
GDF6 Knockdown in a Family with Multiple Synostosis Syndrome and Speech Impairment




Abstract
1. Introduction
2. Materials and Methods
3. Results
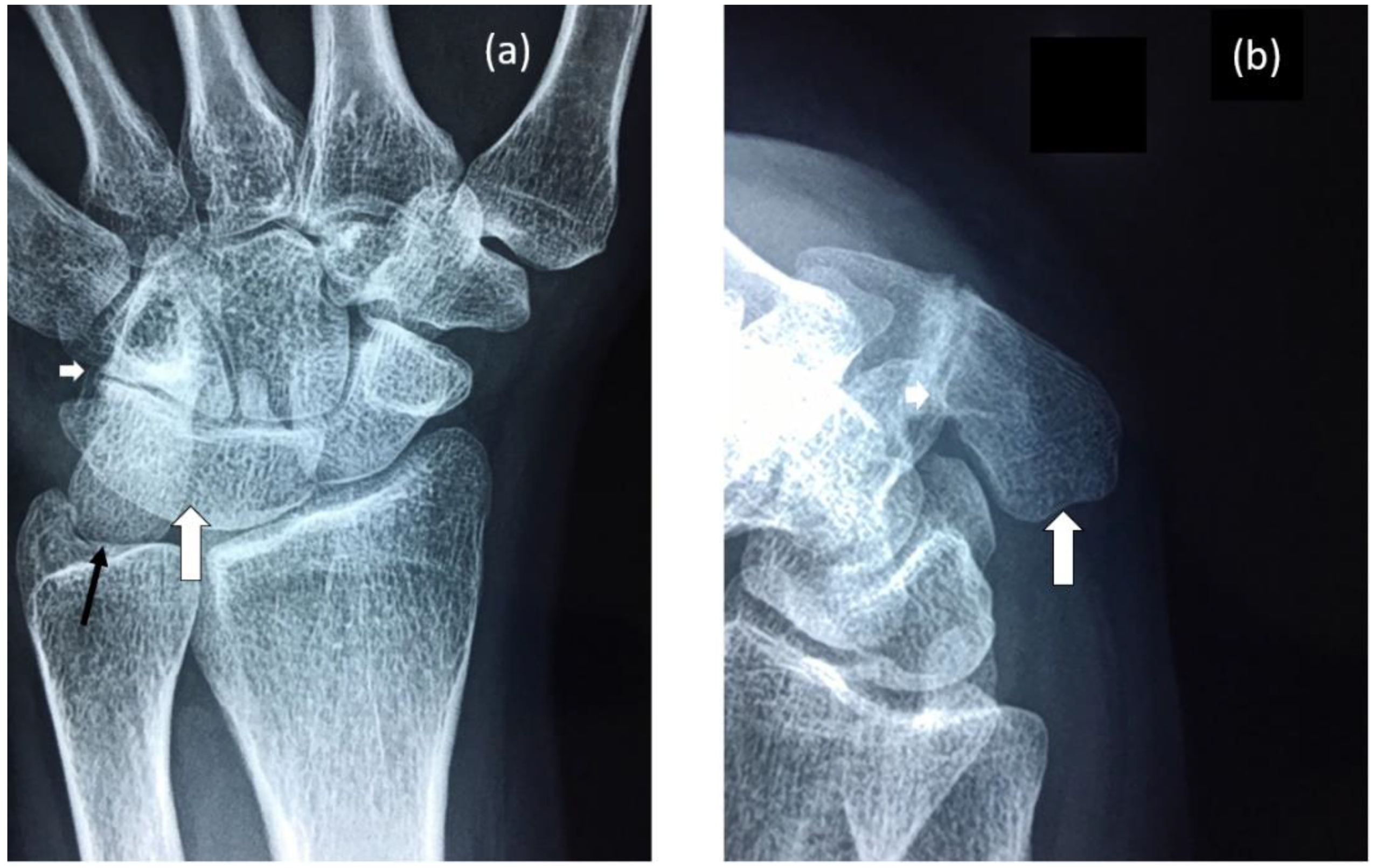
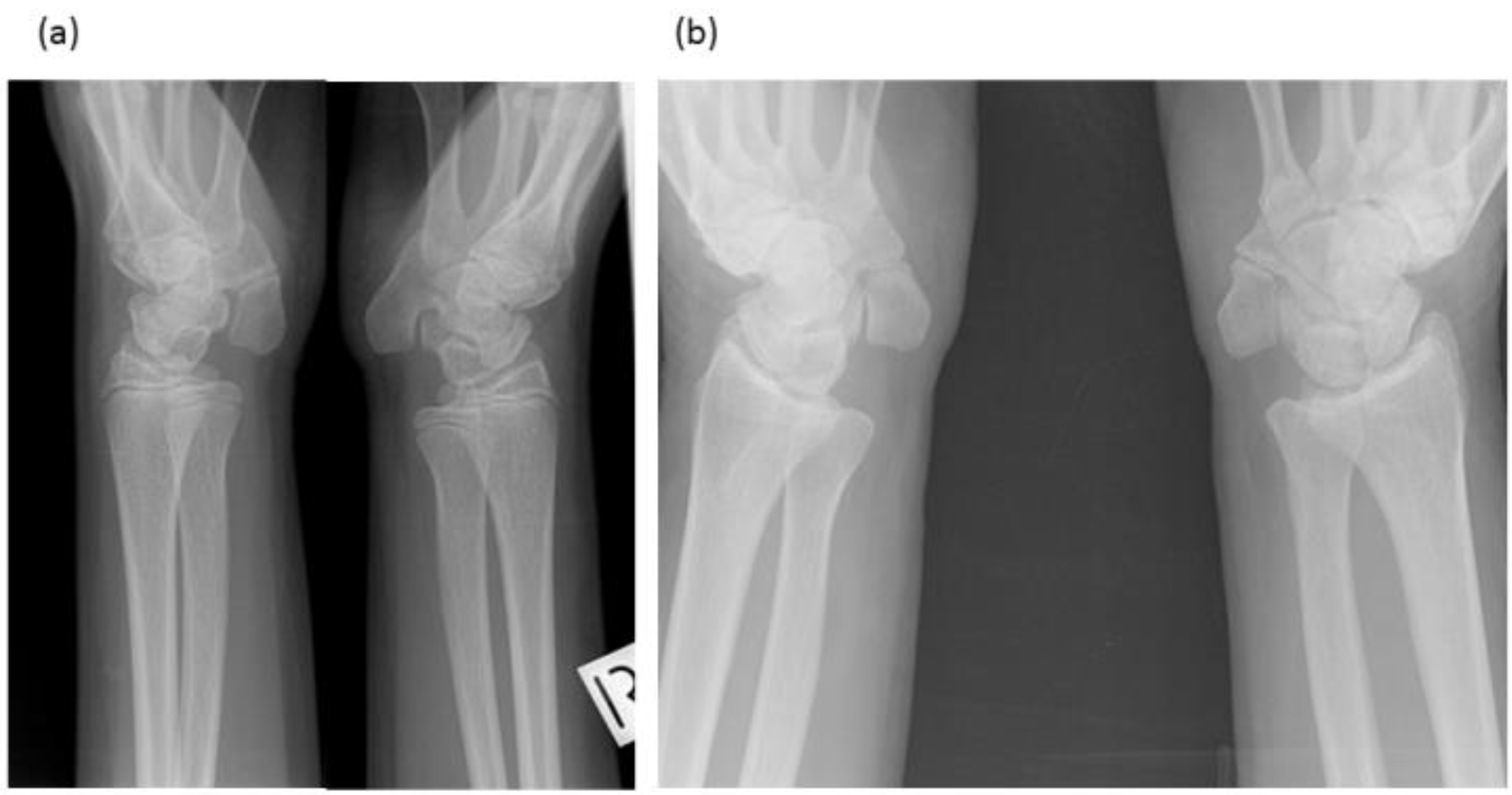
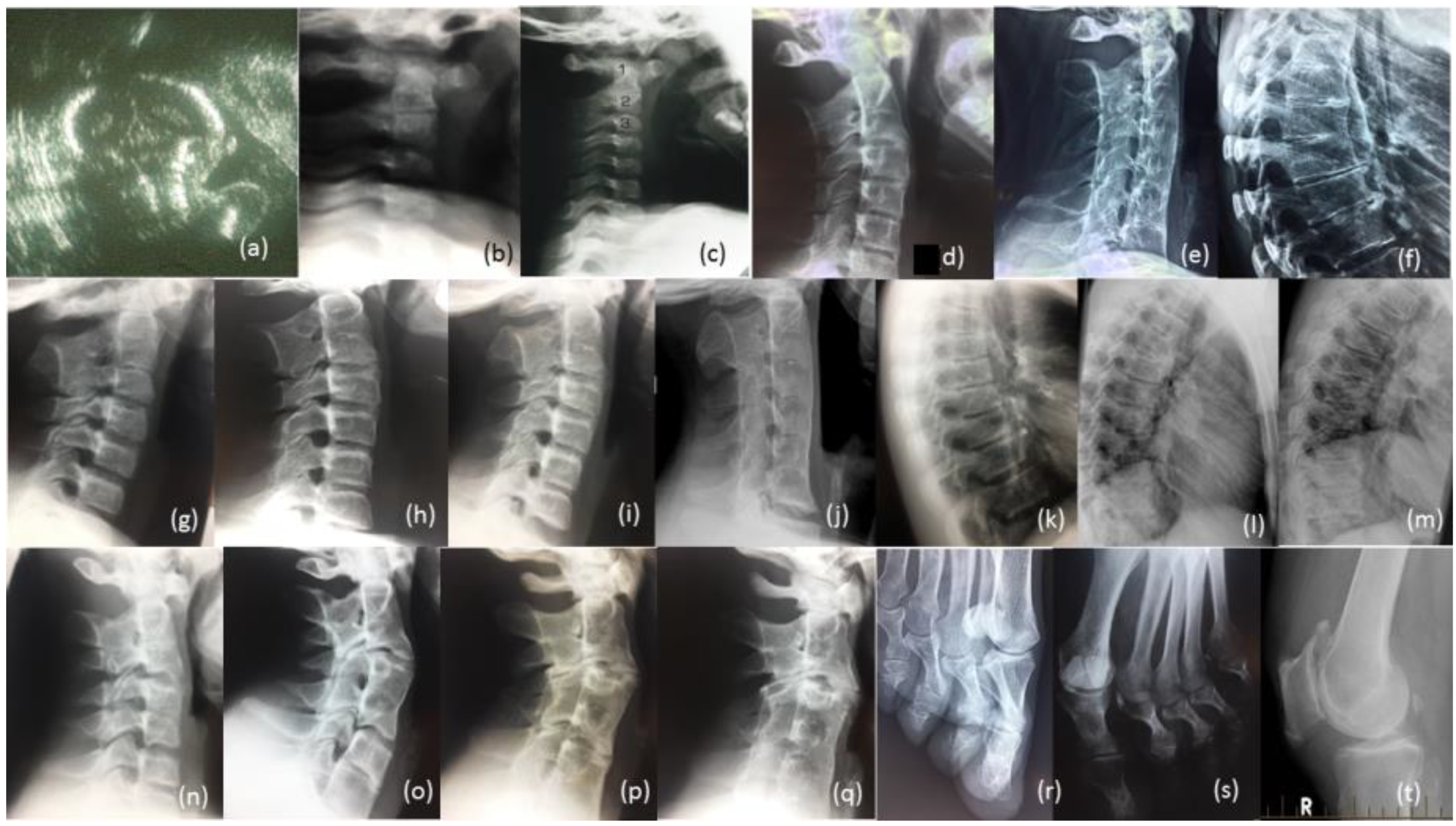
3.1. Radiological Review
3.1.1. Female Proband (IV-12)
3.1.2. Brother (IV-10)
3.1.3. Brother (IV-9)
3.1.4. Male Cousin (IV-5)
3.1.5. Familial Skeletal Anomalies
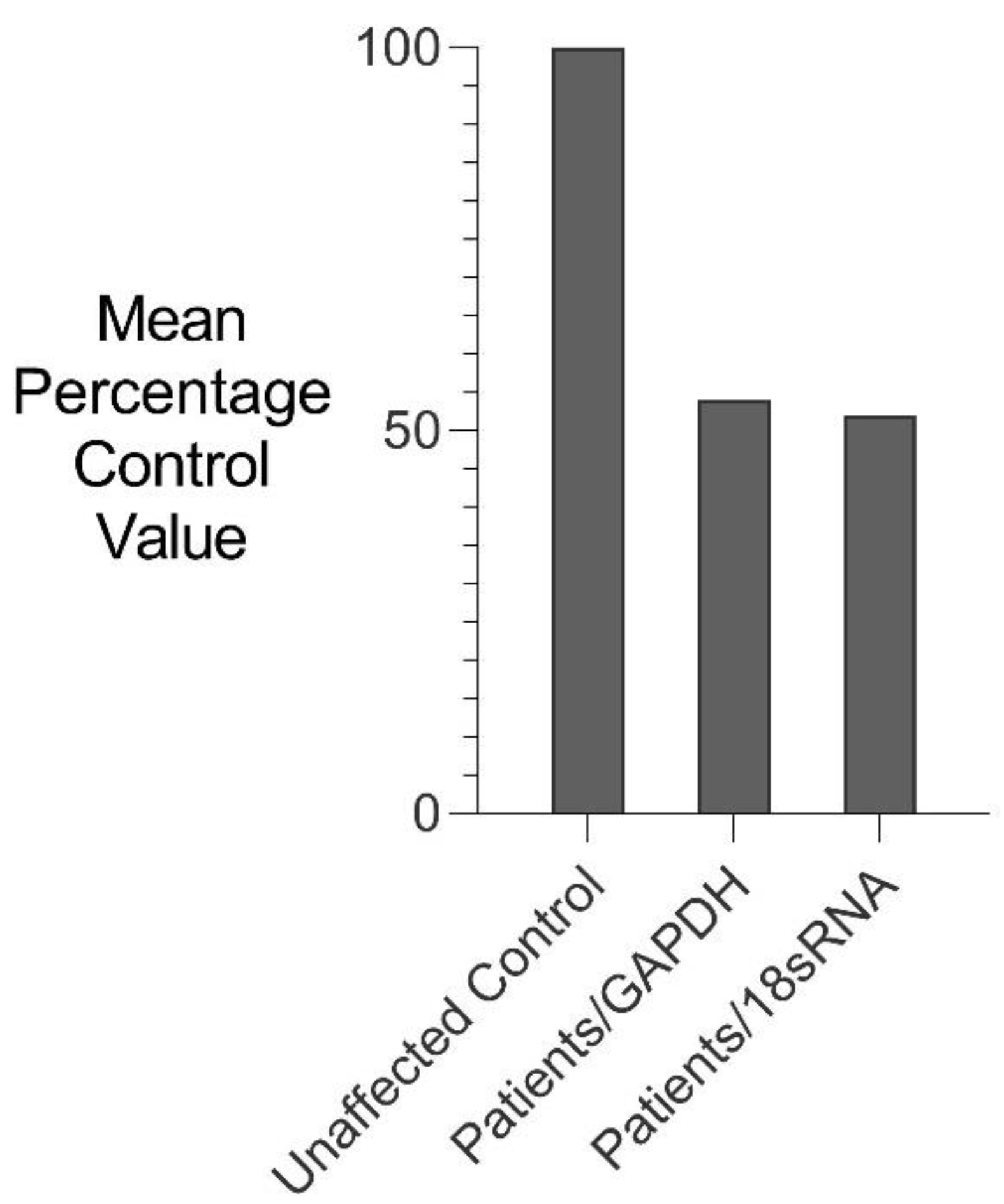
3.2. Gene Expression Analyses
3.2.1. Comparative rtPCR Gene Expression Analysis
3.2.2. RNA Sequencing Gene Expression Analysis (RNAseq)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Settle, S.H.; Rountree, R.B.; Sinha, A.; Thacker, A.; Higgins, K.; Kingsley, D.M. Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev. Biol. 2003, 254, 116–130. [Google Scholar] [CrossRef]
- Mortlock, D.P.; Guenther, C.; Kingsley, D.M. A General Approach for Identifying Distant Regulatory Elements Applied to the Gdf6 Gene. Genome Res. 2003, 13, 2069–2081. [Google Scholar] [CrossRef]
- Wang, J.; Yu, T.; Wang, Z.; Ohte, S.; Yao, R.-E.; Zheng, Z.; Geng, J.; Cai, H.; Ge, Y.; Li, Y.; et al. A New Subtype of Multiple Synostoses Syndrome Is Caused by a Mutation inGDF6That Decreases Its Sensitivity to Noggin and Enhances Its Potency as a BMP Signal. J. Bone Miner. Res. 2016, 31, 882–889. [Google Scholar] [CrossRef]
