Predicting Functions of Uncharacterized Human Proteins: From Canonical to Proteoforms


Abstract
1. Introduction
2. Materials and Methods
2.1. Gene Sets
2.2. Re-Analysis of MS Data
2.3. Building PPI Network
2.4. Determination of Optimum Threshold
- TP = number of interactions observed both in CORUM and in our dataset.
- FP = number of interactions observed in our dataset but not in CORUM.
- FN = number of interactions observed in CORUM but not in our dataset.
2.5. GO-Annotations for Characterized Proteins
2.6. Prediction of Unknown Protein Functions
2.7. Analysis of Interactome Profiles
2.8. Software Implementation of Algorithms
3. Results
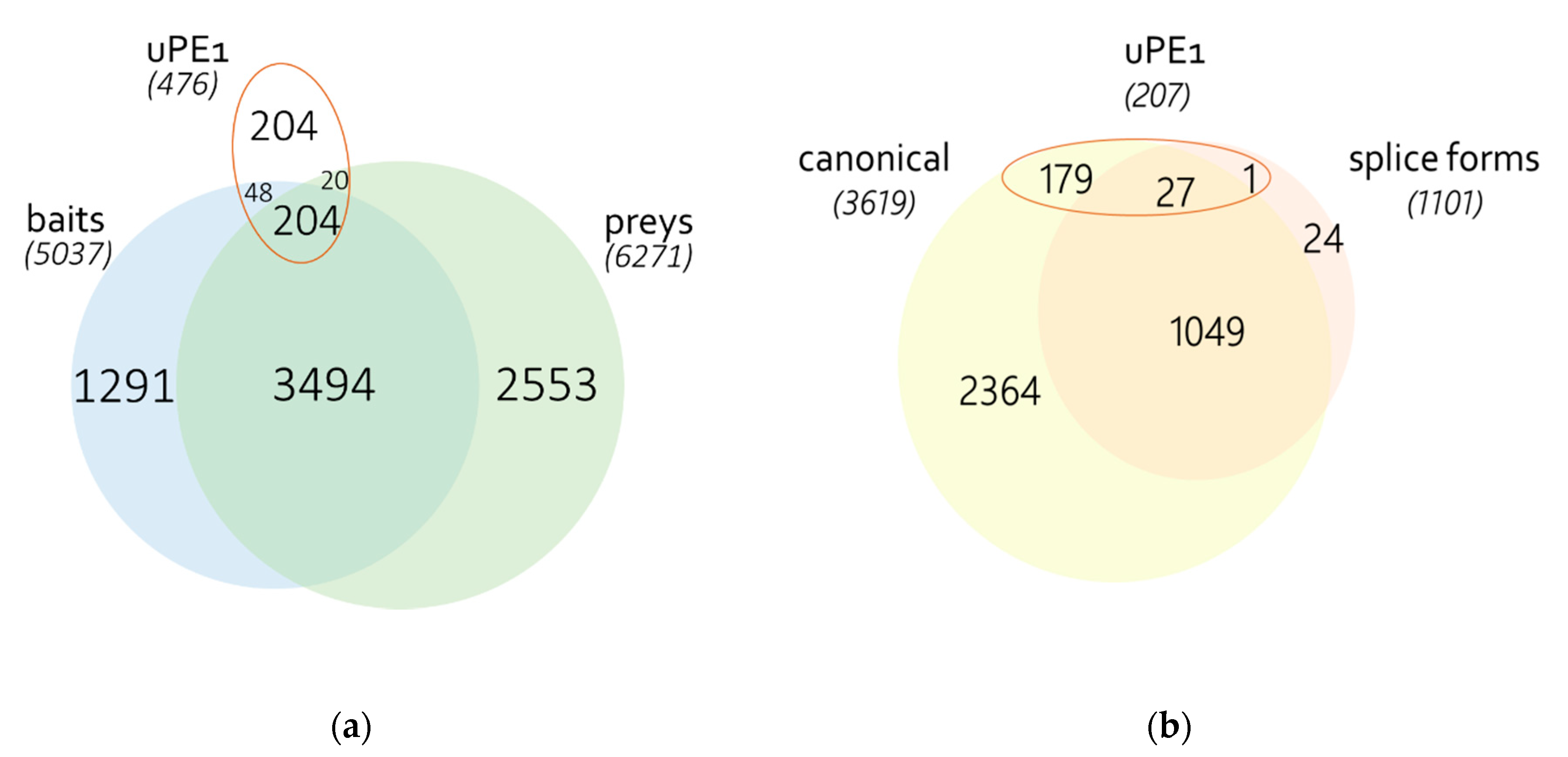
3.1. Identification of Proteins
3.2. Human PPI Network
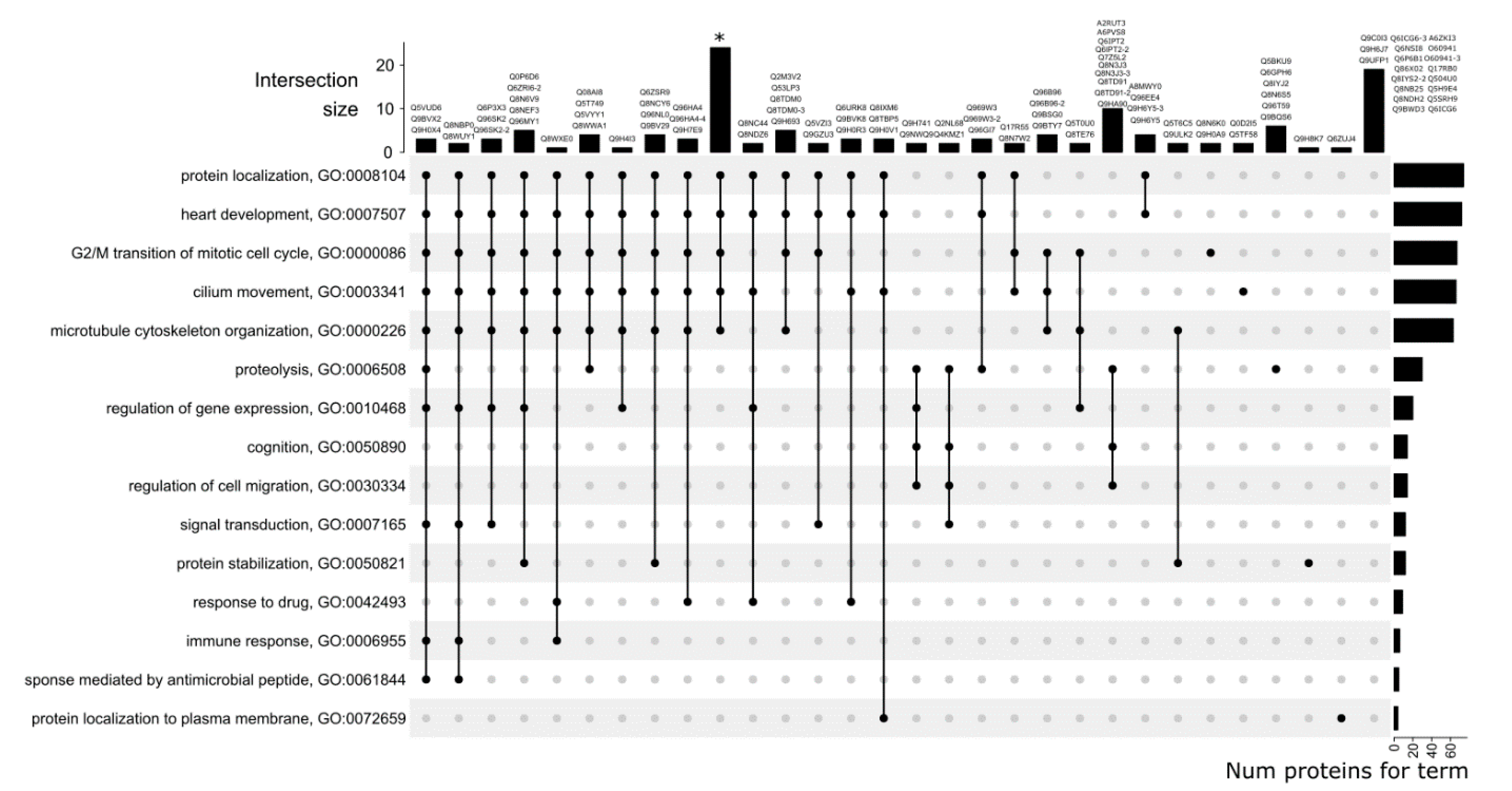
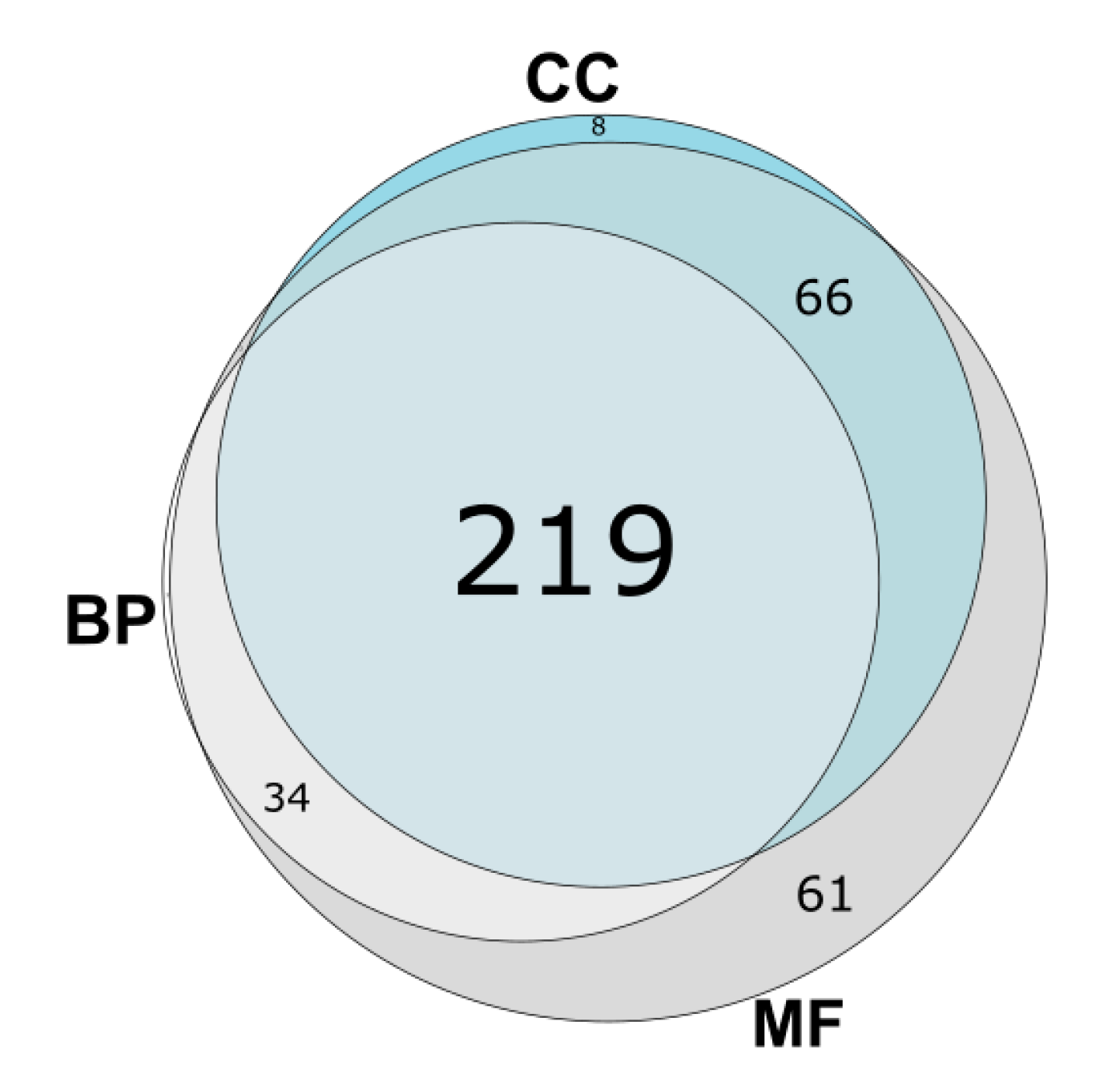
3.3. GO Category Prediction for uPE1 Proteins—Biological Processes
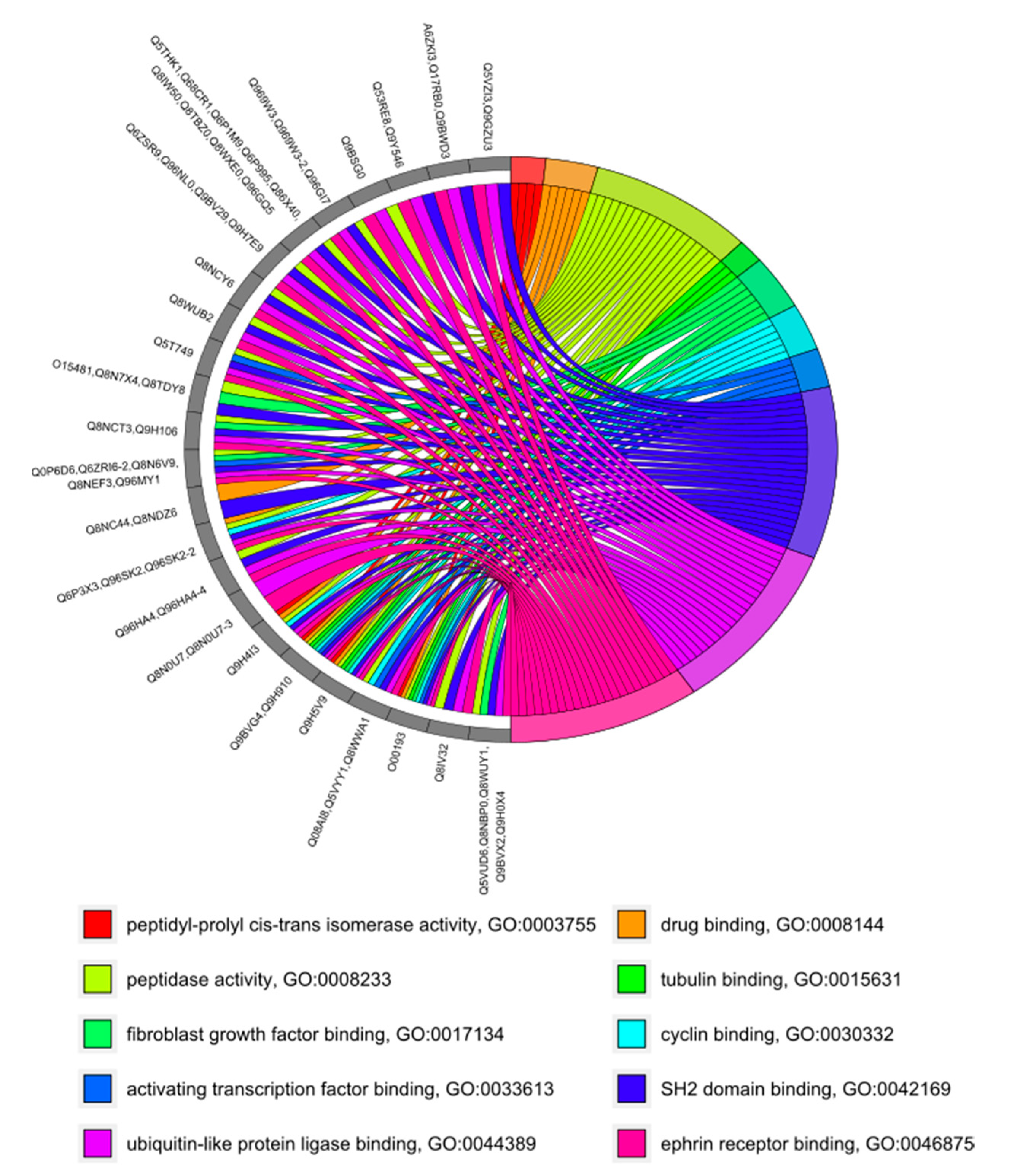
3.4. GO Category Prediction for uPE1 Proteins—Molecular Functions
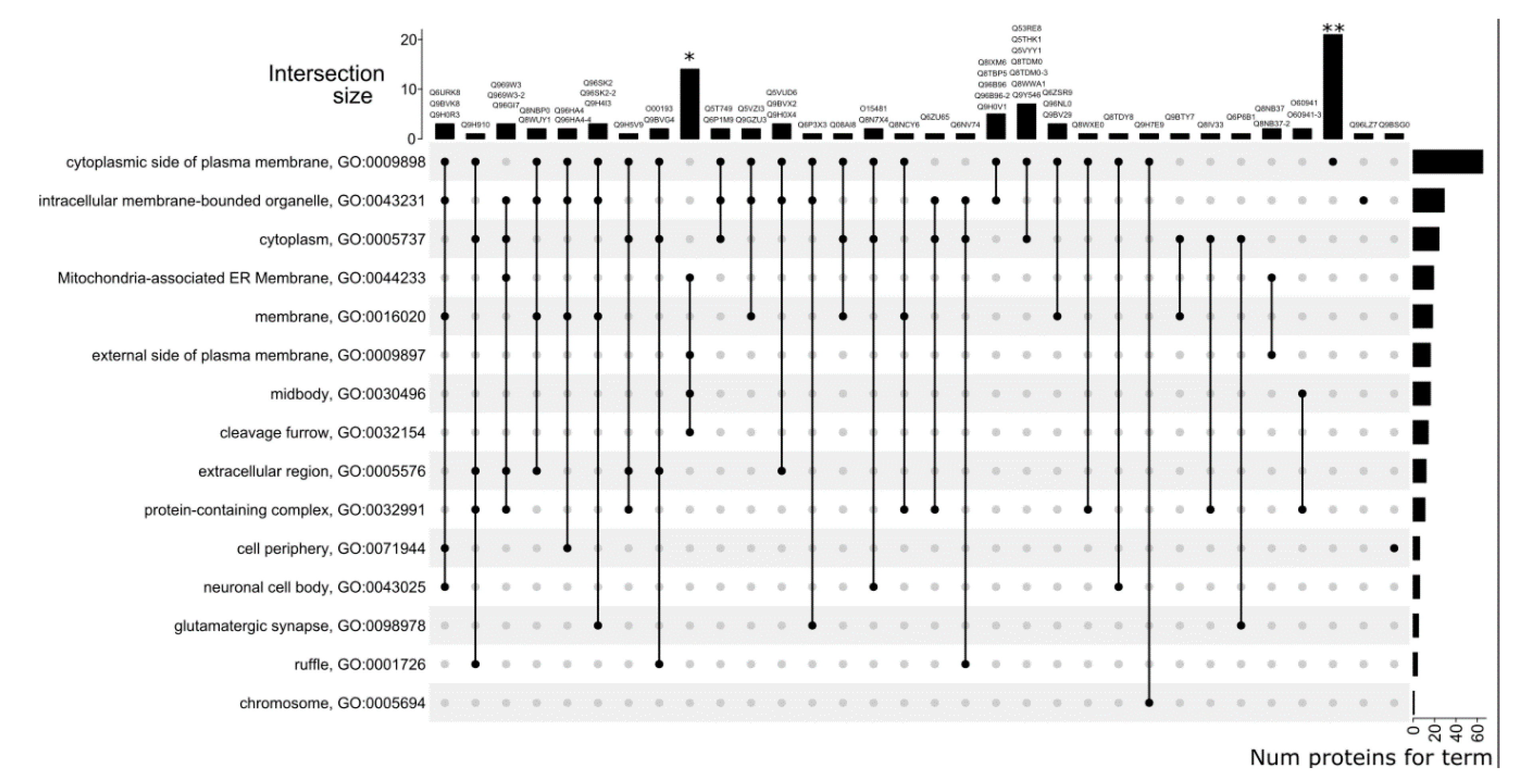
3.5. GO Category Prediction for uPE1 Proteins—Cellular Components
3.6. Differences of Splice-Forms Interactomic Profiles
3.7. Impact of Data Sources
3.8. Comparison with UniProt Predictions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fields, C.; Adams, M.D.; White, O.; Venter, J.C. How many genes in the human genome? Nat. Genet. 1994, 7, 345–346. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, S.L. Open questions: How many genes do we have? BMC Boil. 2018, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Methods 2018, 14, 206–214. [Google Scholar] [CrossRef]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 1–6. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [CrossRef]
