
Expanding the Neurological Phenotype of Ring Chromosome 10 Syndrome: A Case Report and Review of the Literature

 , ,
, ,
Abstract
1. Introduction
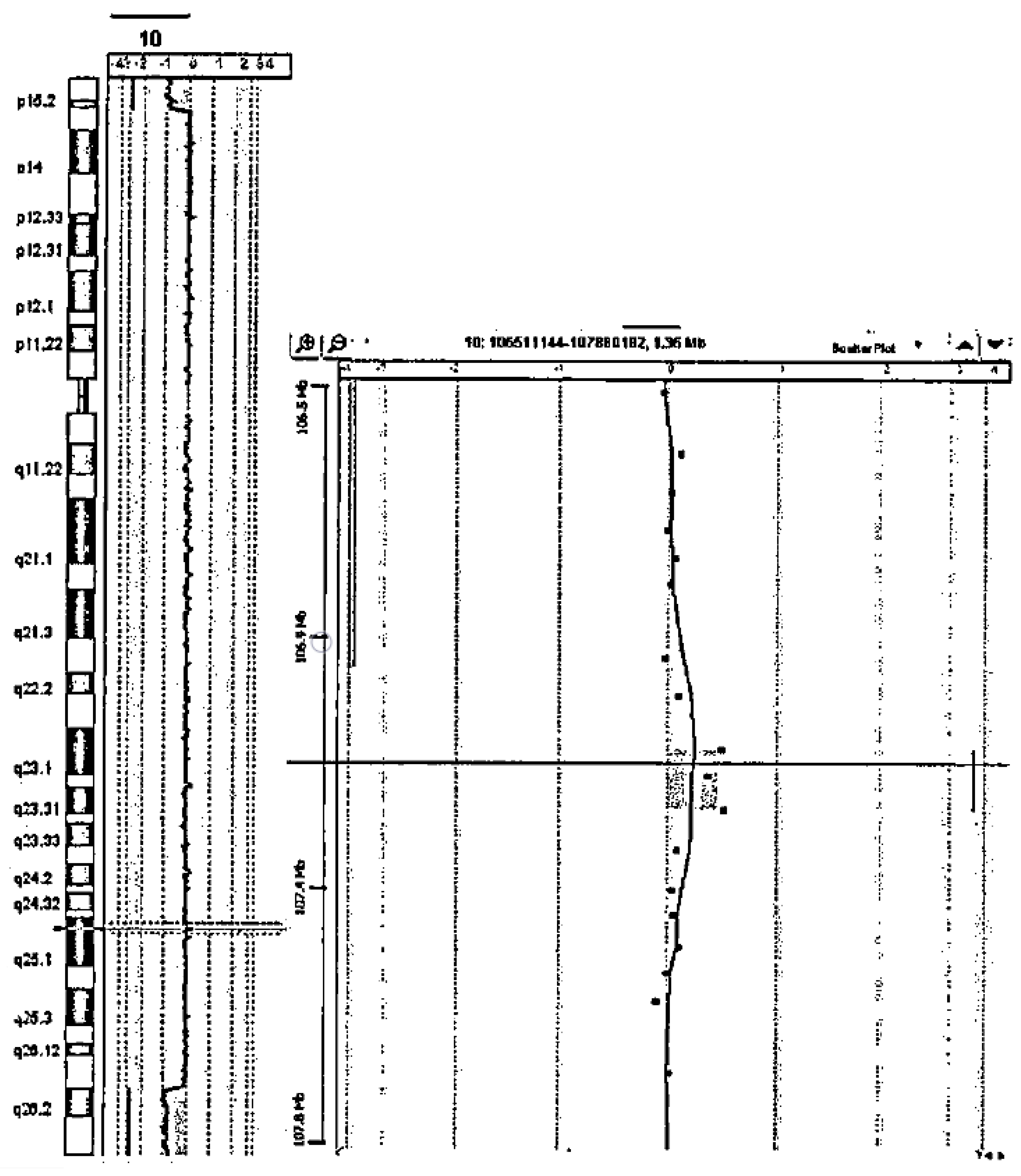
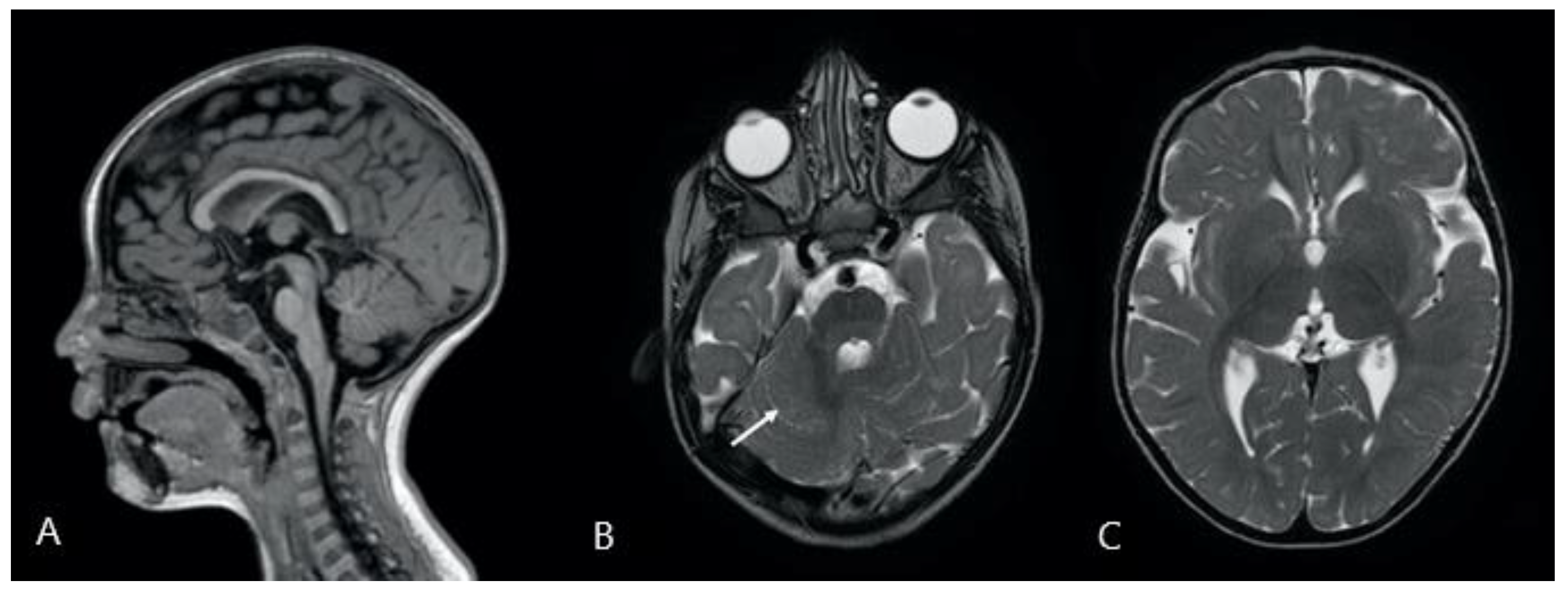
2. Case Presentation
3. Search Strategy and Analysis of Literature
4. Results
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lansky, S.; Daniel, W.; Fleiszar, K. Physical retardation is associated with ring chromosome mosaicism: 46, XX,r(10)/45, XX,10 minus. J. Med. Genet. 1977, 14, 61–63. [Google Scholar] [CrossRef]
- Sparkes, R.S.; Ling, S.M.; Muller, H. Ring 10 chromosome: 46,XX,r10(p15q26). Qual. Life Res. 1978, 43, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Fryns, J.P.; De Boeck, K.; Jaeken, J.; Berghe, H.V.D. Malformative syndrome associated with a ring 10 chromosome and a translocated 10q/19 chromosome. Qual. Life Res. 1978, 43, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Simoni, G.; Rossella, F.; Visconti, G.; Piria-Schwarz, C. Ring chromosome 10 Associated with multiple congenital malformations. Qual. Life Res. 1979, 51, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Tsukino, R.; Tsuda, N.; Dezawa, T.; Ishii, T.; Koike, M. Ring chromosome 10: 46,XX,r(10)(p15->q26). J. Med. Genet. 1980, 17, 148–151. [Google Scholar] [CrossRef]
- Michels, V.V.; Driscoll, D.J.; Ledbetter, D.H.; Riccardi, V.M.; Opitz, J.M. Phenotype associated with ring 10 chromosome: Report of patient and review of literature. Am. J. Med. Genet. 1981, 9, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Serville, F.; Briault, R.; Taillemite, J.L.; Despoisse, S.; Cotoni, P.; Broustet, A. Chromosome 10 en anneau: 46,XX,r(10)(p15q26) [Ring chromosome 10: 46,XX,r(10)(p15q26)]. Ann. Genet. 1982, 25, 168–171. [Google Scholar]