- Terhal, P.A.; Verbeek, N.E.; Knoers, N.; Nievelstein, R.J.A.J.; Ouweland, A.V.D.; Sakkers, R.J.; Speleman, L.; Van Haaften, G. Further delineation of the GDF6 related multiple synostoses syndrome. Am. J. Med. Genet. Part A 2018, 176, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, R.D.; Haukanes, B.I.; Júlíusson, P.B.; Rosendahl, K.; Houge, G. A Novel GDF6 Mutation in a Family with Multiple Synostoses Syndrome without Hearing Loss. Mol. Syndr. 2018, 9, 228–234. [Google Scholar] [CrossRef]
- Schwaerzer, G.K.; Hiepen, C.; Schrewe, H.; Nickel, J.; Ploeger, F.; Sebald, W.; Mueller, T.; Knaus, P. New insights into the molecular mechanism of multiple synostoses syndrome (SYNS): Mutation within the GDF5 knuckle epitope causes noggin-resistance. J. Bone Miner. Res. 2011, 27, 429–442. [Google Scholar] [CrossRef]
- Dawson, K.; Seeman, P.; Sebald, E.; King, L.; Edwards, M.; Williams, J.; Mundlos, S.; Krakow, D. GDF5 Is a Second Locus for Multiple-Synostosis Syndrome. Am. J. Hum. Genet. 2006, 78, 708–712. [Google Scholar] [CrossRef]
- Seemann, P.; Brehm, A.; König, J.; Reissner, C.; Stricker, S.; Kuss, P.; Haupt, J.; Renninger, S.; Nickel, J.; Sebald, W.; et al. Mutations in GDF5 Reveal a Key Residue Mediating BMP Inhibition by NOGGIN. PLoS Genet. 2009, 5, e1000747. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Cao, L.; Liu, W.; Jiang, L.; Sun, M.; Zhang, D.; Wang, S.; Lo, W.H.Y.; Luo, Y.; Zhang, X. Novel point mutations in GDF5 associated with two distinct limb malformations in Chinese: Brachydactyly type C and proximal symphalangism. J. Hum. Genet. 2008, 53, 368–374. [Google Scholar] [CrossRef]
