Altered miRNA and mRNA Expression in Sika Deer Skeletal Muscle with Age

Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Tissues Collection
2.2. RNA Sequencing and Data Analysis
2.3. Small RNA Sequencing and Data Analysis
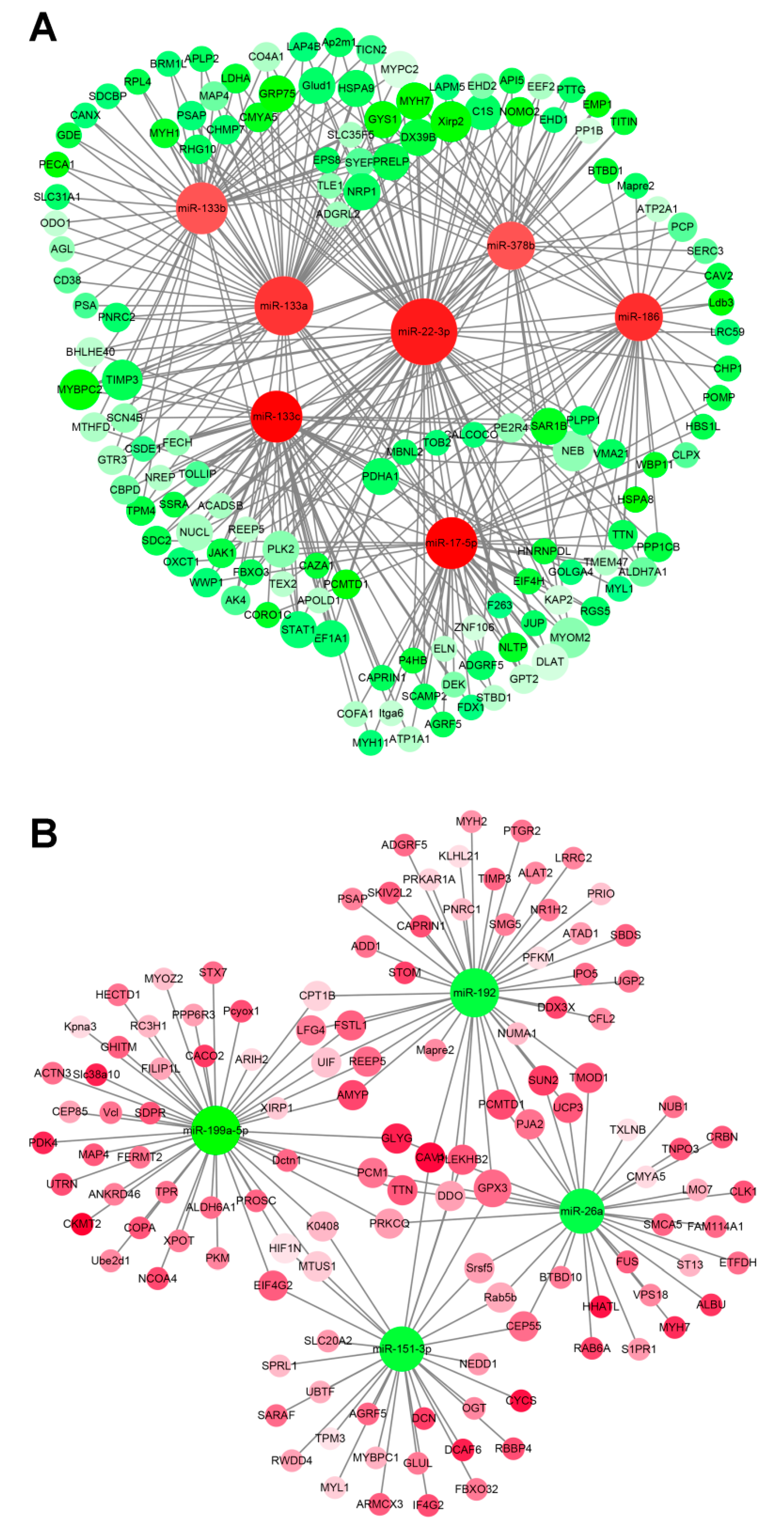
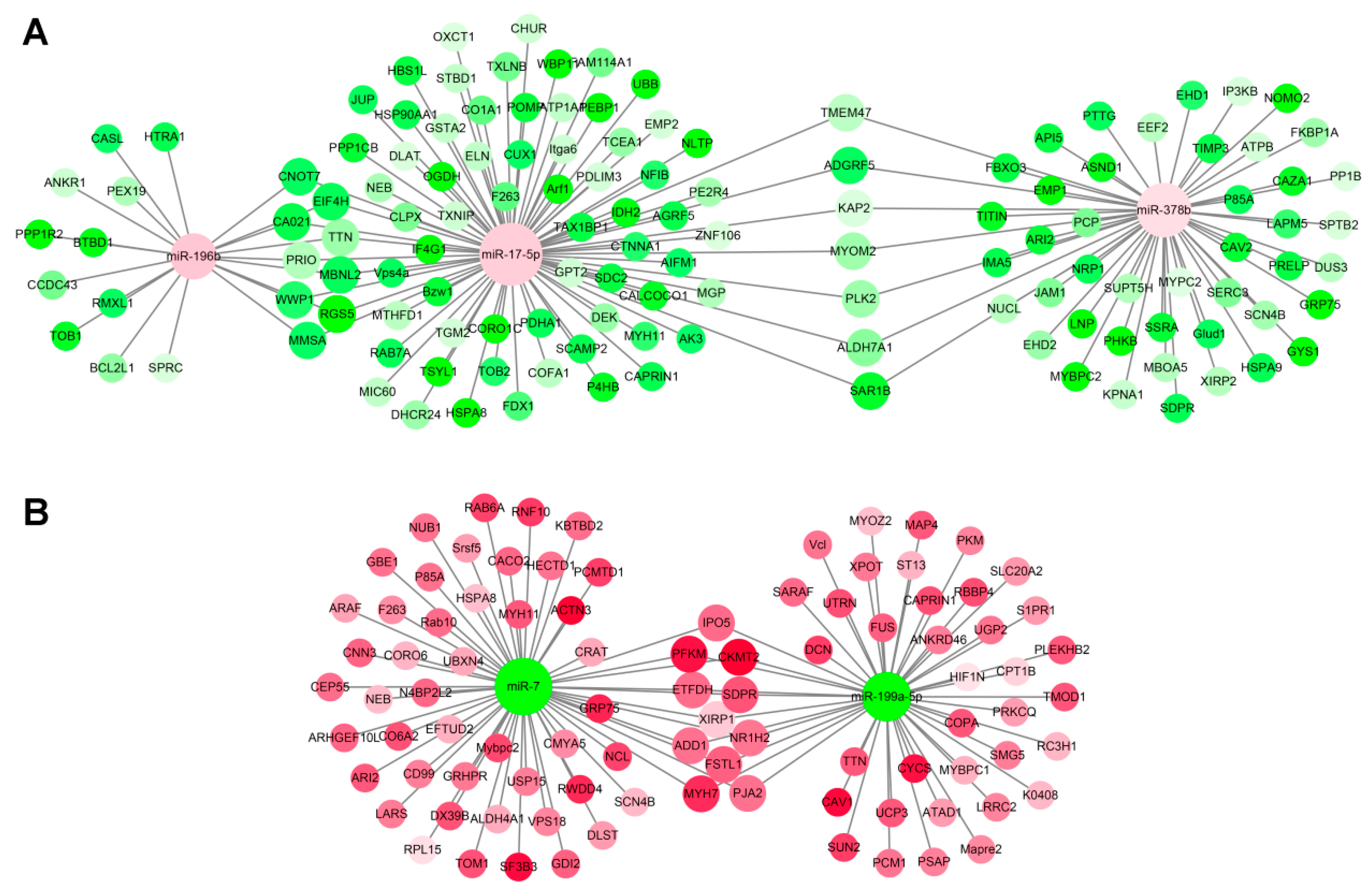
2.4. miRNA–mRNA Interaction Network Construction
2.5. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Analyses
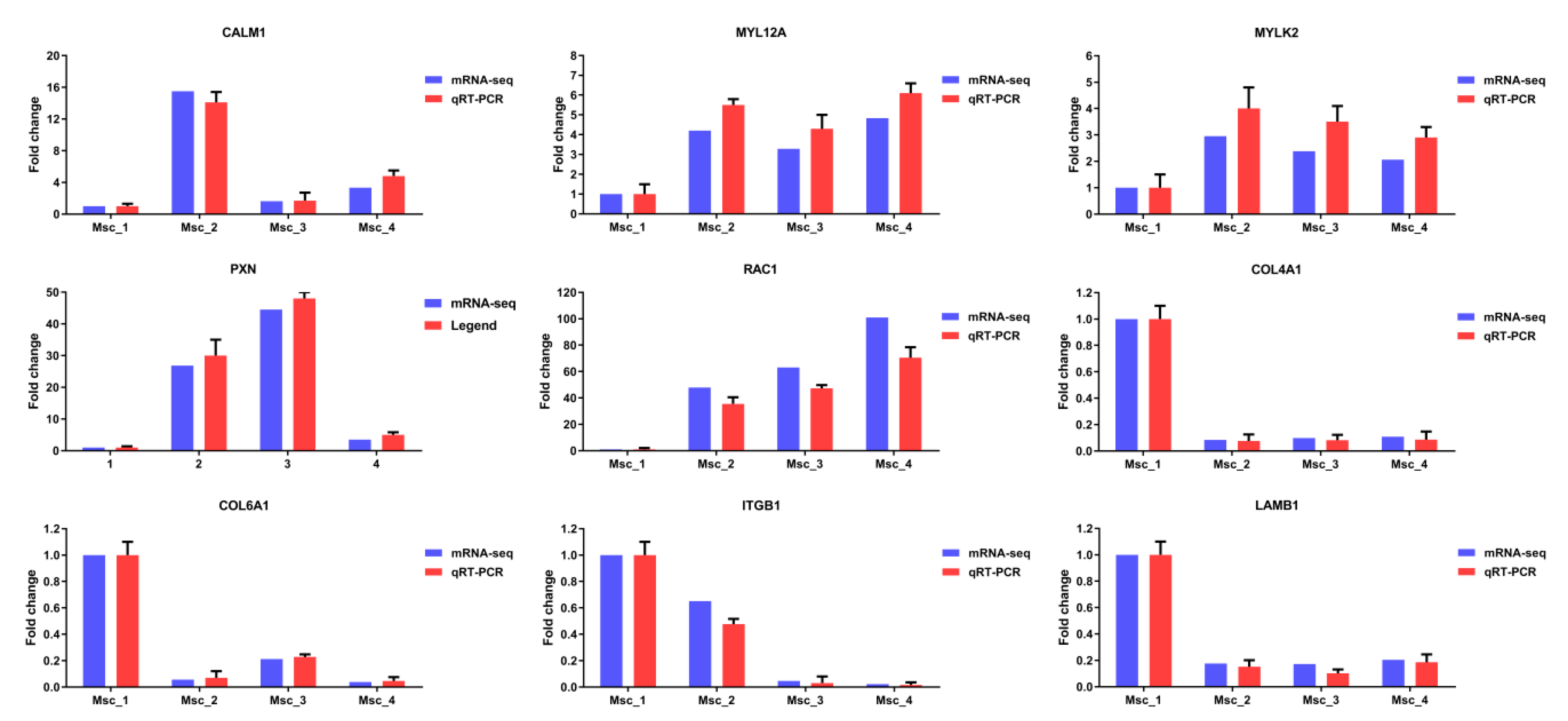
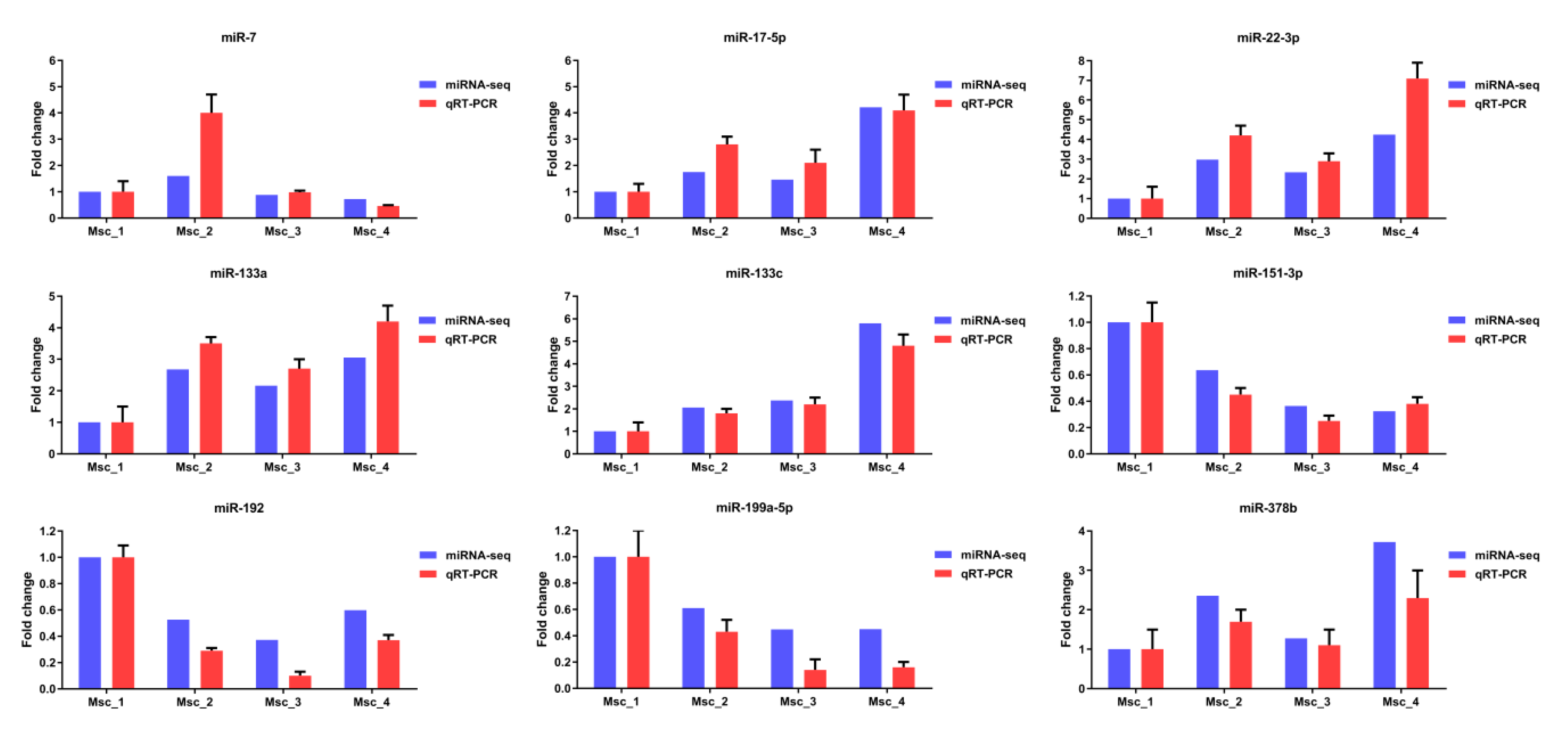
2.6. Real-Time qPCR Validation of Differentially Expressed Unigenes and miRNAs
2.7. Supporting Data Information
3. Results
3.1. Overview of the Transcriptome and the miRNAome
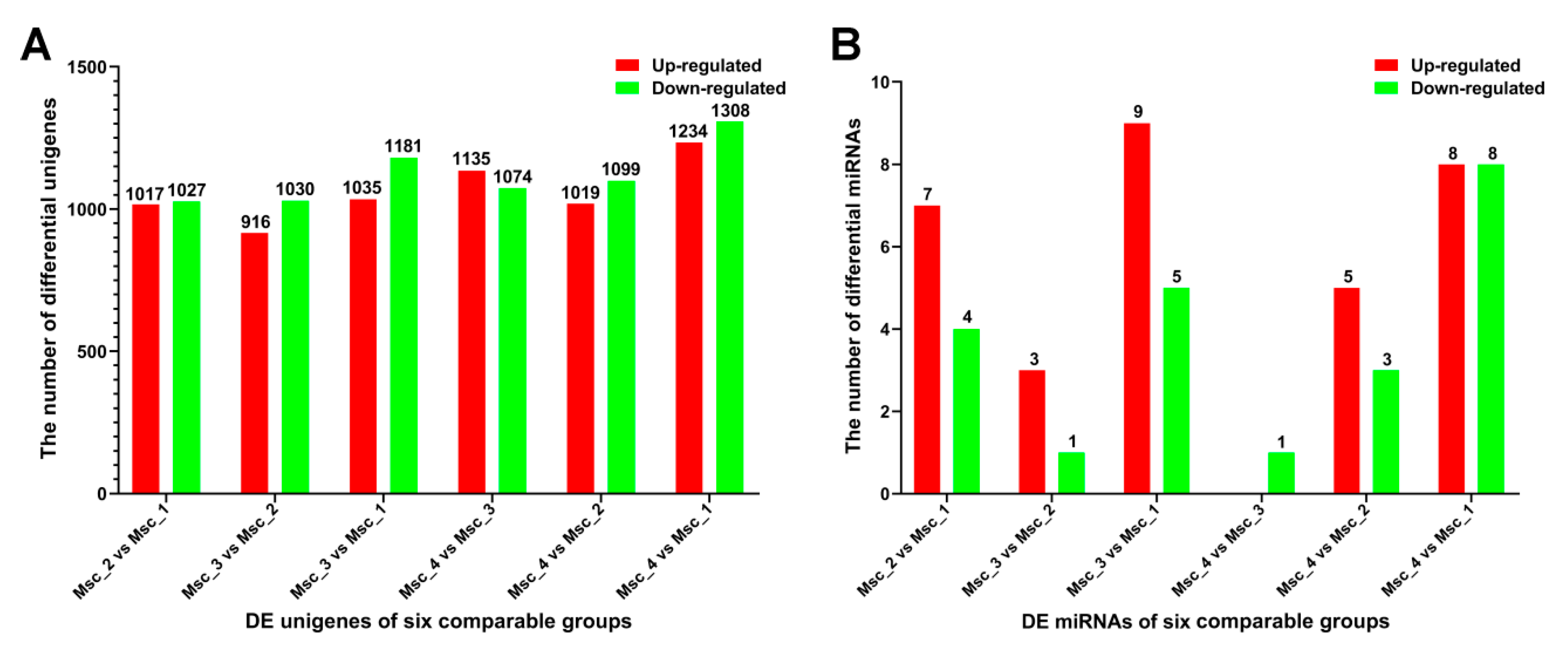
3.2. DE Unigenes, GO and KEGG Analysis
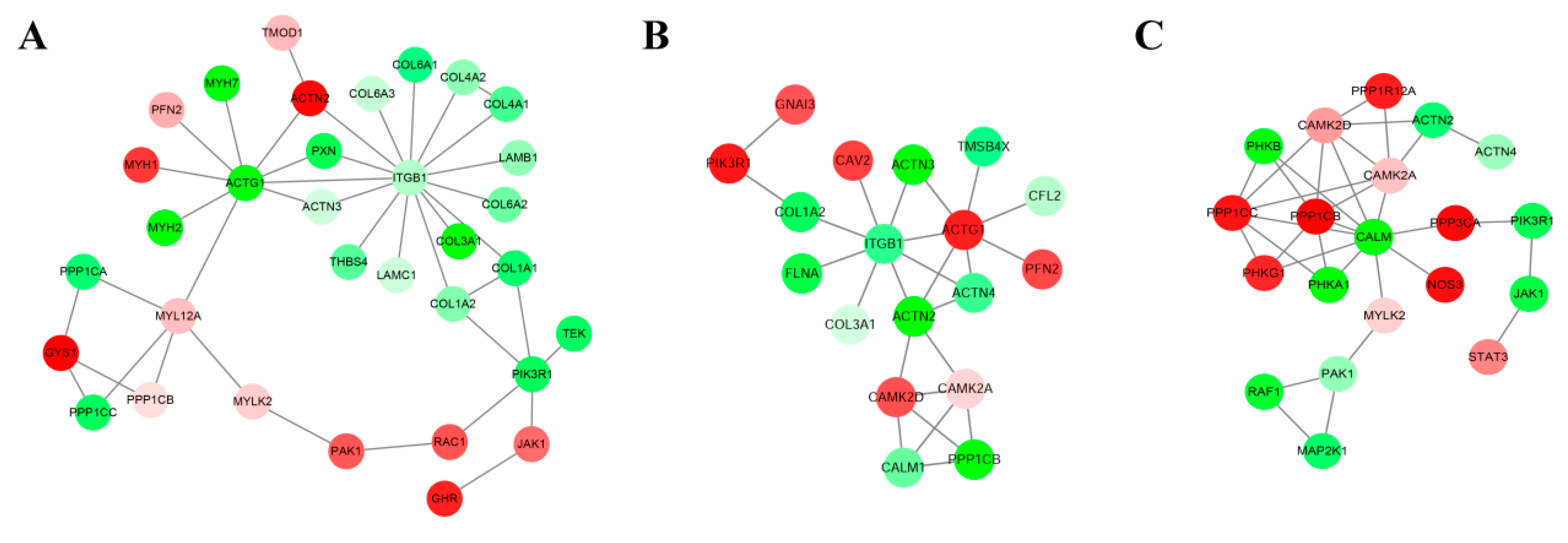
3.3. DE Unigenes Regulatory Network Analysis
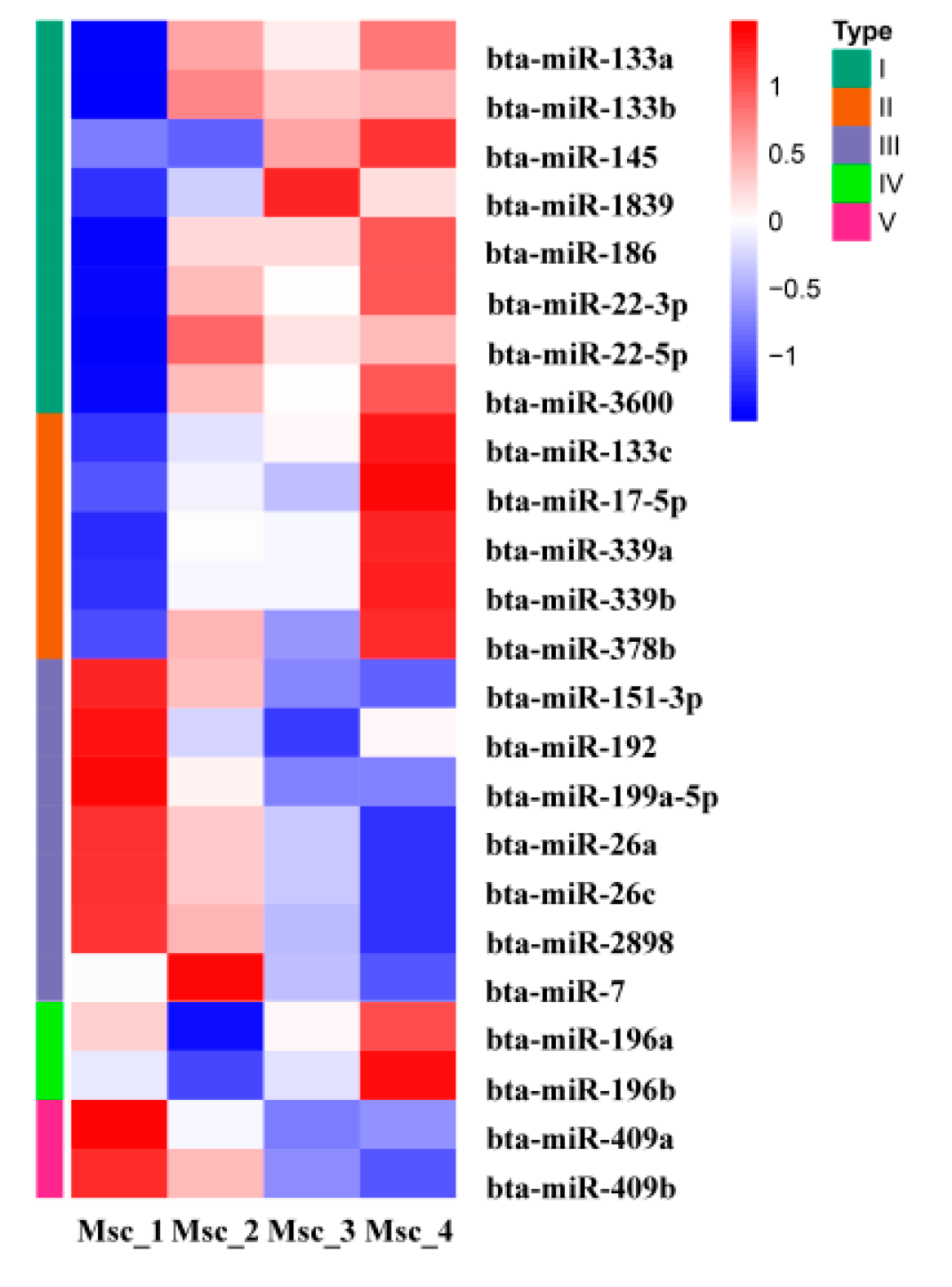
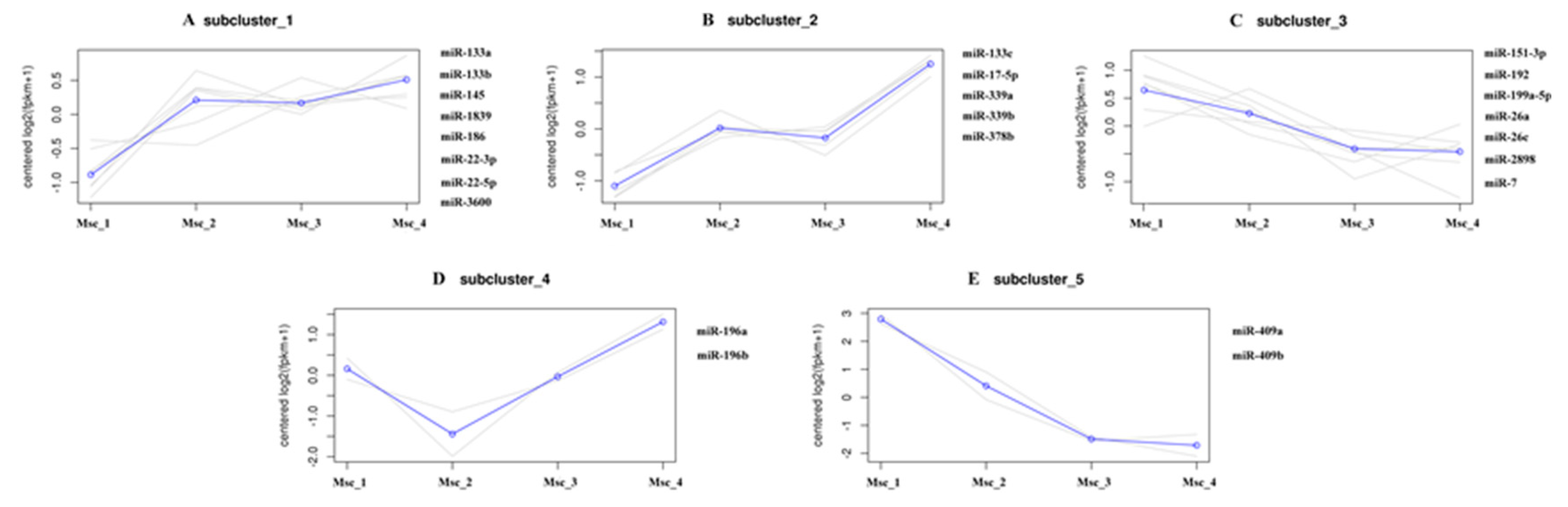
3.4. DE miRNAs, GO, and KEGG Analysis
3.5. miRNA–mRNA Interaction Analysis
3.6. Validation of mRNA-seq and miRNA-seq Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ludwiczak, A.; Stanisz, M.; Bykowska, M.; Skladanowska, J.; Slosarz, P. Effect of storage on quality traits of the semimembranosus muscle of farmed fallow deer (Dama dama) bucks and does. Anim. Sci. J. 2017, 88, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Ju, G.; Zhang, A. Nutritional properties and production situation of venison. J. Econ. Anim. 2014, 18, 228–237. [Google Scholar]
- Triumf, E.C.; Purchas, R.W.; Mielnik, M.; Maehre, H.K.; Elvevoll, E.; Slinde, E.; Egelandsdal, B. Composition and some quality characteristics of the longissimus muscle of reindeer in Norway compared to farmed New Zealand red deer. Meat Sci. 2012, 90, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Sonstegard, T.S.; Li, C.; Huang, Y.; Li, Z.; Lan, X.; Zhang, C.; Lei, C.; Zhao, X.; Chen, H. Altered microRNA expression in bovine skeletal muscle with age. Anim. Genet. 2015, 46, 227–238. [Google Scholar] [CrossRef]
- Mercken, E.M.; Majounie, E.; Ding, J.; Guo, R.; Kim, J.; Bernier, M.; Mattison, J.; Cookson, M.R.; Gorospe, M.; de Cabo, R.; et al. Age-associated miRNA alterations in skeletal muscle from rhesus monkeys reversed by caloric restriction. Aging (Albany NY) 2013, 5, 692–703. [Google Scholar] [CrossRef]
