1. Introduction
Knobloch syndrome (KS, MIM# 267750) is a rare syndromic autosomal recessive disorder, characterized by eye abnormalities and occipital defects. The ocular manifestations include high myopia, retinal detachment, congenital cataracts, lens subluxation, vitreoretinal degeneration, and retinitis pigmentosa-like features [
1,
2,
3]. Numerous extraocular abnormalities have been reported, including occipital defects, encephalocele, hydrocephalus, polymicrogyria, bifid ureter, dextrocardia, and seizures [
4,
5,
6].
The most common disease-causing molecular defects are mutations of the
COL18A1 gene (OMIM# 120328), located on chromosome 21q22.3. The gene comprises of 41 exons and encodes the collagen alpha-1 (XVIII) chain, a basement membrane constituent highly expressed throughout the eye. Collagen XVIII has an essential role in ocular development, including angiogenesis and structural maintenance [
7,
8]. To date, there have been 48 genetically confirmed KS families of various ethnicities; nevertheless, KS is rarely reported in east Asian populations. Only one Chinese family was reported to have a homozygous
COL18A1 mutation [
9]. However, the clinical appearance in this family was limited, due to the opacity of the optical media (
Supplementary Table S1,
COL18A1 mutation of patients with KS) [
3,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21].
Here, we present the extensive study of six affected individuals with KS, from four unrelated Chinese families, using comprehensive multimodal imaging and genetic testing, in which we identified five novel pathogenic mutations of COL18A1. This study could help to further explore the genotypic and phenotypic spectrum of KS.
2. Materials and Methods
Five KS patients, from four unrelated families, were referred to Zhongshan ophthalmic center and included in this study. The diagnoses of KS were based on characteristic clinical features and confirmation by identification of pathogenic COL18A1 variants. This study was conducted following the tenets of the Declaration of Helsinki and was approved by the institutional review board of Zhongshan Ophthalmic Center, Sun Yat-sen University. Informed written consent was obtained from the patients or their legal guardians.
Clinical information, including age, sex, history, pedigree, symptoms, and clinical diagnosis, were recorded. Detailed ophthalmic examinations included the best corrected visual acuity (BCVA), intraocular pressure, refractive errors, axial length (IoL Master, Carl Zeiss, Meditec, Jena, Germany), slit lamp biomicroscopy, wild field scanning laser ophthalmoscope (Optomap 200Tx, Optos pls, Dunfermline, UK), or RetCam imaging (RetCam, Clarity Medical Systems Inc., Pleasanton, CA, USA). Full-field flicker ERGs were recorded using the RETeval system (LKC Technologies, Gaithersburg, MD, USA); ISCEV standard flicker parameters were followed. Four patients underwent magnetic resonance imaging of the brain.
Genomic DNA of the proband and available family members was extracted from peripheral blood using the TIANamp Blood DNA Kit (DP348-03, Tiangen Biotech, Beijing, China), as instructed by the manufacturer. The quantity and quality of DNA were verified by using NanoDrop. Whole-exome sequencing (WES) was performed for four probands, and identified mutations were validated using Sanger sequencing of the family members. The Human Gene Mutation Database, Genome Aggregation Database, and Exome Aggregation Consortium were used to identify the reported pathogenic variants. Online algorithms, including MutationTaster, sorting intolerant from tolerant (SIFT), and Polymorphism Phenotyping v2 (Polyphen2), were used to evaluate the pathogenicity of missense mutations.
3. Results
3.1. Clinical Features
Homozygous or compound heterozygous mutations were identified in six patients diagnosed with KS, from four unrelated families. The clinical manifestations are shown in
Figure 1 and
Figure 2. The clinical data are summarized in
Table 1.