- Gaudet, P.; Michel, P.-A.; Zahn-Zabal, M.; Britan, A.; Cusin, I.; Domagalski, M.; Duek, P.D.; Gateau, A.; Gleizes, A.; Hinard, V.; et al. The neXtProt knowledgebase on human proteins: 2017 Update. Nucleic Acids Res. 2016, 45, D177–D182. [Google Scholar] [CrossRef]
- Legrain, P.; Aebersold, R.; Archakov, A.; Bairoch, A.; Bala, K.; Beretta, L.; Bergeron, J.; Borchers, C.H.; Corthals, G.L.; Costello, C.E.; et al. The Human Proteome Project: Current State and Future Direction. Mol. Cell. Proteomics 2011, 10, M111.009993. [Google Scholar] [CrossRef]
- Paik, Y.-K.; Overall, C.M.; Corrales, F.; Deutsch, E.W.; Lane, L.; Omenn, G.S. Advances in Identifying and Characterizing the Human Proteome. J. Proteome Res. 2019, 18, 4079–4084. [Google Scholar] [CrossRef]
- Kulmanov, M.; Khan, M.A.; Hoehndorf, R. DeepGO: Predicting protein functions from sequence and interactions using a deep ontology-aware classifier. Bioinformatics 2017, 34, 660–668. [Google Scholar] [CrossRef]
- Zhou, N.; Jiang, Y.; Bergquist, T.R.; Lee, A.J.; Kacsoh, B.Z.; Crocker, A.W.; Lewis, K.A.; Georghiou, G.; Nguyen, H.N.; Hamid, N.; et al. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Boil. 2019, 20, 1–23. [Google Scholar] [CrossRef]
- Piovesan, D.; E Tosatto, S.C. INGA 2.0: Improving protein function prediction for the dark proteome. Nucleic Acids Res. 2019, 47, W373–W378. [Google Scholar] [CrossRef] [PubMed]