- Goto, M. Inflammaging (inflammation + aging): A driving force for human aging based on an evolutionarily antagonistic pleiotropy theory? Biosci. Trends 2008, 2, 218–230. [Google Scholar]
- Carrington, J.C.; Ambros, V. Role of microRNAs in plant and animal development. Science 2003, 301, 336–338. [Google Scholar] [CrossRef]
- Rajewsky, N. microRNA target predictions in animals. Nat. Genet. 2006, 38, S8–S13. [Google Scholar] [CrossRef]
- Chen, J.F.; Tao, Y.; Li, J.; Deng, Z.; Yan, Z.; Xiao, X.; Wang, D.Z. Microrna-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J. Cell Biol. 2010, 190, 867–879. [Google Scholar] [CrossRef]
- Huang, M.B.; Xu, H.; Xie, S.J.; Zhou, H.; Qu, L.H. Insulin-like growth factor-1 receptor is regulated by microRNA-133 during skeletal myogenesis. PLoS ONE 2011, 6, e29173. [Google Scholar] [CrossRef]
- Chen, X.; Wang, K.; Chen, J.; Guo, J.; Yin, Y.; Cai, X.; Guo, X.; Wang, G.; Yang, R.; Zhu, L.; et al. In vitro evidence suggests that miR-133a-mediated regulation of uncoupling protein 2 (UCP2) is an indispensable step in myogenic differentiation. J. Biol. Chem. 2009, 284, 5362–5369. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; He, M.; Yang, L.; Zhou, R.; Yan, Y.Q.; Liang, Y.; Teng, C.B. MiR-17 and miR-19 cooperatively promote skeletal muscle cell differentiation. Cell Mol. Life Sci. 2019, 76, 5041–5054. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.J.; Xie, J.; Shu, W.J.; Wang, H.Y.; Bi, J.; Jiang, W.; Du, H.N. MiR-15b and miR-322 inhibit SETD3 expression to repress muscle cell differentiation. Cell Death Dis. 2019, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, C.; Cao, X.; Li, H.; Cai, H.; Ma, Y.; Huang, Y.; Lan, X.; Lei, C.; Ma, Y.; et al. MiR-208b regulates cell cycle and promotes skeletal muscle cell proliferation by targeting CDKN1A. J. Cell Physiol. 2019, 234, 3720–3729. [Google Scholar] [CrossRef] [PubMed]
- Mok, G.F.; Lozano-Velasco, E.; Munsterberg, A. MicroRNAs in skeletal muscle development. Semin. Cell Dev. Biol. 2017, 72, 67–76. [Google Scholar] [CrossRef]