3.1.1. Patient 1
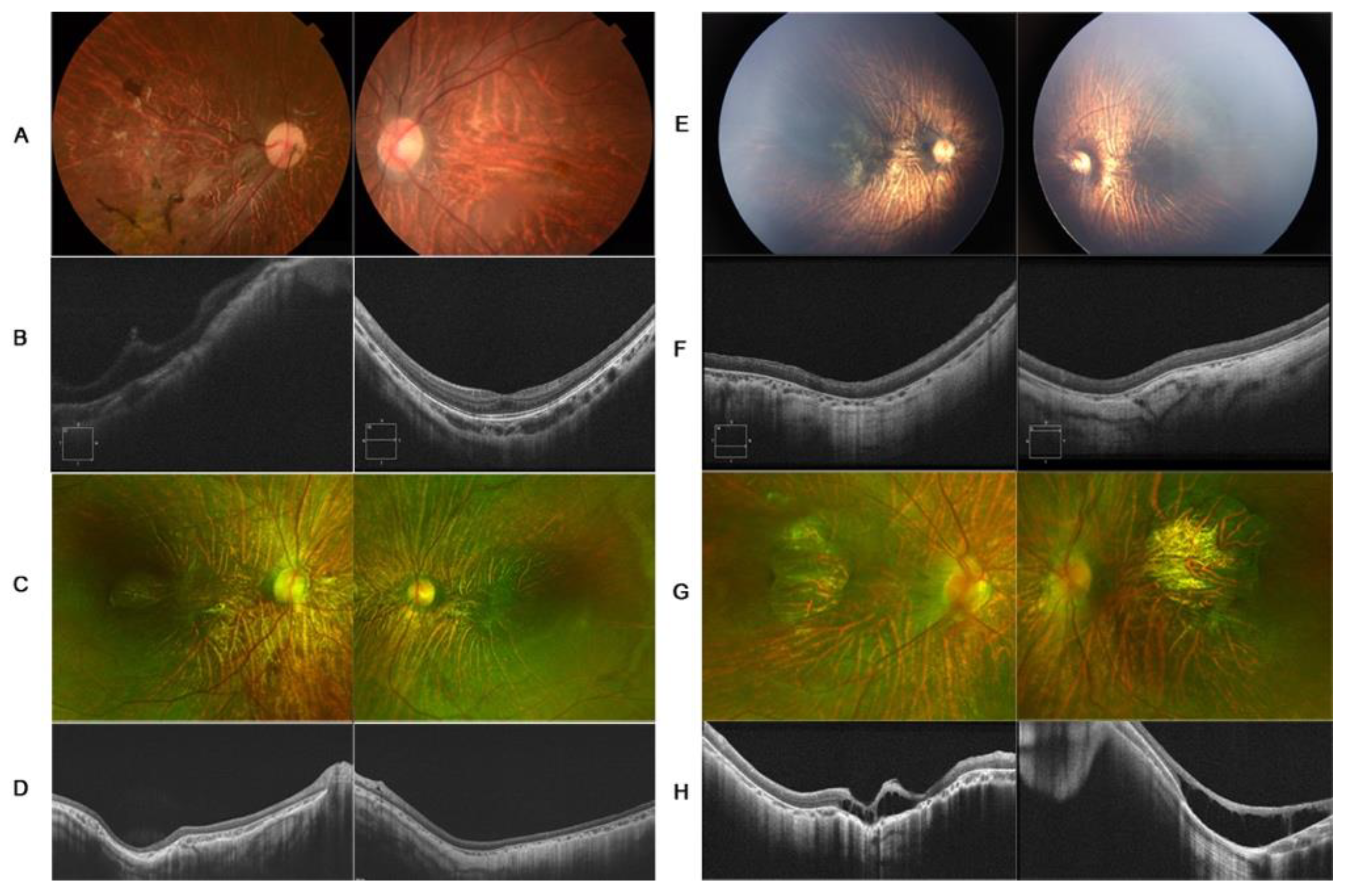
A 7-year-old girl, with a history of rhegmatogenous retinal detachment and pars plana vitrectomy in her right eye at 4 years of age, was referred to Zhongshan Ophthalmology Center with symptoms of blurred vision in the left eye. The BCVA of the proband was hand move/before eye OD and 0.3 logMAR OS. The axial lengths of the right and left eyes were 27.08 mm and 26.92 mm, respectively, with spherical equivalents of −15.0D OD and −14.25D OS. The anterior segment was unremarkable. Fundus examination revealed clumped pigmentary deposition in the right eye and tessellated funds in the left eye. OCT was mildly abnormal in her left eye, with slightly macular hypoplasia. The retina remained attached in her right eye (
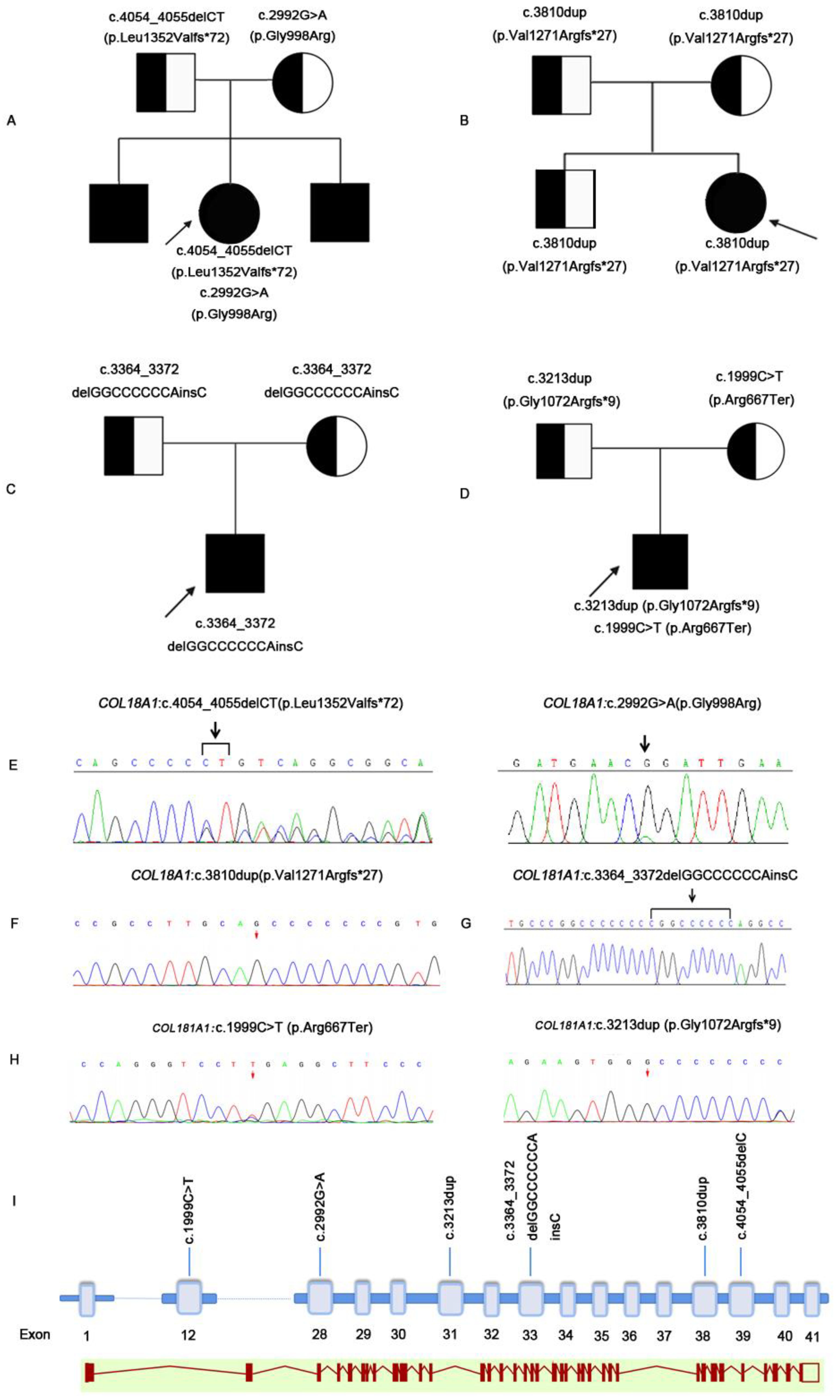
Figure 1A,B). A compound heterozygous mutation of the
COL18A1 gene was detected, c.4054_4055delCT (p.Leu1352Valfs*72) in exon39 and c.2992G>A (p.Gly998Arg) in exon 28. The same mutations were identified in her two brothers. Her 11-year-old brother had a diagnosis of congenital cataracts in both eyes at 1 year of age and underwent cataract surgery. Retinal detachment in the left eye was noted at 10 years of age, and pars plana vitrectomy was performed as treatment. The BCVA was hand move/30 cm OD and no light perception OS. Examination revealed a longstanding retinal detachment in his right eye and corneal leukoma of his left eye. Her 1-year-old brother underwent a fundus examination by RetCam for poor vision and nystagmus, since 3 months of age, revealing tessellated fundus and macular dysplasia of both eyes (
Supplementary Figure S1A–D).