- Nakai, H.; Adachi, M.; Katsushima, N.; Yamazaki, N.; Sakamoto, M.; Tada, K. Ring chromosome 10 and its clinical features. J. Med. Genet. 1983, 20, 142–144. [Google Scholar] [CrossRef][Green Version]
- Kondo, I.; Shimakura, Y.; Hirano, T.; Kaneko, M.; Yabuta, K. Ring chromosome 10 syndrome: Case report and the possibility of clinical diagnosis. Clin. Genet. 1984, 25, 196–200. [Google Scholar] [CrossRef]
- Shapiro, S.D.; Hansen, K.L.; Pasztor, L.M.; DiLiberti, J.H.; Jorgenson, R.J.; Young, R.S.; Moore, C.M.; Opitz, J.M.; Reynolds, J.F. Deletions of the long arm of chromosome 10. Am. J. Med. Genet. 1985, 20, 181–196. [Google Scholar] [CrossRef]
- Kishi, K.; Ikfuchi, T.; Yamamoto, K.; Tonomura, A.; Sakurada, N.; Satoh, Y. Report of a patient with a ring chromosome 10: mos45,XY,−10/46,XY/46,XY,r(10) (p15.3q26.3). J. Hum. Genet. 1985, 30, 233–238. [Google Scholar] [CrossRef]
- Calabrese, G.; Franchi, P.G.; Stuppia, L.; Mingarelli, R.; Rossi, C.; Ramenghi, L.A.; Marino, M.; Morizio, E.; Peila, R.; Antonucci, A. A newborn with ring chromosome 10, aganglionic megacolon, and renal hypoplasia. J. Med. Genet. 1994, 31, 804–806. [Google Scholar] [CrossRef] [PubMed]
- Concolino, D.; Iembo, M.; Marotta, R.; Rossi, E.; Moricca, M.; Giglio, S.; Strisciuglio, P. Ring chromosome 10 (p15q26) in a patient with unipolar affective disorder, multiple minor anomalies, and mental retardation. Am. J. Med. Genet. 2003, 123A, 201–203. [Google Scholar] [CrossRef]
