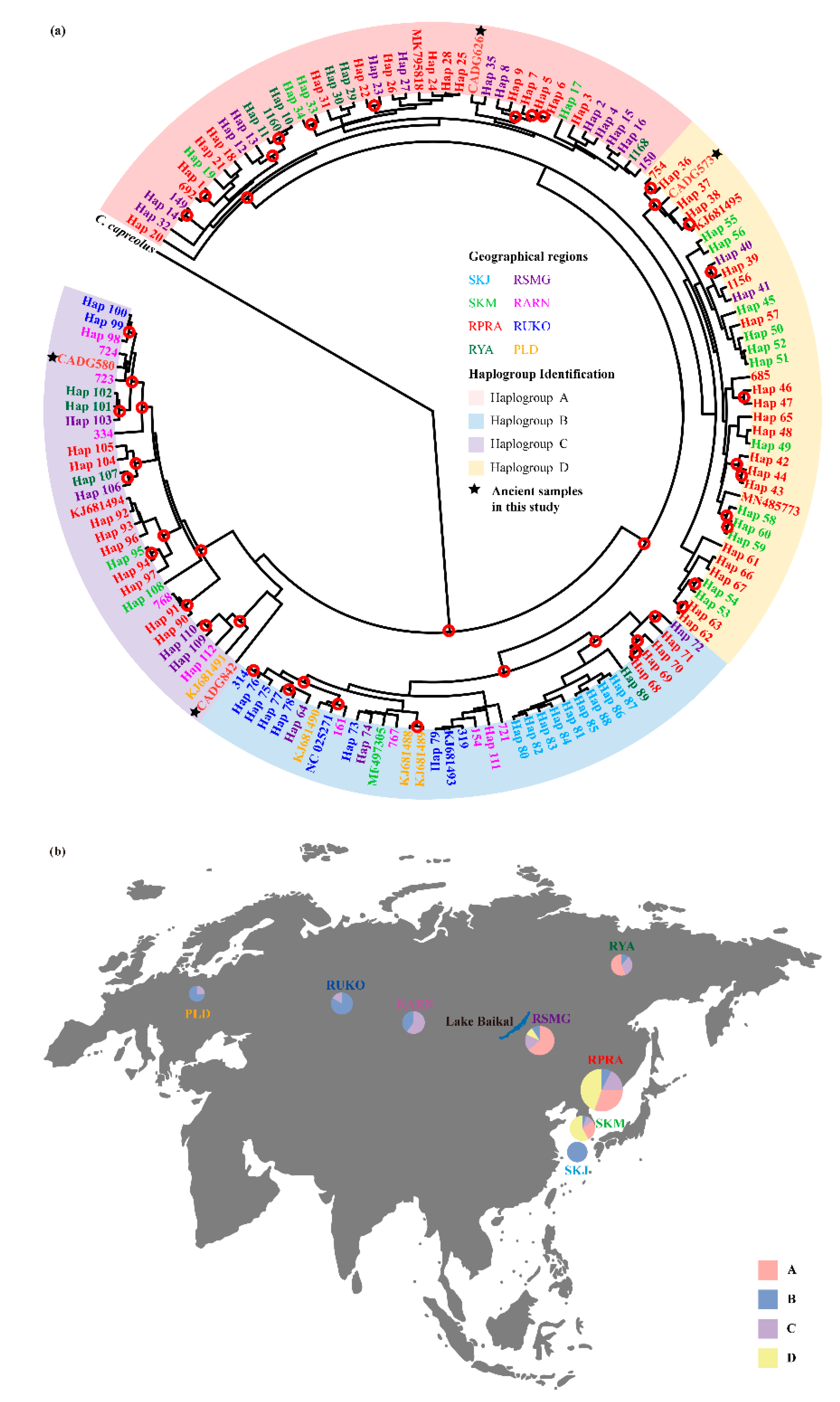
3. Results
4. Discussion
4.1. Phylogeny and Phylogeography of the Roe Deer in Northeastern China
4.2. Divergence among Capreolus
4.3. Maternal Demographic History of the Roe Deer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vislobokova, I.; Dmitrieva, E.; Kalmykov, N. Artiodactyls from the Late Pliocene of Udunga, Western Trans-Baikal, Russia. J. Vertebr. Paléontol. 1995, 15, 146–159. [Google Scholar] [CrossRef]
- Randi, E.; Pierpaoli, M.; Danilkin, A. Mitochondrial DNA polymorphism in populations of Siberian and European roe deer (Capreolus pygargus and C. capreolus). Heredity 1998, 80, 429–437. [Google Scholar] [CrossRef]
- Sokolov, V.; Gromov, V. The contemporary ideas on roe deer (Capreolus Gray, 1821) systematization: Morphological, ethological and hybridological analysis. Mammalia 1990, 54, 431–444. [Google Scholar] [CrossRef]
- Danilkin, A.A. Capreolus pygargus. Mamm. Species 1995, 512, 1–7. [Google Scholar] [CrossRef]
- Sheremetyeva, I.N.; Sheremetyev, I.S. Skull variation in the Siberian roe deer Capreolus pygargus from the Far East: A revision of the distribution of the subspecies. Eur. J. Wildl. Res. 2008, 54, 557–569. [Google Scholar] [CrossRef]
- Koh, H.S.; Bayarlkhagva, D.; Jang, K.H.; Han, E.D.; Jo, J.E.; Ham, E.J.; Jeong, S.K.; Lee, J.H.; Kim, K.S.; Kweon, G.H.; et al. Genetic divergence of the Siberian roe deer from Korean Jeju Island (Capreolus pygargus ochraceus), reexamined from nuclear IRBP and mitochondrial cytochrome b and control region sequences of C. pygargus. J. Biol. Res. 2013, 19, 46–55. [Google Scholar]
- Sokolov, V.E.; Shurkhal, A.V.; Danilkin, A.A.; Podogas, A.V.; Rakitskaya, T.A.; Markov, G.G. A comparative analysis of electrophoretic spectra of blood and muscle tissue proteins of European (Capreolus capreolus L.) and Siberian (Capreolus pygargus Pall.) roe deer. Dokl. Akad. Nauk 1986, 288, 1274–1276. [Google Scholar]
- Danilkin, A.A.; Markov, G.G. On the taxonomy of roe deer (Capreolus pygargus Pall.) of the Far East, East Siberia and Tien Shan. Izv. Akad. Nauk. SSSR 1987, 2, 315–318. [Google Scholar]
- Vorobieva, N.V.; Sherbakov, D.; Druzhkova, A.S.; Stanyon, R.; Tsybankov, A.A.; Vasil’Ev, S.K.; Shunkov, M.V.; Trifonov, V.; Graphodatsky, A. Genotyping of Capreolus pygargus Fossil DNA from Denisova Cave Reveals Phylogenetic Relationships between Ancient and Modern Populations. PLoS ONE 2011, 6, e24045. [Google Scholar] [CrossRef]