- Frasca, M.; Cesa-Bianchi, N.A. Multitask Protein Function Prediction through Task Dissimilarity. IEEE/ACM Trans. Comput. Boil. Bioinform. 2019, 16, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Luo, Y.; Zhang, Y.; Ying, J.; Xue, W.; Xie, T.; Tao, L.; Zhu, F. Protein functional annotation of simultaneously improved stability, accuracy and false discovery rate achieved by a sequence-based deep learning. Brief. Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Prasad, A.; Chatterjee, P.; Basu, S.; Nasipuri, M. Protein function prediction from dynamic protein interaction network using gene expression data. J. Bioinform. Comput. Boil. 2019, 17, 1950025. [Google Scholar] [CrossRef]
- Paik, Y.-K.; Lane, L.; Kawamura, T.; Chen, Y.-J.; Cho, J.-Y.; LaBaer, J.; Yoo, J.S.; Domont, G.B.; Corrales, F.; Omenn, G.S.; et al. Launching the C-HPP neXt-CP50 Pilot Project for Functional Characterization of Identified Proteins with No Known Function. J. Proteome Res. 2018, 17, 4042–4050. [Google Scholar] [CrossRef] [PubMed]
- Duek, P.; Gateau, A.; Bairoch, A.; Lane, L. Exploring the Uncharacterized Human Proteome Using neXtProt. J. Proteome Res. 2018, 17, 4211–4226. [Google Scholar] [CrossRef]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2010, 12, 56–68. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Z.-P. Analysis of Topological Parameters of Complex Disease Genes Reveals the Importance of Location in a Biomolecular Network. Genes 2019, 10, 143. [Google Scholar] [CrossRef]