- Guilherme, R.S.; Meloni, V.F.A.; Kim, C.A.; Pellegrino, R.; Takeno, S.S.; Spinner, N.B.; Conlin, L.K.; Christofolini, D.M.; Kulikowski, L.D.; Melaragno, M.I. Mechanisms of ring chromosome formation, ring instability and clinical consequences. BMC Med. Genet. 2011, 12, 171. [Google Scholar] [CrossRef]
- Christopoulou, G.; Tzetis, M.; Konstantinidou, A.; Tsezou, A.; Kanavakis, E.; Kitsiou-Tzeli, S.; Velissariou, V. Clinical and molecular description of a fetus in prenatal diagnosis with a rare de novo ring 10 and deletions of 12.59Mb in 10p15.3–p14 and 4.22Mb in 10q26.3. Eur. J. Med. Genet. 2012, 55, 75–79. [Google Scholar] [CrossRef]
- Guilherme, R.S.; Kim, C.A.; Alonso, L.G.; Honjo, R.S.; Meloni, V.A.; Christofolini, D.M.; Kulikowski, L.D.; Melaragno, M.I. Ring chromosome 10: Report on two patients and review of the literature. J. Appl. Genet. 2012, 54, 35–41. [Google Scholar] [CrossRef]
- Čiuladaitė, Ž.; Burnytė, B.; Vansevičiūtė, D.; Dagytė, E.; Kučinskas, V.; Utkus, A. Clinical, cytogenetic and molecular study of a case of ring chromosome 10. Mol. Cytogenet. 2015, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Kosztolányi, G. The genetics and clinical characteristics of constitutional ring chromosomes. J. Assoc. Genet. Technol. 2009, 35, 44–48. [Google Scholar]
- Bertino, E.; Spada, E.; Occhi, L.; Coscia, A.; Giuliani, F.; Gagliardi, L.; Gilli, G.; Bona, G.; Fabris, C.; De Curtis, M.; et al. Neonatal Anthropometric Charts: The Italian Neonatal Study Compared With Other European Studies. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man (OMIM). Available online: https://www.omim.org (accessed on 19 September 2021).