- Asai-Coakwell, M.; French, C.R.; Ye, M.; Garcha, K.; Bigot, K.; Perera, A.G.; Staehling-Hampton, K.; Mema, S.; Chanda, B.; Mushegian, A.; et al. Incomplete penetrance and phenotypic variability characterize Gdf6-attributable oculo-skeletal phenotypes. Hum. Mol. Genet. 2009, 18, 1110–1121. [Google Scholar] [CrossRef]
- Ye, M.; Berry-Wynne, K.M.; Asai-Coakwell, M.; Sundaresan, P.; Footz, T.; French, C.R.; Abitbol, M.; Fleisch, V.C.; Corbett, N.; Allison, T.; et al. Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum. Mol. Genet. 2009, 19, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Banka, S.; Cain, S.A.; Carim, S.; Daly, S.B.; Urquhart, J.; Erdem, G.; Harris, J.; Bottomley, M.; Donnai, D.; Kerr, B.; et al. Leri’s pleonosteosis, a congenital rheumatic disease, results from microduplication at 8q22.1 encompassing GDF6 and SDC2 and provides insight into systemic sclerosis pathogenesis. Ann. Rheum. Dis. 2014, 74, 1249–1256. [Google Scholar] [CrossRef]
- Clarke, R.; Singh, S.; McKenzie, H.; Kearsley, J.H.; Yip, M.Y. Familial Klippel-Feil syndrome and paracentric inversion inv(8)(q22.2q23.3). Am. J. Hum. Genet. 1995, 57, 1364–1370. [Google Scholar]
- Fang, Z.; Eapen, V.; Clarke, R.A. CTNNA3 discordant regulation of nested LRRTM3, implications for autism spectrum disorder and Tourette syndrome. Meta Gene 2017, 11, 43–48. [Google Scholar] [CrossRef]