- Ponomarenko, E.; Kopylov, A.; Lisitsa, A.V.; Radko, S.P.; Kiseleva, Y.Y.; Kurbatov, L.K.; Ptitsyn, K.G.; Tikhonova, O.V.; Moisa, A.A.; Novikova, S.; et al. Chromosome 18 Transcriptoproteome of Liver Tissue and HepG2 Cells and Targeted Proteome Mapping in Depleted Plasma: Update 2013. J. Proteome Res. 2013, 13, 183–190. [Google Scholar] [CrossRef]
- Cafarelli, T.; Desbuleux, A.; Wang, Y.; Choi, S.G.; De Ridder, D.; Vidal, M. Mapping, modeling, and characterization of protein–protein interactions on a proteomic scale. Curr. Opin. Struct. Boil. 2017, 44, 201–210. [Google Scholar] [CrossRef]
- Yang, X.; Coulombe-Huntington, J.; Kang, S.; Sheynkman, G.M.; Hao, T.; Richardson, A.; Sun, S.; Yang, F.; Shen, Y.A.; Murray, R.R.; et al. Widespread Expansion of Protein Interaction Capabilities by Alternative Splicing. Cell 2016, 164, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.V.; Das, J.; Meyer, M.J.; Cordero, N.A.; Akturk, N.; Wei, X.; Fair, B.J.; Degatano, A.G.; Fragoza, R.; Liu, L.G.; et al. A Proteome-wide Fission Yeast Interactome Reveals Network Evolution Principles from Yeasts to Human. Cell 2016, 164, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J.; Barabási, A.-L. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 1257601. [Google Scholar] [CrossRef] [PubMed]
- Sahni, N.; Yi, S.; Taipale, M.; Bass, J.I.F.; Coulombe-Huntington, J.; Yang, F.; Peng, J.; Weile, J.; Karras, G.; Wang, Y.; et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015, 161, 647–660. [Google Scholar] [CrossRef]
- Feng, S.; Zhou, L.; Huang, C.; Xie, K.; Nice, E.C. Interactomics: Toward protein function and regulation. Expert Rev. Proteom. 2015, 12, 37–60. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Kim, D.-K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef]