- Lee, Y.S.; Markov, N.; Argunov, A.; Voloshina, I.; Bayarlkhagva, D.; Kim, B.; Min, M.; Lee, H.; Kim, K.S. Genetic diversity and phylogeography of Siberian roe deer, Caproulus pygargus, in central and peripheral populations. Ecol. Evol. 2016, 6, 7286–7297. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, V.G.; Tokarskaya, O.N.; Danilkin, A.A.; Ryskov, A. Quantitative Analysis of Genetic Parameters in Populations of European (Capreolus capreolus L.) and Siberian (Capreolus pygargus Pall.) Roe Deer with RAPD Markers. Russ. J. Genet. 2002, 38, 676–683. [Google Scholar] [CrossRef]
- Sheremet’Eva, I.N.; Sheremet’Ev, I.S.; Kartavtseva, I.V.; Zhuravlev, I.N. Polymorphism of a short fragment of the mitochondrial genome control region (D-loop) in the Siberian roe deer Capreolus pygargus Pallas, 1771 (Artiodactyla, Cervidae) from the Russian Far East. Генетика 2010, 46, 677–684. [Google Scholar] [CrossRef]
- ZvychaĬnaia, E.I.; A Danilkin, A.; Kholodova, M.V.; Sipko, T.P.; Berber, A.P. Analysis of the variability of the control region and cytochrome b gene of mtDNA of Capreolus pygargus Pall. Izv. Akad. Nauk. Seriia Boil. 2011, 5, 511–517. [Google Scholar] [CrossRef]
- Lorenzini, R.; Garofalo, L.; Qin, X.; Voloshina, I.; Lovari, S. Global phylogeography of the genusCapreolus(Artiodactyla: Cervidae), a Palaearctic meso-mammal. Zool. J. Linn. Soc. 2014, 170, 209–221. [Google Scholar] [CrossRef]
- Zong, G.; Tang, Y.; Xu, Q. The Early Pleistocene of Tunlius County, Shanxi Province. Vertebr. PalAsiatica 1982, 20, 236–247. [Google Scholar]
- Valli, A.M. Dispersion of the genus Procapreolus and the relationships between Procapreolus cusanus and the roe deer (Capreolus). Quat. Int. 2008, 212, 80–85. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Zou, B.K.; Zhang, L.K. The discovery of fossil mammals at Anping, Liaoyang. Vertebr. PalAsiatica 1980, 18, 74–109. [Google Scholar]
- Zhou, X.X.; Sun, Y.F.; Wang, J.M. A new mammalian fauna found in Gulongshan, Dalian. Vertebr. PalAsiatica 1984, 22, 73–98. [Google Scholar]
- Hu, S.M.; Zhang, P.C.; Yuan, M. A Study on the faunal remains from the Huoshiliang site in Yulin, Shaanxi. Acta Anthropol. Sin. 2008, 27, 232–248. [Google Scholar]
- Wei, H.B.; Liu, Y.H. The pleistocene mammalian faunas in Northeastern China and related paleoenvironment changes. In Proceedings of the Twelfth Annual Meeting of the Chinese Society of Vertebrate Paleontology, Linyi, China, 13–15 September 2010; pp. 53–60. [Google Scholar]