- Dey, B.K.; Gagan, J.; Yan, Z.; Dutta, A. MiR-26a is required for skeletal muscle differentiation and regeneration in mice. Genes Dev. 2012, 26, 2180–2191. [Google Scholar] [CrossRef]
- Lin, C.Y.; Chen, J.S.; Loo, M.R.; Hsiao, C.C.; Chang, W.Y.; Tsai, H.J. MicroRNA-3906 regulates fast muscle differentiation through modulating the target gene homer-1b in zebrafish embryos. PLoS ONE 2013, 8, e70187. [Google Scholar] [CrossRef]
- Lu, C.; Wu, J.; Xiong, S.; Zhang, X.; Zhang, J.; Mei, J. MicroRNA-203a regulates fast muscle differentiation by targeting dmrt2a in zebrafish embryos. Gene 2017, 625, 49–54. [Google Scholar] [CrossRef]
- Hu, Z.; Klein, J.D.; Mitch, W.E.; Zhang, L.; Martinez, I.; Wang, X.H. MicroRNA-29 induces cellular senescence in aging muscle through multiple signaling pathways. Aging (Albany NY) 2014, 6, 160–175. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, Y.K.; Lee, K.P.; Lee, S.M.; Kang, T.W.; Kim, H.J.; Dho, S.H.; Kim, S.Y.; Kwon, K.S. Genome-wide profiling of the microRNA-mRNA regulatory network in skeletal muscle with aging. Aging (Albany NY) 2014, 6, 524–544. [Google Scholar] [CrossRef]
- Mitchell, C.J.; D’Souza, R.F.; Schierding, W.; Zeng, N.; Ramzan, F.; O’Sullivan, J.M.; Poppitt, S.D.; Cameron-Smith, D. Identification of human skeletal muscle miRNA related to strength by high-throughput sequencing. Physiol. Genom. 2018, 50, 416–424. [Google Scholar] [CrossRef]
- Drummond, M.J.; McCarthy, J.J.; Sinha, M.; Spratt, H.M.; Volpi, E.; Esser, K.A.; Rasmussen, B.B. Aging and microRNA expression in human skeletal muscle: A microarray and bioinformatics analysis. Physiol. Genom. 2011, 43, 595–603. [Google Scholar] [CrossRef]
- Sun, L.; Lu, S.; Bai, M.; Xiang, L.; Li, J.; Jia, C.; Jiang, H. Integrative microRNA-mRNA Analysis of Muscle Tissues in Qianhua Mutton Merino and Small Tail Han Sheep Reveals Key Roles for oar-miR-655-3p and oar-miR-381-5p. DNA Cell Biol. 2019, 38, 423–435. [Google Scholar] [CrossRef]
- Li, Z.; Abdalla, B.A.; Zheng, M.; He, X.; Cai, B.; Han, P.; Ouyang, H.; Chen, B.; Nie, Q.; Zhang, X. Systematic transcriptome-wide analysis of mRNA-miRNA interactions reveals the involvement of miR-142-5p and its target (FOXO3) in skeletal muscle growth in chickens. Mol. Genet. Genom. 2018, 293, 69–80. [Google Scholar] [CrossRef]