- DECIPHER. Available online: https://www.deciphergenomics.org (accessed on 19 September 2021).
- GeneCards. Available online: https://www.genecards.org (accessed on 19 September 2021).
- Jandeaux, C.; Kuchcinski, G.; Ternynck, C.; Riquet, A.; Leclerc, X.; Pruvo, J.-P.; Soto-Ares, G. Biometry of the Cerebellar Vermis and Brain Stem in Children: MR Imaging Reference Data from Measurements in 718 Children. Am. J. Neuroradiol. 2019, 40, 1835–1841. [Google Scholar] [CrossRef]
- Garel, C.; Cont, I.; Alberti, C.; Josserand, E.; Moutard, M.; Le Pointe, H.D. Biometry of the Corpus Callosum in Children: MR Imaging Reference Data. Am. J. Neuroradiol. 2011, 32, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Rajput, R.; Mishra, S.; Ahlawat, P.; Yadav, P.K. Dandy-Walker variant with precocious puberty: A rare association. BMJ Case Rep. 2018, 11, e226281. [Google Scholar] [CrossRef] [PubMed]
- DeScipio, C.; Conlin, L.; Rosenfeld, J.; Tepperberg, J.; Pasion, R.; Patel, A.; McDonald, M.T.; Aradhya, S.; Ho, D.; Goldstein, J.; et al. Subtelomeric deletion of chromosome 10p15.3: Clinical findings and molecular cytogenetic characterization. Am. J. Med. Genet. Part A 2012, 158A, 2152–2161. [Google Scholar] [CrossRef]
- Yates, T.M.; Drucker, M.; Barnicoat, A.; Low, K.; Gerkes, E.H.; Fry, A.E.; Parker, M.J.; O’Driscoll, M.; Charles, P.; Cox, H.; et al. ZMYND11 -related syndromic intellectual disability: 16 patients delineating and expanding the phenotypic spectrum. Hum. Mutat. 2020, 41, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Tumiene, B.; Čiuladaitė, Ž.; Preikšaitienė, E.; Mameniškienė, R.; Utkus, A.; Kučinskas, V. Phenotype comparison confirms ZMYND11 as a critical gene for 10p15.3 microdeletion syndrome. J. Appl. Genet. 2017, 58, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Harms, F.L.; Girisha, K.M.; Hardigan, A.A.; Kortüm, F.; Shukla, A.; Alawi, M.; Dalal, A.; Brady, L.; Tarnopolsky, M.; Bird, L.; et al. Mutations in EBF3 Disturb Transcriptional Profiles and Cause Intellectual Disability, Ataxia, and Facial Dysmorphism. Am. J. Hum. Genet. 2016, 100, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Lopes, F.; Soares, G.; Gonçalves-Rocha, M.; Pinto-Basto, J.; Maciel, P. Whole Gene Deletion of EBF3 Supporting Haploinsufficiency of This Gene as a Mechanism of Neurodevelopmental Disease. Front. Genet. 2017, 8, 143. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Deletion | Gene a | Gene OMIM Number | Transmission | Function | Phenotype and Phenotype OMIM Number | Discussion |
|---|---|---|---|---|---|---|
| 1 | ZMYND11 | 608668 | AD | Zinc finger | OMIM 616083 Mental retardation, autosomal dominant 30 | ID/DD and behavioral problems documented in the present case; differences in MRI abnormalities |
| 1 | WDR37 | 618586 | AD | Protein involved in cell cycle, transduction and apoptosis | OMIM 618652 Neuro-oculo-cardio-genito-urinary syndrome | ID/DD with prevalent language impairment, hypotonia, microcrania and CNS abnormailities partially overlap the present case major heart and eye abnormalities |
| 1 | PITRM1 | 618211 | AR | Metallopeptidase | OMIM 619405 Spinocerebellar ataxia, autosomal recessive 30 | DD +/− ID, ataxia, behavioral abnormalities. Cerebellar atrophy. Progressive condition. |
| 2 | MMP21 | 608416 | AR | Matrix metalloproteinase | OMIM 616749 Autosomal visceral heterotaxy-7 | Major cardiac abnormalitites (minor cardiac abnormalities in the present case). Situs inversus |