- Lee, C.-M.; Adamchek, C.; Feke, A.; Nusinow, D.A.; Gendron, J.M. Mapping Protein–Protein Interactions Using Affinity Purification and Mass Spectrometry. Adv. Struct. Saf. Stud. 2017, 1610, 231–249. [Google Scholar] [CrossRef]
- Dunham, W.H.; Mullin, M.; Gingras, A.-C. Affinity-purification coupled to mass spectrometry: Basic principles and strategies. Proteomics 2012, 12, 1576–1590. [Google Scholar] [CrossRef]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef]
- Ghadie, M.; Xia, Y. Estimating dispensable content in the human interactome. Nat. Commun. 2019, 10, 3205. [Google Scholar] [CrossRef]
- Vidal, M.; Cusick, M.E.; Barabási, A.-L. Interactome Networks and Human Disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Zorin, A.; Dass, G.; Vu, M.-T.; Xu, P.; Glont, M.; Vizcaíno, J.A.; Jarnuczak, A.; Petryszak, R.; Ping, P.; et al. Quantifying the impact of public omics data. Nat. Commun. 2019, 10, 3512–3610. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Sheynkman, G.M.; Zhang, I.; Vidal, M. Proteome-scale human interactomics. Trends Biochem. Sci. 2017, 42, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Lapek, J.D.; Greninger, P.; Morris, R.; Amzallag, A.; Pruteanu-Malinici, I.; Benes, C.H.; Haas, W. Detection of dysregulated protein-association networks by high-throughput proteomics predicts cancer vulnerabilities. Nat. Biotechnol. 2017, 35, 983–989. [Google Scholar] [CrossRef]