- Chen, Q.J.; Zhao, H.L.; Wang, F.G.; Wang, C.X. A preliminary report of animal fossils and pollen from the Xianrendong paleolithic site in Huadian City, Jilin Province. Acta Anthropol. Sin. 2013, 32, 52–62. [Google Scholar]
- Dong, W.; Liu, W.H.; Bai, W.P. Cladistic approach on chronological relationship of the Pleistocene mammalian faunas from China. Vertebr. PalAsiatica 2020, 58, 67–81. [Google Scholar]
- Meyer, M.; Kircher, M. Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing. Cold Spring Harb. Protoc. 2010, 2010, 5448. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Margaryan, A.; Sinding, M.H.S.; Liu, S.; Vieira, F.G.; Chan, Y.L.; Nathan, S.K.S.S.; Moodley, Y.; Bruford, M.W.; Gilbert, M.T.P. Recent mitochondrial lineage extinction in the critically endangered Javan rhinoceros. Zool. J. Linn. Soc. 2020, 190, 372–383. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, J.; Yu, J.; Gibbs, R.A.; Yu, F. An integrative variant analysis pipeline for accurate genotype/haplotype inference in population NGS data. Genome Res. 2013, 23, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.F.; Orlando, L. mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef]
- Hassanin, A.; Delsuc, F.; Ropiquet, A.; Hammer, C.; van Vuuren, B.J.; Matthee, C.; Ruiz-Garcia, M.; Catzeflis, F.; Areskoug, V.; Nguyen, T.T.; et al. Pattern and timing of diversification of Cetartiodactyla (Mammalia, Laurasiatheria), as revealed by a comprehensive analysis of mitochondrial genomes. Comptes Rendus. Biol. 2012, 335, 32–50. [Google Scholar] [CrossRef]
- Matosiuk, M.; Sheremetyeva, I.N.; Saveljev, A.; Borkowska, A. Evolutionary neutrality of mtDNA introgression: Evidence from complete mitogenome analysis in roe deer. J. Evol. Biol. 2014, 27, 2483–2494. [Google Scholar] [CrossRef]
- Liu, H.; Jiang, G. Complete mitochondrial genome sequence of Ussurian moose, Alces alces cameloides. Mitochondrial DNA Part A 2015, 27, 4199–4200. [Google Scholar] [CrossRef]
- Kim, H.R.; Jeon, M.G.; Min, J.H.; Kim, H.J.; Park, Y.C. Complete mitochondrial genome of the roe deer Capreolus pygargus tianschanicus (Cervidae) from Korea. Mitochondrial DNA Part B 2017, 2, 558–559. [Google Scholar] [CrossRef]
- Li, T.; Liang, Y.-K.; Zhang, J.-J.; Ren, Z.-M. Complete mitochondrial genome of Capreolus pygargus (Cervidae: Capreolinae), a protected and threatened species in China. Mitochondrial DNA Part B 2019, 4, 2469–2470. [Google Scholar] [CrossRef]
- Ao, D.; Yao, Y.; Li, D.; Xie, M.; Ni, Q.; Zhang, M.; Xu, H. The complete mitochondrial genome of Siberian roe deer (Capreolus pygargus bedfordi) and its phylogenetic analysis. Mitochondrial DNA Part B 2020, 5, 1122–1123. [Google Scholar] [CrossRef] [PubMed]