- Zhang, X.; Cai, S.; Chen, L.; Yuan, R.; Nie, Y.; Ding, S.; Fang, Y.; Zhu, Q.; Chen, K.; Wei, H.; et al. Integrated miRNA-mRNA transcriptomic analysis reveals epigenetic-mediated embryonic muscle growth differences between Wuzhishan and Landrace pigs1. J. Anim. Sci. 2019, 97, 1967–1978. [Google Scholar] [CrossRef]
- Jia, B.Y.; Ba, H.X.; Wang, G.W.; Yang, Y.; Cui, X.Z.; Peng, Y.H.; Zheng, J.J.; Xing, X.M.; Yang, F.H. Transcriptome analysis of sika deer in China. Mol. Genet. Genom. 2016, 291, 1941–1953. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Ono, S. Regulation of structure and function of sarcomeric actin filaments in striated muscle of the nematode Caenorhabditis elegans. Anat. Rec. (Hoboken) 2014, 297, 1548–1559. [Google Scholar] [CrossRef]
- Wang, X.; Cao, X.; Dong, D.; Shen, X.; Cheng, J.; Jiang, R.; Yang, Z.; Peng, S.; Huang, Y.; Lan, X.; et al. Circular RNA TTN Acts As a miR-432 Sponge to Facilitate Proliferation and Differentiation of Myoblasts via the IGF2/PI3K/AKT Signaling Pathway. Mol. Ther. Nucleic Acids 2019, 18, 966–980. [Google Scholar] [CrossRef]
- Zheng, L.F.; Chen, P.J.; Xiao, W.H. Signaling pathways controlling skeletal muscle mass. Sheng Li Xue Bao 2019, 71, 671–679. [Google Scholar]
- Ropka-Molik, K.; Bereta, A.; Zukowski, K.; Piorkowska, K.; Gurgul, A.; Zak, G. Transcriptomic gene profiling of porcine muscle tissue depending on histological properties. Anim. Sci. J. 2017, 88, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Hamezah, H.S.; Durani, L.W.; Yanagisawa, D.; Ibrahim, N.F.; Aizat, W.M.; Bellier, J.P.; Makpol, S.; Ngah, W.Z.W.; Damanhuri, H.A.; Tooyama, I. Proteome profiling in the hippocampus, medial prefrontal cortex, and striatum of aging rat. Exp. Gerontol. 2018, 111, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Kanai, S.; Hosoya, H.; Ohta, M.; Miyasaka, K. Decreased hydrogen-potassium-activated ATPase (H+-K+-ATPase) expression and gastric acid secretory capacity in aged mice. Arch. Gerontol. Geriatr. 2007, 45, 243–252. [Google Scholar] [CrossRef]
- Logan, S.; Cameron, J.A.; Vig, P.J. Calmodulin activity in aging rat heart. Biomed. Instrum. 2003, 39, 561–566. [Google Scholar]