- Drew, K.; Lee, C.; Huizar, R.L.; Tu, F.; Borgeson, B.; McWhite, C.D.; Ma, Y.; Wallingford, J.B.; Salemi, M. Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes. Mol. Syst. Boil. 2017, 13, 932. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Gao, X.; Qi, R. Integrated bioinformatic analysis of differentially expressed genes and signaling pathways in plaque psoriasis. Mol. Med. Rep. 2019, 20, 225–235. [Google Scholar] [CrossRef]
- Shatsky, M.; Allen, S.; Gold, B.L.; Liu, N.L.; Juba, T.R.; Reveco, S.A.; Elias, D.A.; Prathapam, R.; He, J.; Yang, W.; et al. Bacterial Interactomes: Interacting Protein Partners Share Similar Function and Are Validated in Independent Assays More Frequently Than Previously Reported. Mol. Cell. Proteom. 2016, 15, 1539–1555. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Kiseleva, O.; Poverennaya, E.; Shargunov, A.; Lisitsa, A. Proteomic Cinderella: Customized analysis of bulky MS/MS data in one night. J. Bioinform. Comput. Boil. 2018, 16, 1740011. [Google Scholar] [CrossRef] [PubMed]
- Barsnes, H.; Vaudel, M. SearchGUI: A Highly Adaptable Common Interface for Proteomics Search and de Novo Engines. J. Proteome Res. 2018, 17, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification–mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [PubMed]
- He, Z. PPI network inference from AP-MS data. Data Min. Bioinform. Appl. 2015, 16, 51–59. [Google Scholar] [CrossRef]
- Qingzhou Zhang SMAD: Statistical Modelling of AP-MS Data (SMAD), R package. Available online: https://www.bioconductor.org/packages/SMAD (accessed on 21 June 2020).
- Hart, T.; Lee, I.; Salemi, M. A high-accuracy consensus map of yeast protein complexes reveals modular nature of gene essentiality. Bmc Bioinform. 2007, 8, 236. [Google Scholar] [CrossRef]