- Kazutaka, K.; Misakwa, K.; Kei-ichi, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A Rapid Bootstrap Algorithm for the RAxML Web Servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Mennecart, B.; DeMiguel, D.; Bibi, F.; Rössner, G.E.; Métais, G.; Neenan, J.M.; Wang, S.; Schulz, G.; Müller, B.; Costeur, L. Bony labyrinth morphology clarifies the origin and evolution of deer. Sci. Rep. 2017, 7, 13176. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Xiao, C.-T.; Zhang, M.-H.; Fu, Y.; Koh, H.-S. Mitochondrial DNA Distinction of Northeastern China Roe Deer, Siberian Roe Deer, and European Roe Deer, to Clarify the Taxonomic Status of Northeastern China Roe Deer. Biochem. Genet. 2007, 45, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Matosiuk, M.; Borkowska, A.; Świsłocka, M.; Mirski, P.; Borowski, Z.; Krysiuk, K.; Danilkin, A.A.; Zvychaynaya, E.Y.; Saveljev, A.; Ratkiewicz, M. Unexpected population genetic structure of European roe deer in Poland: An invasion of the mtDNA genome from Siberian roe deer. Mol. Ecol. 2014, 23, 2559–2572. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Markov, N.; Voloshina, I.; Argunov, A.; Bayarlkhagva, D.; Oh, J.G.; Park, Y.-S.; Min, M.-S.; Lee, H.; Kim, K.S. Genetic diversity and genetic structure of the Siberian roe deer (Capreolus pygargus) populations from Asia. BMC Genet. 2015, 16, 100. [Google Scholar] [CrossRef]
- Zhang, M.H.; Xiao, C.T.; Koh, H.S. Taxonomic status of roe deer in Northeastern China based on mitochondrial DNA sequences. Acta Theriol. Sin. 2005, 25, 14–19. [Google Scholar]
- Zhang, M.H.; Liu, Y.H.; Jia, J.B. Population genetic diversity of roe deer (Capreolus pygargus) in Northeastern China. Acta Theriol. Sin. 2010, 30, 58–64. [Google Scholar]
- Liu, M.Y.; Xie, Y.H.; JI, D.M.; Gao, Z.X.; Li, S.Z.; Gao, W. Complete Vertebrate of China; Liaoning University Press: Shenyang, China, 2000; ISBN 978-7-5610-3904-5. [Google Scholar]
- Ellerman, J.R.; Morrisonscott, T.C.S. Checklist of Palaearctic and Indian Mammals 1758 to 1946; Brtish Museum (Natural History): London, UK, 2011; ISBN 978-1-1749-0774-6. [Google Scholar]
- Geptner, V.G.; Nasimovich, A.A.; Bannikoc, A.G.; Hoffmann, R.S. Mammals of the Soviet Union; Smithsonian Institution Libraries and National Science Foundation: Washington, DC, USA, 1988; Available online: https://library.si.edu/digital-library/book/mammalsofsovietu11988gept (accessed on 3 January 2022).