- Zhu, L.; Hou, L.; Ou, J.; Xu, G.; Jiang, F.; Hu, C.; Wang, C. MiR-199b represses porcine muscle satellite cells proliferation by targeting JAG1. Gene 2019, 691, 24–33. [Google Scholar] [CrossRef]
- Wang, L.; Chen, X.; Zheng, Y.; Li, F.; Lu, Z.; Chen, C.; Liu, J.; Wang, Y.; Peng, Y.; Shen, Z.; et al. MiR-23a inhibits myogenic differentiation through down regulation of fast myosin heavy chain isoforms. Exp. Cell Res. 2012, 318, 2324–2334. [Google Scholar] [CrossRef]
- Chemello, F.; Grespi, F.; Zulian, A.; Cancellara, P.; Hebert-Chatelain, E.; Martini, P.; Bean, C.; Alessio, E.; Buson, L.; Bazzega, M.; et al. Transcriptomic Analysis of Single Isolated Myofibers Identifies miR-27a-3p and miR-142-3p as Regulators of Metabolism in Skeletal Muscle. Cell Rep. 2019, 26, 3784–3797. [Google Scholar] [CrossRef]
- Lena, A.M.; Mancini, M.; Rivetti di Val Cervo, P.; Saintigny, G.; Mahe, C.; Melino, G.; Candi, E. MicroRNA-191 triggers keratinocytes senescence by SATB1 and CDK6 downregulation. Biochem. Biophys. Res. Commun. 2012, 423, 509–514. [Google Scholar] [CrossRef]
- Boon, R.A.; Seeger, T.; Heydt, S.; Fischer, A.; Hergenreider, E.; Horrevoets, A.J.; Vinciguerra, M.; Rosenthal, N.; Sciacca, S.; Pilato, M.; et al. MicroRNA-29 in aortic dilation: Implications for aneurysm formation. Circ. Res. 2011, 109, 1115–1119. [Google Scholar] [CrossRef]
- Sun, J.; Li, M.; Li, Z.; Xue, J.; Lan, X.; Zhang, C.; Lei, C.; Chen, H. Identification and profiling of conserved and novel microRNAs from Chinese Qinchuan bovine longissimus thoracis. BMC Genom. 2013, 14, 42. [Google Scholar] [CrossRef]
- Sheng, X.; Song, X.; Yu, Y.; Niu, L.; Li, S.; Li, H.; Wei, C.; Liu, T.; Zhang, L.; Du, L. Characterization of microRNAs from sheep (Ovis aries) using computational and experimental analyses. Mol. Biol. Rep. 2011, 38, 3161–3171. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S. Muscle fiber type diversity revealed by anti-myosin heavy chain antibodies. FEBS J. 2018, 285, 3688–3694. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.S.; Kim, D.H.; Park, H.B.; Han, S.H.; Hwang, S.; Cho, I.C.; Lee, J.W. Ectopic Overexpression of Porcine Myh1 Increased in Slow Muscle Fibers and Enhanced Endurance Exercise in Transgenic Mice. Int. J. Mol. Sci. 2018, 19, 2959. [Google Scholar] [CrossRef] [PubMed]