- Giurgiu, M.; Reinhard, J.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Ruepp, A. CORUM: The comprehensive resource of mammalian protein complexes—2019. Nucleic Acids Res. 2018, 47, D559–D563. [Google Scholar] [CrossRef]
- Scott, N.E.; Brown, L.M.; Kristensen, A.R.; Foster, L.J. Development of a computational framework for the analysis of protein correlation profiling and spatial proteomics experiments. J. Proteom. 2015, 118, 112–129. [Google Scholar] [CrossRef]
- Scott, N.E.; Rogers, L.D.; Prudova, A.; Brown, N.F.; Fortelny, N.; Overall, C.M.; Foster, L.J. Interactome disassembly during apoptosis occurs independent of caspase cleavage. Mol. Syst. Boil. 2017, 13, 906. [Google Scholar] [CrossRef]
- Brionne, A.; Juanchich, A.; Hennequet-Antier, C. ViSEAGO: A Bioconductor package for clustering biological functions using Gene Ontology and semantic similarity. Biodata Min. 2019, 12, 13–16. [Google Scholar] [CrossRef]
- Frasca, M.; Bertoni, A.; Re, M.; Valentini, G. A neural network algorithm for semi-supervised node label learning from unbalanced data. Neural Netw. 2013, 43, 84–98. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. Bmc Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef]
- (The Gene Ontology Consortium) The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef] [PubMed]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological Network Exploration with Cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 8.13.1–8.13.24. [Google Scholar] [CrossRef] [PubMed]
- (R Core Team). R: A Language and Environment for Statistical Computing.