- Clarke, R.A.; Davis, P.J.; Tonkin, J. Klippel-Feil Syndrome Associated with Malformed Larynx. Case report. Ann. Otol. Rhinol. Laryngol. 1994, 103, 201–207. [Google Scholar] [CrossRef]
- Pregizer, S.; Mortlock, D.P. Control of BMP gene expression by long-range regulatory elements. Cytokine Growth Factor Rev. 2009, 20, 509–515. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reed, N.P.; Mortlock, D.P. Identification of a distant cis-regulatory element controlling pharyngeal arch-specific expression of zebrafish gdf6a/radar. Dev. Dyn. 2010, 239, 1047–1060. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shen, B.; Bhargav, D.; Wei, A.; A Williams, L.; Tao, H.; Ma, D.D.F.; Diwan, A. BMP-13 Emerges as a Potential Inhibitor of Bone Formation. Int. J. Biol. Sci. 2009, 5, 192–200. [Google Scholar] [CrossRef]
- Becker, J.; McCarthy, R.L.; Sidoli, S.; Donahue, G.; Kaeding, K.; He, Z.; Lin, S.; Garcia, B.A.; Zaret, K.S. Genomic and Proteomic Resolution of Heterochromatin and Its Restriction of Alternate Fate Genes. Mol. Cell 2017, 68, 1023–1037.e15. [Google Scholar] [CrossRef]
- Nicot, R.; Hottenstein, M.; Raoul, G.; Ferri, J.; Horton, M.; Tobias, J.W.; Barton, E.; Gelé, P.; Sciote, J.J. Nodal Pathway Genes Are Down-regulated in Facial Asymmetry. J. Craniofacial Surg. 2014, 25, e548–e555. [Google Scholar] [CrossRef]