- Dugdale, H.F.; Hughes, D.C.; Allan, R.; Deane, C.S.; Coxon, C.R.; Morton, J.P.; Stewart, C.E.; Sharples, A.P. The role of resveratrol on skeletal muscle cell differentiation and myotube hypertrophy during glucose restriction. Mol. Cell Biochem. 2018, 444, 109–123. [Google Scholar] [CrossRef]
- Meredith, C.; Herrmann, R.; Parry, C.; Liyanage, K.; Dye, D.E.; Durling, H.J.; Duff, R.M.; Beckman, K.; de Visser, M.; van der Graaff, M.M.; et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1). Am. J. Hum. Genet. 2004, 75, 703–708. [Google Scholar] [CrossRef]
- Chen, R.; Jiang, T.; Lei, S.; She, Y.; Shi, H.; Zhou, S.; Ou, J.; Liu, Y. Expression of circular RNAs during C2C12 myoblast differentiation and prediction of coding potential based on the number of open reading frames and N6-methyladenosine motifs. Cell Cycle 2018, 17, 1832–1845. [Google Scholar] [CrossRef]
- Burguiere, A.C.; Nord, H.; von Hofsten, J. Alkali-like myosin light chain-1 (myl1) is an early marker for differentiating fast muscle cells in zebrafish. Dev. Dyn. 2011, 240, 1856–1863. [Google Scholar] [CrossRef]
- Ravenscroft, G.; Zaharieva, I.T.; Bortolotti, C.A.; Lambrughi, M.; Pignataro, M.; Borsari, M.; Sewry, C.A.; Phadke, R.; Haliloglu, G.; Ong, R.; et al. Bi-allelic mutations in MYL1 cause a severe congenital myopathy. Hum. Mol. Genet. 2018, 27, 4263–4272. [Google Scholar] [CrossRef]
- Nawrotzki, R.; Fischman, D.A.; Mikawa, T. Antisense suppression of skeletal muscle myosin light chain-1 biosynthesis impairs myofibrillogenesis in cultured myotubes. J. Muscle Res. Cell Motil. 1995, 16, 45–56. [Google Scholar] [CrossRef]
- Cheung, T.H.; Quach, N.L.; Charville, G.W.; Liu, L.; Park, L.; Edalati, A.; Yoo, B.; Hoang, P.; Rando, T.A. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature 2012, 482, 524–528. [Google Scholar] [CrossRef]