| 2 | EBF3 | 607407 | AD | Trasciption factor inducing apoptosis and cell cycle arrest | OMIM 617330 Hypotonia, ataxia, and delayed development syndrome | Hypotonia, ID/DD, ataxia; in a few cases, cerebellar vermian hypoplasia, partially overlapping the present case |
| 2 | ECHS1 | 602292 | AR | short-chain enoyl-CoA hydratase, catalyzing mitochondrial fatty acid β-oxidation | OMIM 616277 Mitochondrial short-chain enoyl-CoA hydratase-1 deficiency | DD, hyptonia, brain atrophy and brain lesions in the basal ganglia. Progressive condition |
| Case | Breakpoints (10p and 10q) | ID/DD | Microcephaly | Strabismus | Hypotonia | Other | EEG | Seizures | Brain Imaging | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | p14, q25 | + | + | Lansky et al. (1977) | ||||||
| 2 | p15, q26 | + | + | + | Hyperactivity | Sparkes et al. (1978) | ||||
| 3 | p15, q25 | + | + | Normal CT | Fryns et al. (1978) | |||||
| 4 | p15, q26 | + | + | + | Simoni et al. (1979) | |||||
| 5 | p15, q26 | + | + | + | Normal | FS | Normal US | Tsukino et al. (1980) | ||
| 6 | p15.3, q26.1 | + | + | + | Occipital SW; diffuse fast activity; paroxysmal generalized SW | Normal CT | Michels et al. (1981) | |||
| 7 | p15, q26 | + | + | Serville et al. (1982) | ||||||
| 8 | p15, q26 | + | + | Normal | FS | Nakai et al. (1983) | ||||
| 9 | p15.3, q26.3 | + | + | Normal | Normal CT | Kondo et al. (1984) | ||||
| 10 | + | Zorn (1984) | ||||||||
| 11 | p15, q26 | + | + | + | Shapiro et al. (1985) | |||||
| 12 | p15.3, q26.3 | + | Normal | Kishi et al. (1985) | ||||||
| 13 | p13-15, q26 | + | + | + | Convulsions | Calabrese et al. (1994) | ||||
| 14 | p15, q26 | + | + | MDD | Normal CT | Concolino et al. (2003) | ||||
| 15 | p15.3, q26.12 | + | + | Horner Syndrome | Normal MRI | Gunnarsson et al. (2009) | ||||
| 16 | p14, q26.3 | Macrocephaly | Christopoulou et al. (2012) | |||||||
| 17 | p15.3, q26.2 | + | + | Guilherme et al. (2013) | ||||||
| 18 | p15.3, q26.13 | + | + | + | FS | Dandy-Walker variant at CT | Guilherme et al. (2013) | |||
| 19 | p15.1, q26.1 | + | + | + | + | Stereotypies | US: dilated ventricles, then normal | Čiuladaitė et al. (2015) | ||
| 20 | p15, q26.1 | + | + | + | + | Stereotypies | Temporo-parietal BL rapid rhythms, posterior slowing | None | MRI: posterior cranial fossa and CC abnormalities | Present case |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pruccoli, J.; Graziano, C.; Locatelli, C.; Maltoni, L.; Sheikh Maye, H.A.; Cordelli, D.M. Expanding the Neurological Phenotype of Ring Chromosome 10 Syndrome: A Case Report and Review of the Literature. Genes 2021, 12, 1513. https://doi.org/10.3390/genes12101513
Pruccoli J, Graziano C, Locatelli C, Maltoni L, Sheikh Maye HA, Cordelli DM. Expanding the Neurological Phenotype of Ring Chromosome 10 Syndrome: A Case Report and Review of the Literature. Genes. 2021; 12(10):1513. https://doi.org/10.3390/genes12101513
Chicago/Turabian StylePruccoli, Jacopo, Claudio Graziano, Chiara Locatelli, Lucia Maltoni, Hodman Ahmed Sheikh Maye, and Duccio Maria Cordelli. 2021. "Expanding the Neurological Phenotype of Ring Chromosome 10 Syndrome: A Case Report and Review of the Literature" Genes 12, no. 10: 1513. https://doi.org/10.3390/genes12101513
APA StylePruccoli, J., Graziano, C., Locatelli, C., Maltoni, L., Sheikh Maye, H. A., & Cordelli, D. M. (2021). Expanding the Neurological Phenotype of Ring Chromosome 10 Syndrome: A Case Report and Review of the Literature. Genes, 12(10), 1513. https://doi.org/10.3390/genes12101513