- Scheffler, K.; Jalland, C.M.; Benestad, S.L.; Moldal, T.; Ersdal, C.; Gunnes, G.; Suganthan, R.; Bjørås, M.; Tranulis, M.A. DNA glycosylase Neil2 contributes to genomic responses in the spleen during clinical prion disease. Free. Radic. Biol. Med. 2020, 152, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Sarker, A.H.; Chatterjee, A.; Williams, M.; Lin, S.; Havel, C.; Iii, P.J.; Boldogh, I.; Hazra, T.K.; Talbot, P.; Hang, B. NEIL2 Protects against Oxidative DNA Damage Induced by Sidestream Smoke in Human Cells. PLoS ONE 2014, 9, e90261. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Levin, M. A unified model for left–right asymmetry? Comparison and synthesis of molecular models of embryonic laterality. Dev. Biol. 2013, 379, 1–15. [Google Scholar] [CrossRef] [PubMed]
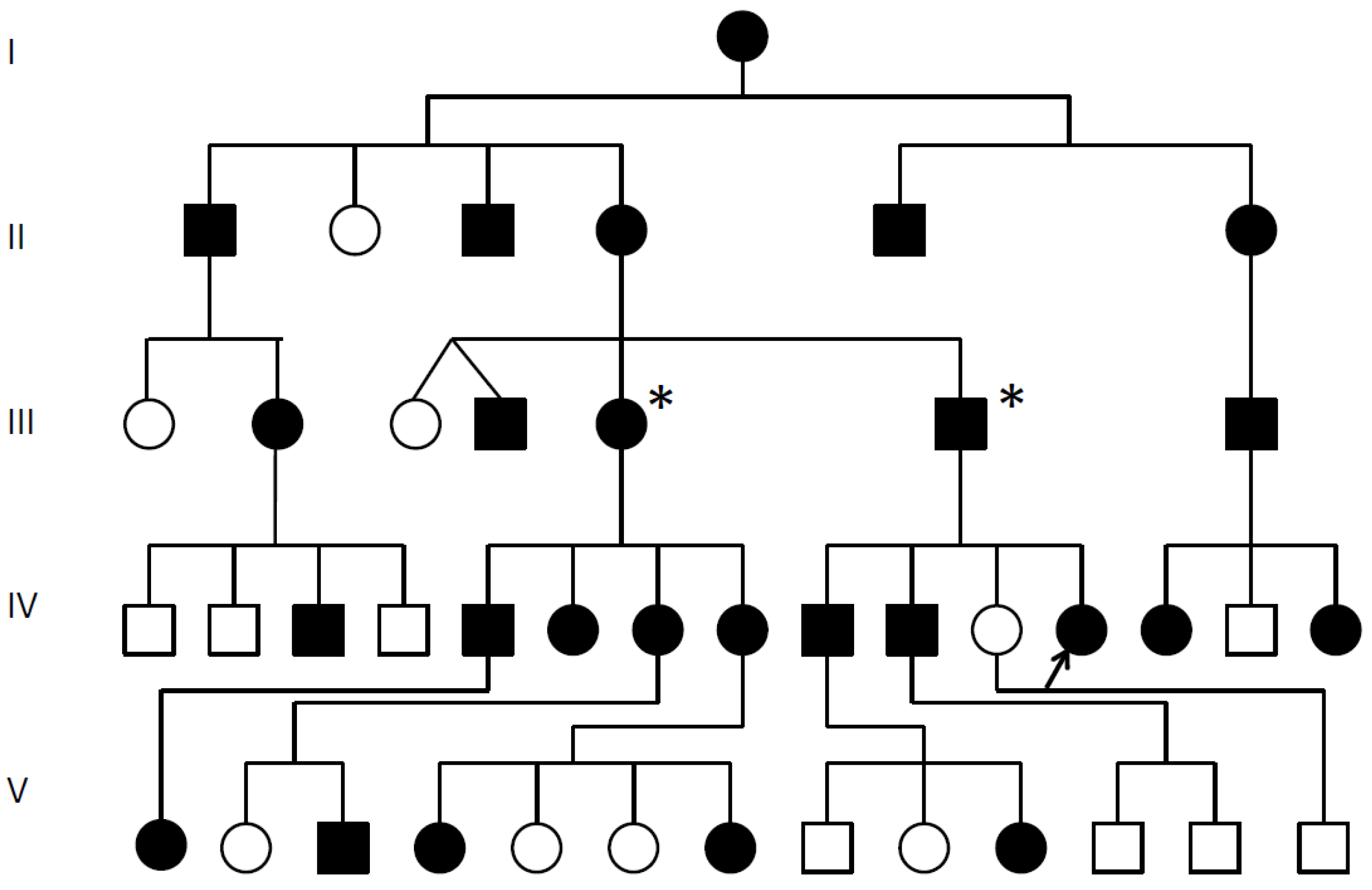
 Multiple synostoses, vertebral fusion, speech impairment; chromosomal breakpoint 3′ of GDF6; * Decreased GDF6 expression confirmed; Proband arrowed;
Multiple synostoses, vertebral fusion, speech impairment; chromosomal breakpoint 3′ of GDF6; * Decreased GDF6 expression confirmed; Proband arrowed;  Unaffected.
Multiple synostoses, vertebral fusion, speech impairment; chromosomal breakpoint 3′ of GDF6; * Decreased GDF6 expression confirmed; Proband arrowed; Unaffected.
Unaffected.
Multiple synostoses, vertebral fusion, speech impairment; chromosomal breakpoint 3′ of GDF6; * Decreased GDF6 expression confirmed; Proband arrowed; Unaffected.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family Member | Sex | Ages | Anomalies |