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Int.J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinform. 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Schoch, D. graphlayouts: Additional Layout Algorithms for Network Visualizations. In Educational Technology Research and Development; Springer: Berlin, Germany, 2020; pp. 1–32. [Google Scholar]
- Lewis, B.W. threejs: Interactive 3D Scatter Plots, Networks and Globes, R package. Available online: https://CRAN.R-project.org/package=threejs (accessed on 21 June 2020).
- Morris, J.H.; Knudsen, G.M.; Verschueren, E.; Johnson, J.R.; Cimermancic, P.; Greninger, A.L.; Pico, A.R. Affinity purification–mass spectrometry and network analysis to understand protein-protein interactions. Nat. Protoc. 2014, 9, 2539–2554. [Google Scholar] [CrossRef]
- Yang, X.; Boehm, J.S.; Yang, X.; Salehi-Ashtiani, K.; Hao, T.; Shen, Y.; Lubonja, R.; Thomas, S.R.; Alkan, O.; Bhimdi, T.; et al. A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 2011, 8, 659–661. [Google Scholar] [CrossRef]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallström, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Boil. 2019, 15, e8503. [Google Scholar] [CrossRef]
- Zhang, B.; Park, B.-H.; Karpinets, T.; Samatova, N.F. From pull-down data to protein interaction networks and complexes with biological relevance. Bioinformatics 2008, 24, 979–986. [Google Scholar] [CrossRef]
- Walter, W.; Sanchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis: Figure 1. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2011, 40, D841–D846. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Li, F.; Qin, Y.; Bo, X.; Wu, Y.; Wang, S. GOSemSim: An R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 2010, 26, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mi, Y.; Mueller, T.; Kreibich, S.; Williams, E.G.; Van Drogen, A.; Borel, C.; Frank, M.; Germain, P.-L.; Bludau, I.; et al. Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nat. Biotechnol. 2019, 37, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Bin Goh, W.W.; Wang, W.; Wong, L. Why Batch Effects Matter in Omics Data, and How to Avoid Them. Trends Biotechnol. 2017, 35, 498–507. [Google Scholar] [CrossRef]
- Zhang, C.; Lane, L.; Omenn, G.S.; Zhang, Y. Blinded Testing of Function Annotation for uPE1 Proteins by I-TASSER/COFACTOR Pipeline Using the 2018–2019 Additions to neXtProt and the CAFA3 Challenge. J. Proteome Res. 2019, 18, 4154–4166. [Google Scholar] [CrossRef]
- Gligorijevic, V.; Barot, M.; Bonneau, R. deepNF: Deep network fusion for protein function prediction. Bioinformatics 2018, 34, 3873–3881. [Google Scholar] [CrossRef]
- Peng, J.; Xue, H.; Wei, Z.; Tuncali, I.; Hao, J.-Y.; Shang, X. Integrating multi-network topology for gene function prediction using deep neural networks. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Gámez-Valero, A.; Beyer, K. Alternative Splicing of α- and β-Synuclein Genes Plays Differential Roles in Synucleinopathies. Genes 2018, 9, 63. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Genes | <50% | ≥50% | ≥75% | ≥90% |
|---|---|---|---|---|
| 62 | 24 | 11 | 3 | 24 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poverennaya, E.; Kiseleva, O.; Romanova, A.; Pyatnitskiy, M. Predicting Functions of Uncharacterized Human Proteins: From Canonical to Proteoforms. Genes 2020, 11, 677. https://doi.org/10.3390/genes11060677
Poverennaya E, Kiseleva O, Romanova A, Pyatnitskiy M. Predicting Functions of Uncharacterized Human Proteins: From Canonical to Proteoforms. Genes. 2020; 11(6):677. https://doi.org/10.3390/genes11060677
Chicago/Turabian StylePoverennaya, Ekaterina, Olga Kiseleva, Anastasia Romanova, and Mikhail Pyatnitskiy. 2020. "Predicting Functions of Uncharacterized Human Proteins: From Canonical to Proteoforms" Genes 11, no. 6: 677. https://doi.org/10.3390/genes11060677
APA StylePoverennaya, E., Kiseleva, O., Romanova, A., & Pyatnitskiy, M. (2020). Predicting Functions of Uncharacterized Human Proteins: From Canonical to Proteoforms. Genes, 11(6), 677. https://doi.org/10.3390/genes11060677