- Kremneva, E.; Makkonen, M.H.; Skwarek-Maruszewska, A.; Gateva, G.; Michelot, A.; Dominguez, R.; Lappalainen, P. Cofilin-2 controls actin filament length in muscle sarcomeres. Dev. Cell. 2014, 31, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Yang, H.; Zhao, S.; Zhang, J.; Liu, D.; Tian, Y.; Shen, Z.; Su, Y. Role of the cofilin 2 gene in regulating the myosin heavy chain genes in mouse myoblast C2C12 cells. Int. J. Mol. Med. 2018, 41, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Del Coso, J.; Valero, M.; Salinero, J.J.; Lara, B.; Diaz, G.; Gallo-Salazar, C.; Ruiz-Vicente, D.; Areces, F.; Puente, C.; Carril, J.C.; et al. ACTN3 genotype influences exercise-induced muscle damage during a marathon competition. Eur. J. Appl. Physiol. 2017, 117, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Musial, A.D.; Ropka-Molik, K.; Piorkowska, K.; Jaworska, J.; Stefaniuk-Szmukier, M. ACTN3 genotype distribution across horses representing different utility types and breeds. Mol. Biol. Rep. 2019, 46, 5795–5803. [Google Scholar] [CrossRef] [PubMed]
- Garton, F.C.; North, K.N. The Effect of Heterozygosity for the ACTN3 Null Allele on Human Muscle Performance. Med. Sci. Sports Exerc. 2016, 48, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Staff, P.G. Correction: Altered Ca2+ Kinetics Associated with alpha-Actinin-3 Deficiency May Explain Positive Selection for ACTN3 Null Allele in Human Evolution. PLoS Genet. 2015, 11, e1005312. [Google Scholar]
- Head, S.I.; Chan, S.; Houweling, P.J.; Quinlan, K.G.; Murphy, R.; Wagner, S.; Friedrich, O.; North, K.N. Altered Ca2+ kinetics associated with alpha-actinin-3 deficiency may explain positive selection for ACTN3 null allele in human evolution. PLoS Genet. 2015, 11, e1004862. [Google Scholar] [CrossRef]
- Lightfoot, A.P.; Cooper, R.G. The role of myokines in muscle health and disease. Curr. Opin. Rheumatol. 2016, 28, 661–666. [Google Scholar] [CrossRef]
- Lai, J.; Chen, F.; Chen, J.; Ruan, G.; He, M.; Chen, C.; Tang, J.; Wang, D.W. Overexpression of decorin promoted angiogenesis in diabetic cardiomyopathy via IGF1R-AKT-VEGF signaling. Sci.Rep. 2017, 7, 44473. [Google Scholar] [CrossRef]
- Murata, K.; Morino, K.; Ida, S.; Ohashi, N.; Lemecha, M.; Park, S.Y.; Ishikado, A.; Kume, S.; Choi, C.S.; Sekine, O.; et al. Lack of O-GlcNAcylation enhances exercise-dependent glucose utilization potentially through AMP-activated protein kinase activation in skeletal muscle. Biochem. Biophys. Res. Commun. 2018, 495, 2098–2104. [Google Scholar] [CrossRef]