|---|---|---|---|
| Proband (IV-12) | F | 0–27 | Carpal Tarsal Coalition, Pisiform elongated No hearing impairment or congenital vertebral fusion, Postnatal vertebral fusion, speech impaired, short tongue and microstomia. |
| Brother (IV-10) | M | 7–26 | Carpal Tarsal Coalition, Pisiform elongated No hearing impairment or congenital vertebral fusion, Postnatal vertebral fusion, No speech impairment, short tongue and microstomia. |
| Brother (IV-9) | M | 12–19 | Carpal Tarsal Coalition, Pisiform elongated No hearing impairment, Postnatal vertebral fusion, Severe speech impairment, short tongue and microstomia. |
| Cousin (IV-5) | M | 17–50 | Carpal Tarsal Coalition, Pisiform not tested No hearing impairment, Progressive vertebral fusion, Severe speech impairment, short tongue and microstomia. |
| Primer Direction | Primer Sequence |
|---|---|
| GDF6—forward | CCTGTTGCTTGTTTGGTTCA |
| GDF6—reverse | GCTGTCCATTTCCTCTTTGC |
| 18S rRNA—forward | GTAACCCGTTGAACCCCATT |
| 18S rRNA—reverse | CCATCCAATCGGTAGTAGCG |
| GAPDH—forward | CCACCCATGGCAAATTCCATGGCA |
| GAPDH—reverse | TCTAGACGGCAGGTCAGGTCCACC |
| Stealth control—sense | CAAGAACAGCGAGAAGCAGCCGUCA |
| Stealth control—antisense | UGACGGCUGCUUCUCGCUGUUCUUG |
| Fibroblast Cell Lines | White Blood Cells | ||||||
|---|---|---|---|---|---|---|---|
| Down-Regulation | Up-Regulation | Down-Regulation | Up-Regulation | ||||
| Change | Gene | Change | Gene | Change | Gene | Change | Gene |