3.1.2. Patient 2
A 3-year-old girl, from a non-consanguineous family, was referred to our hospital for high myopia and low vision detected on a routine examination. Her family history was unremarkable. Examination revealed horizontal conjugate nystagmus in both eyes. The spherical equivalent was −8.25 diopters in the right eye and −8.50 diopters in the left eye, with axial lengths of 25.16 mm and 25.19 mm, respectively. The intraocular pressure and anterior segment were unremarkable. Dilated fundus examination revealed empty vitreous, tessellate fundus, retinal pigmental changes, and punched-out macular in both eyes. The features in OCT included outer segment retina atrophy, significant thinning of the retina and choroid, macular hypoplasia, and posterior staphyloma. A homozygous
COL18A1 variant, c.3810dup (p.Val1271Arg fs*27), was identified by WES. Sanger sequencing showed that both parents and her older sister were heterozygous carriers (
Figure 1C,D).
3.1.3. Patient 3
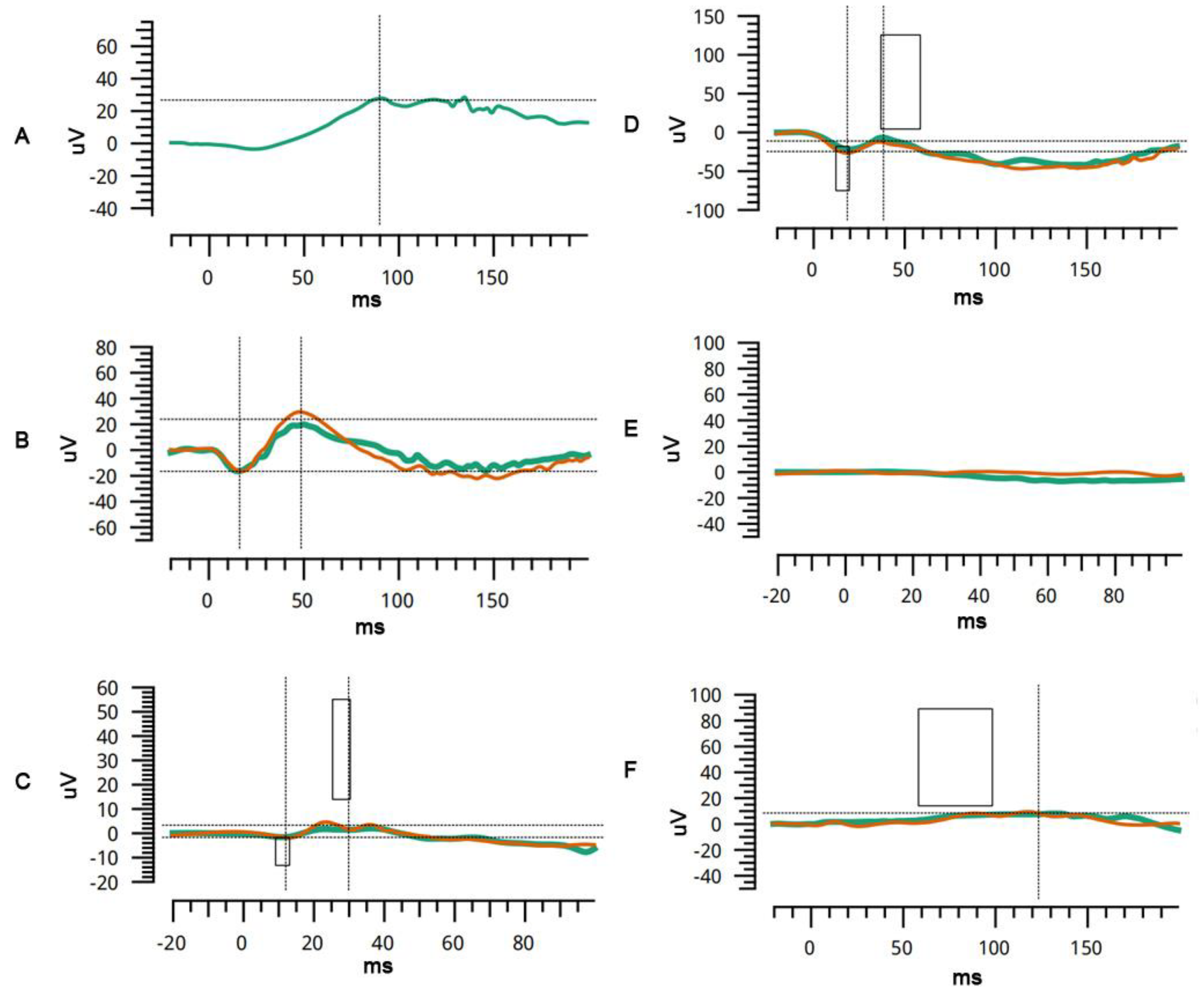
A 6-month-old boy, from a non-consanguineous family, presented with nystagmus since birth. The family history was unremarkable. The patient exhibited horizontal conjugate nystagmus in both eyes. Examination revealed axial lengths of 23.55 mm OD and 24.00 mm OS, with spherical equivalents of −9D OD and −10D OS. The anterior segment was unremarkable. Fundus examination by RetCam revealed widespread pigmentary changes, chorioretinal atrophy, and macular hypoplasia in both eyes. SD-OCT confirmed macular hypoplasia with the non-identifiable foveal pits. A poorly laminated retinal layer and absence of IZ-EZ (interdigitation zone and ellipsoid zone) were also identified. Flash electroretinogram showed a slightly decreased amplitude under scotopic conditions and dramatically decreased amplitude under photopic conditions. Genetic testing revealed a homozygous mutation for the
COL18A1 gene, c.3364_3372 delGGCCCCCCAinsC. Sanger sequencing showed that both parents and her older sister were heterozygous carriers (