- Dussex, N.; Alberti, F.; Heino, M.T.; Olsen, R.-A.; Van Der Valk, T.; Ryman, N.; Laikre, L.; Ahlgren, H.; Askeyev, I.V.; Askeyev, O.V.; et al. Moose genomes reveal past glacial demography and the origin of modern lineages. BMC Genom. 2020, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Groves, C.; Grubb, P. Ungulate Taxonomy; The Johns Hopkins University Press: Baltimore, MD, USA, 2011; pp. 59–84. ISBN 978-1-4214-0093-8. [Google Scholar]
- Kryukov, A.P. Comparative phylogeographic patterns of several vertebrates in the east Palearctic. Mosc. Univ. Biol. Sci. Bull. 2010, 65, 184–186. [Google Scholar] [CrossRef]
- Courtin, J.; Andreev, A.A.; Raschke, E.; Bala, S.; Biskaborn, B.K.; Liu, S.; Zimmermann, H.; Diekmann, B.; Stoof-Leichsenring, K.R.; Pestryakova, L.A.; et al. Vegetation Changes in Southeastern Siberia During the Late Pleistocene and the Holocene. Front. Ecol. Evol. 2021, 9, 625096. [Google Scholar] [CrossRef]
- De Jong, M.J.; Li, Z.; Qin, Y.; Quéméré, E.; Baker, K.; Wang, W.; Hoelzel, A.R. Demography and adaptation promoting evolutionary transitions in a mammalian genus that diversified during the Pleistocene. Mol. Ecol. 2020, 29, 2777–2792. [Google Scholar] [CrossRef]
- Sommer, R.S.; Fahlke, J.M.; Schmölcke, U.; Benecke, N.; Zachos, F.E. Quaternary history of the European roe deer Capreolus capreolus. Mamm. Rev. 2009, 39, 1–16. [Google Scholar] [CrossRef]
- Randi, E.; Alves, P.C.; Carranza, J.; Milošević-Zlatanović, S.; Sfougaris, A.; Mucci, N. Phylogeography of roe deer (Capreolus capreolus) populations: The effects of historical genetic subdivisions and recent nonequilibrium dynamics. Mol. Ecol. 2004, 13, 3071–3083. [Google Scholar] [CrossRef] [PubMed]
- Lord, E.; Dussex, N.; Kierczak, M.; Díez-Del-Molino, D.; Ryder, O.A.; Stanton, D.W.; Gilbert, M.T.P.; Sánchez-Barreiro, F.; Zhang, G.; Sinding, M.-H.S.; et al. Pre-extinction Demographic Stability and Genomic Signatures of Adaptation in the Woolly Rhinoceros. Curr. Biol. 2020, 30, 3871–3879.e7. [Google Scholar] [CrossRef] [PubMed]
- Meiri, M.; Lister, A.; Higham, T.; Stewart, J.R.; Straus, L.G.; Obermaier, H.; Morales, M.R.G.; Marin-Arroyo, A.B.; Barnes, I. Late-glacial recolonization and phylogeography of European red deer (Cervus elaphusL.). Mol. Ecol. 2013, 22, 4711–4722. [Google Scholar] [CrossRef]
- Li, X.L. Pleistocene climate change and human evolution in Eurasia. Fossil 2019, 1, 8–15. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, M.-X.; Xiao, B.; Yuan, J.-X.; Hu, J.-M.; Kim, K.S.; Westbury, M.V.; Lai, X.-L.; Sheng, G.-L. Ancient Mitogenomes Suggest Stable Mitochondrial Clades of the Siberian Roe Deer. Genes 2022, 13, 114. https://doi.org/10.3390/genes13010114
Deng M-X, Xiao B, Yuan J-X, Hu J-M, Kim KS, Westbury MV, Lai X-L, Sheng G-L. Ancient Mitogenomes Suggest Stable Mitochondrial Clades of the Siberian Roe Deer. Genes. 2022; 13(1):114. https://doi.org/10.3390/genes13010114
Chicago/Turabian StyleDeng, Miao-Xuan, Bo Xiao, Jun-Xia Yuan, Jia-Ming Hu, Kyung Seok Kim, Michael V. Westbury, Xu-Long Lai, and Gui-Lian Sheng. 2022. "Ancient Mitogenomes Suggest Stable Mitochondrial Clades of the Siberian Roe Deer" Genes 13, no. 1: 114. https://doi.org/10.3390/genes13010114
APA StyleDeng, M.-X., Xiao, B., Yuan, J.-X., Hu, J.-M., Kim, K. S., Westbury, M. V., Lai, X.-L., & Sheng, G.-L. (2022). Ancient Mitogenomes Suggest Stable Mitochondrial Clades of the Siberian Roe Deer. Genes, 13(1), 114. https://doi.org/10.3390/genes13010114