| −15.3854 −15.0439 −14.9457 −14.7973 −14.7586 −14.6305 −14.5996 −14.4498 −14.1642 −14.0789 −14.0487 −14.0126 −13.7795 −13.7398 −13.6138 −13.3585 −13.2922 −13.2741 −13.2696 −13.2425 −13.2212 −13.1889 −13.1802 −13.104 −12.7709 −12.6132 −11.8528 −11.8236 −11.801 | NOMO3 * DNAH1 PRDM10 IDO1 SP140L MYSM1 GRB10 PTGS2BLOC1S2 TMEM260 SNX13 RCBTB1 CCP110 RBMXL1 * EPHX2 NF2 GZF1 CDCA8 AMZ2 POLR1B TTI1 WDR19 ABCB7 SMC6 DERA ZNF33B POLR3B NEIL2 * BEGAIN | 10.4092 11.4755 11.9372 12.1726 12.301 12.4321 12.5031 12.5352 12.7603 12.833 12.9472 13.1595 13.1675 13.2051 13.2154 13.2714 13.276 13.3216 13.3411 13.7474 13.7502 14.072 14.0828 14.1284 14.1698 14.341 14.5617 14.7657 14.7712 14.8012 15.2457 15.7457 16.0123 | MDGA1 GMEB1 CSTF2 RPGRIP1 TRIT1 ADGRA2 PCGF1 GAL3ST4 POM121 HTATIP2 PHETA1 MAP2K6 AP5S1 TBP KIAA023 2 ZNF550 SOCS2 ZNF280D EML3 FECH GZF1 ZNF331 DAG1 MKS1 AKAP7 COG6 ZNF302 PAX5 ZNF628 ZNF133 POT1 ZNF57 CACNB3 | −15.077 −15.0329 −14.6255 −14.4338 −14.2377 −14.0202 −13.9483 −13.9278 −13.7308 −13.6908 −13.6735 −13.2039 −12.9068 −12.7789 −12.676 −12.624 −12.6236 −12.603 −12.5258 −12.4878 −12.4472 −12.1771 −12.1662 −12.0355 −11.5972 −11.497 −11.2932 −11.0185 −10.4318 | AK4 GPR68 CDK19 PHETA1 ABHD16B ZNF229 TSC1 BTBD3 TMEM134 AFMID LIG1 GRAMD1A TRIT1 ZBTB43 MTR NOMO3 * RBMXL1 * RPF2 SP100 KRBA1 MORC2 UBE2F LRRN3 MAP3K4 NEIL2 * EPHA4 NT5M REEP2 ZFYVE27 | 11.0154 11.1429 11.8143 11.8187 12.0527 12.1243 12.1532 