Figure 1E,F and
Figure 2A–C).
3.1.4. Patient 4
A 2-year-old boy, born from a non-consanguineous family, presented with poor vision for six months. His personal history and family history were unremarkable. Cycloplegic refraction showed high myopia: −12.5 and −12 spherical equivalent diopters for the right and left eye, respectively. The axial lengths of the right and left eyes were 25.67 mm and 25.38 mm, respectively. The anterior segment was unremarkable. Fundus examination revealed the tessellate fundus, posterior scleral staphyloma, and macular pseudocolobomas. OCT revealed apparent scleral staphyloma, chorioretinal atrophy, macular retinoschisis, and poor retinal lamination. Flash electroretinogram showed an almost extinguished pattern under both scotopic and photopic conditions, which indicated the loss of function of cone and rod photoreceptors (
Figure 1G,H and
Figure 2D–F). A focal occipital skin defect was observed on the occipital scalp (
Supplementary Figure S1). MRI revealed a normal occipital skull. A compound heterozygous mutation was identified by genetic analysis: c.1999C>T (p.Arg667Ter) and c.3213dup(p.Gly1072ArgfsTer9). Sanger sequencing showed that both parents were heterozygous carriers.
3.2. Genetic Finding
Pedigree graphs of the four families are shown in
Figure 2. Six
COL18A1 pathogenic mutations were detected in four families. The coding impact of one of the mutations was missense: c.2992G>A(p.Gly998Arg); one of the mutations was nonsense: c.1999C>T(p.Arg667Ter); and four of the mutations were frameshift: c.4054_4055delC(p.Leu1352ValTer72), c.3810dup(p.Val1271ArgfsTer27), c.3364_3372delGGCCCCCCAinsC(p.Gly1122ArgfsTer142), and c.3213dup(Gly1072ArgfsTer9). The mutated sequence in the identified pathogenic variants was highly conserved in the homologs of
COL18A1 (
Supplementary Figure S1E). Of the six mutations, five (83.3%) were novel (
Table 2 and
Figure 3).
4. Discussion
Considering the rarity of this disease, the spectrum of the genetic variability remains to be discovered. Since being shown to be associated with KS in 2000 [
22],
COL18A1 remains the only gene to be definitively, causally related to the disease. Although
ADAMTS18 was once reported to be disease-causing in KS, a splicing mutation of
COL18A1 was later reported in the same patients. To date, 22
COL18A1 mutations have been reported to be likely pathogenic [
13]. In the present study, we identified five new mutations of
COL18A1, in four distinct KS families. Three of these were frameshift mutations, one was missense mutation, and one was nonsense mutation. The mutations were compound heterozygous in two families and homozygous in the other two families for the respective sites. Our results accord with previous studies, in which the majority of
COL18A1 variants (5/6, 83.3%) that led to KS were protein-truncating variants [
13]. In this study, one missense mutation has been detected. Although patient 1, with the heterozygous missense mutation and frameshift mutation, presented as a mild type. Considering the clinically heterogeneous of this disease, the genotype–phenotype correlation still needs further exploration.
Since being first reported by Knobloch in 1971, 48 genetically confirmed KS families have been reported. KS is classically defined as a triad of high myopia, retinal detachment, and occipital defect. The ophthalmic features are diverse, on account of the severity and course of the disease. In the present study, all patients presented with early-onset high myopia, with refractive diopters higher than −8 D. All patients showed retina changes, including tessellated fundus and macular abnormalities. Two patients had retinal detachment, due to the peripheral retinal tear before age 10. Because of the tendency of retinal detachment, prophylactic laser photocoagulation might be considered to improve visual outcomes.
Interestingly, occipital change was noticed in only one patient. Our results suggest that the eye phenotype alone can be pathognomonic. Children with early-onset high myopia, presenting with retinal changes at a very young age, should be suspected of having KS and undergo genetic testing, even without occipital changes.
In addition to high myopia and retinal detachment, other ocular manifestations have been described as characteristic features of KS. Hull et al. [
13] reported cone–rod dysfunction, revealed by electrophysiologic examination. Chorioretinal atrophy and colobomas have also been reported [
17,
23]. In the present study, we observed macular abnormalities in all KS patients, from mild macular hypoplasia to completely undetectable macular and retinoschisis. One proband had macular hypoplasia grade I; two probands had posterior staphyloma and macular hypoplasia grade IV; one proband had macular retinoschisis and macular hypoplasia grade IV. Although posterior staphyloma and macular atrophy are not specific and could occur secondary to high myopia, secondary changes in high myopia require long periods to occur. Thus, we suggest that macular changes, especially macular hypoplasia, could be a primary and key feature of KS.
The limitations of our study are as follows: (1) the sample size was limited to six, due to the rarity of KS; (2) all patients were recruited from the pediatric ophthalmology department of Zhongshan ophthalmic center, which might introduce selection bias; and (3) although characteristic clinical manifestations were observed in all patients, novel mutations will be further confirmed in future studies.
5. Conclusions
In conclusion, we identified novel COL18A1 mutations as causes of KS. Considering the variable manifestations, the diagnosis of KS might be considered in any patient with early-onset high myopia and macular abnormalities.
Supplementary Materials
The following are available online at
https://www.mdpi.com/article/10.3390/genes12101512/s1, Figure S1: The clinical feature and amino acid sequences of two brother of family A and skin defect of proband of family D. The fundus photograph or anterior segmental photography of two brothers from family A (A–D) and the occipital skin defect of proband D (F). The old brother presented with visual loss and uniocular blindness. The fundus images of right eye showed tessellated changes and retinal detachment occurred soon after presence (A). The anterior segmental image showed corneal leukoma of left eye (B). The fundus photograph of younger brother showed tessellated fundus appearance and macular dysplasia (C, D). The result of amino acid sequences of c.2992g>A from nine species reported in the mutation taster is shown (E). A focal occipital skin defect was observed on the occipital scalp in the proband of family D (F). Table S1:
COL18A1 mutation of patients with KS.