12.1681 12.3948 12.4649 12.6268 12.6402 12.8589 13.0328 13.0354 13.0485 13.0628 13.086 13.1499 13.1582 13.2509 13.2561 13.3067 13.3068 13.3209 13.4586 13.617 13.6751 13.8247 13.8448 13.9705 14.1731 14.3364 14.5402 14.8302 14.9429 15.0595 15.5187 15.8286 16.3832 | TMEM201 ARNTL SLC25A38 ADARB1 ZCCHC4 BBS5 UCP2 ATP6V0A1 CEP83 NMNAT1 TMEM209 IQCE PHTF1 UBL4A GOLPH3L MED19 GLI2 XKR8 ZBED8 SCML1 NEDD9 ROR1 ZC3H8 CEP44 ZNF274 POGZ TENT5A TNIK TOP3A BTRC INPP4A PIK3R1 ELAPOR2 NAV3 GALNT15 NUP155 ADGRB2 SLC4A3 MPDZ ZNF510 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clarke, R.A.; Fang, Z.; Murrell, D.; Sheriff, T.; Eapen, V. GDF6 Knockdown in a Family with Multiple Synostosis Syndrome and Speech Impairment. Genes 2021, 12, 1354. https://doi.org/10.3390/genes12091354
Clarke RA, Fang Z, Murrell D, Sheriff T, Eapen V. GDF6 Knockdown in a Family with Multiple Synostosis Syndrome and Speech Impairment. Genes. 2021; 12(9):1354. https://doi.org/10.3390/genes12091354
Chicago/Turabian StyleClarke, Raymond A., Zhiming Fang, Dedee Murrell, Tabrez Sheriff, and Valsamma Eapen. 2021. "GDF6 Knockdown in a Family with Multiple Synostosis Syndrome and Speech Impairment" Genes 12, no. 9: 1354. https://doi.org/10.3390/genes12091354
APA StyleClarke, R. A., Fang, Z., Murrell, D., Sheriff, T., & Eapen, V. (2021). GDF6 Knockdown in a Family with Multiple Synostosis Syndrome and Speech Impairment. Genes, 12(9), 1354. https://doi.org/10.3390/genes12091354