Author Contributions
Conceptualization, X.D., S.L. and Y.W.; methodology, S.L.; investigation, S.L., Y.W. and W.Y.; resources, X.D., L.S., L.H., T.Z. and Z.Z.; writing—original draft preparation, S.L. and Y.W.; writing—review and editing, X.D.; supervision, X.D.; funding acquisition, X.D. and S.L. All authors have read and agreed to the published version of the manuscript.
Funding
Supported in part by a by grants from the Fundamental Research Funds of State Key Laboratory of Ophthalmology, research funds of Sun Yat-sen University (18zxxt73; Guangzhou, Guangdong, China), Science and Technology Program, Guangdong, China (2018A030310230; Guangzhou, Guangdong, China), and the grant from the National Natural Science Foundation of China (31800873). The sponsors and funding organizations had no role in the design or conduct of this research.
Informed Consent Statement
This study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the institutional review board of Zhongshan Ophthalmic Center, Sun Yat-sen University (2020KYPJ173). Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
All raw data used during the study are available from the corresponding author by request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Czeizel, A.E.; Göblyös, P.; Kustos, G.; Mester, E.; Paraicz, E. The second report of Knobloch syndrome. Am. J. Med. Genet. 1992, 42, 777–779. [Google Scholar] [CrossRef]
- Passos-Bueno, M.R.; Marie, S.K.; Monteiro, M.; Neustein, I.; Whittle, M.R.; Vainzof, M.; Zatz, M. Knobloch syndrome in a large Brazilian consanguineous family: Confirmation of autosomal recessive inheritance. Am. J. Med. Genet. 1994, 52, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Aldahmesh, M.A.; Mohamed, J.Y.; Al-Mesfer, S.; Alkuraya, F.S. The distinct ophthalmic phenotype of Knobloch syndrome in children. Br. J. Ophthalmol. 2012, 96, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Kliemann, S.E.; Waetge, R.T.; Suzuki, O.T.; Passos-Bueno, M.R.; Rosemberg, S. Evidence of neuronal migration disorders in Knobloch syndrome: Clinical and molecular analysis of two novel families. Am. J. Med. Genet. A 2003, 119a, 15–19. [Google Scholar] [CrossRef]
- Caglayan, A.O.; Baranoski, J.F.; Aktar, F.; Han, W.; Tuysuz, B.; Guzel, A.; Guclu, B.; Kaymakcalan, H.; Aktekin, B.; Akgumus, G.T.; et al. Brain malformations associated with Knobloch syndrome--review of literature, expanding clinical spectrum, and identification of novel mutations. Pediatr. Neurol. 2014, 51, 806–813.e8. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.A.; Kirkby, G.R.; Williams, D.; Ainsworth, J.R. A phenotypic variant of Knobloch syndrome. Ophthalmic Genet. 2008, 29, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Määttä, M.; Heljasvaara, R.; Pihlajaniemi, T.; Uusitalo, M. Collagen XVIII/endostatin shows a ubiquitous distribution in human ocular tissues and endostatin-containing fragments accumulate in ocular fluid samples. Graefes Arch. Clin. Exp. Ophthalmol. 2007, 245, 74–81. [Google Scholar] [CrossRef]
- Seppinen, L.; Pihlajaniemi, T. The multiple functions of collagen XVIII in development and disease. Matrix Biol. 2011, 30, 83–92. [Google Scholar] [CrossRef]
- Zhang, L.S.; Li, H.B.; Zeng, J.; Yang, Y.; Ding, C. Knobloch syndrome caused by homozygous frameshift mutation of the COL18A1 gene in a Chinese pedigree. Int. J. Ophthalmol. 2018, 11, 918–922. [Google Scholar]
- Suzuki, O.T.; Sertie, A.L.; Der Kaloustian, V.M.; Kok, F.; Carpenter, M.; Murray, J.; Czeizel, A.E.; Kliemann, S.E.; Rosemberg, S.; Monteiro, M.; et al. Molecular analysis of collagen XVIII reveals novel mutations, presence of a third isoform, and possible genetic heterogeneity in Knobloch syndrome. Am. J. Hum. Genet. 2002, 71, 1320–1329. [Google Scholar] [CrossRef] [Green Version]
- Alsulaiman, S.M.; Al-Abdullah, A.A.; Alakeely, A.; Aldhibi, H.; Engelbrecht, L.; Ghazi, N.G.; Mura, M. Macular Hole-Related Retinal Detachment in Children with Knobloch Syndrome. Ophthalmol. Retin. 2020, 4, 498–503. [Google Scholar] [CrossRef]
- Balikova, I.; Sanak, N.S.; Fanny, D.; Smits, G.; Soblet, J.; de Baere, E.; Cordonnier, M. Three cases of molecularly confirmed Knobloch syndrome. Ophthalmic Genet. 2020, 41, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Hull, S.; Arno, G.; Ku, C.A.; Ge, Z.; Waseem, N.; Chandra, A.; Webster, A.R.; Robson, A.G.; Michaelides, M.; Weleber, R.G.; et al. Molecular and Clinical Findings in Patients With Knobloch Syndrome. JAMA Ophthalmol. 2016, 134, 753–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thau, A.; Tsukikawa, M.; Wangtiraumnuay, N.; Capasso, J.; Affel, E.; Alnabi, W.A.; Adam, M.; Alsulaiman, S.M.; Spirn, M.; Levin, A.V. Optical Coherence Tomography in Knobloch Syndrome. Ophthalmic Surg. Lasers Imaging Retin. 2019, 50, e203–e210. [Google Scholar] [CrossRef]
- White, R.J.; Wang, Y.; Tang, P.; Montezuma, S.R. Knobloch syndrome associated with Polymicrogyria and early onset of retinal detachment: Two case reports. BMC Ophthalmol. 2017, 17, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradstein, L.; Hansen, R.M.; Cox, G.F.; Altschwager, P.; Fulton, A.B. Progressive retinal degeneration in a girl with Knobloch syndrome who presented with signs of ocular albinism. Doc. Ophthalmol. 2017, 134, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, V.B.; Olney, A.H.; Garrett, P.; Chary, A.; Dragan, E.; Lerner, G.; Murray, J.; Bassuk, A.G. Collagen XVIII mutation in Knobloch syndrome with acute lymphoblastic leukemia. Am. J. Med. Genet. A 2010, 152A, 2875–2879. [Google Scholar] [CrossRef] [Green Version]
- Haghighi, A.; Tiwari, A.; Piri, N.; Nurnberg, G.; Saleh-Gohari, N.; Haghighi, A.; Neidhardt, J.; Nurnberg, P.; Berger, W. Homozygosity mapping and whole exome sequencing reveal a novel homozygous COL18A1 mutation causing Knobloch syndrome. PLoS ONE 2014, 9, e112747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wawrzynski, J.; Than, J.; Gillam, M.; Foster, P.J. Acute Angle Closure in Knobloch Syndrome. J. Glaucoma 2021, 30, e265–e268. [Google Scholar] [CrossRef]
- Nakousi-Capurro, N.; Huserman, J.; Castillo, S.; Herrera, L.; Romero, P.; Pizarro, F.; Quezada, C.; Cea, F. Knobloch syndrome in a patient from Chile. Am. J. Med. Genet. A 2020, 182, 2239–2242. [Google Scholar] [CrossRef]
- Wang, P.; Li, S.; Sun, W.; Xiao, X.; Jia, X.; Liu, M.; Xu, L.; Long, Y.; Zhang, Q. An Ophthalmic Targeted Exome Sequencing Panel as a Powerful Tool to Identify Causative Mutations in Patients Suspected of Hereditary Eye Diseases. Transl. Vis. Sci. Technol. 2019, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sertié, A.L.; Sossi, V.; Camargo, A.A.; Zatz, M.; Brahe, C.; Passos-Bueno, M.R. Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome). Hum. Mol. Genet. 2000, 9, 2051–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren, B.; Suzuki, O.T.; Gérard-Blanluet, M.; Brémond-Gignac, D.; Elmaleh, M.; Titomanlio, L.; Delezoide, A.L.; Passos-Bueno, M.R.; Verloes, A. CNS malformations in Knobloch syndrome with splice mutation in COL18A1 gene. Am. J. Med. Genet. A 2007, 143a, 1514–1518. [Google Scholar] [CrossRef] [PubMed]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).







{kind=link}
{kind=link}
{kind=link}