- Bullen, J.W.; Balsbaugh, J.L.; Chanda, D.; Shabanowitz, J.; Hunt, D.F.; Neumann, D.; Hart, G.W. Cross-talk between two essential nutrient-sensitive enzymes: O-GlcNAc transferase (OGT) and AMP-activated protein kinase (AMPK). J. Biol. Chem. 2014, 289, 10592–10606. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Han, W.; Li, C.; Li, H.; Zhu, D.; Zhang, Y.; Liu, X. miR-378 attenuates muscle regeneration by delaying satellite cell activation and differentiation in mice. Acta Biochim. Biophys. Sin. (Shanghai) 2016, 48, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.C.; Liu, Z.; Dillin, A.; Hunter, T. A conserved ubiquitination pathway determines longevity in response to diet restriction. Nature 2009, 460, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Bellier, A.; Kao, C.Y.; Yang, Y.L.; Chen, H.D.; Los, F.C.; Aroian, R.V. WWP-1 is a novel modulator of the DAF-2 insulin-like signaling network involved in pore-forming toxin cellular defenses in Caenorhabditis elegans. PLoS ONE 2010, 5, e9494. [Google Scholar] [CrossRef]
- Cao, X.; Xue, L.; Han, L.; Ma, L.; Chen, T.; Tong, T. WW domain-containing E3 ubiquitin protein ligase 1 (WWP1) delays cellular senescence by promoting p27(Kip1) degradation in human diploid fibroblasts. J. Biol. Chem. 2011, 286, 33447–33456. [Google Scholar] [CrossRef]
- Hao, Q.; Zhang, Q.; Li, C.; Wei, S.; Li, Q.; Song, Y.; Mi, Y. A novel variant translocation (1;9)(p22;q34) resulting in a DEK/NUP214 fusion gene in a patient with acute myeloid leukemia: A case report. Oncol. Lett. 2017, 14, 7021–7024. [Google Scholar] [CrossRef]
- Wise-Draper, T.M.; Allen, H.V.; Thobe, M.N.; Jones, E.E.; Habash, K.B.; Munger, K.; Wells, S.I. The human DEK proto-oncogene is a senescence inhibitor and an upregulated target of high-risk human papillomavirus E7. J. Virol. 2005, 79, 14309–14317. [Google Scholar] [CrossRef][Green Version]
- Wise-Draper, T.M.; Morreale, R.J.; Morris, T.A.; Mintz-Cole, R.A.; Hoskins, E.E.; Balsitis, S.J.; Husseinzadeh, N.; Witte, D.P.; Wikenheiser-Brokamp, K.A.; Lambert, P.F.; et al. DEK proto-oncogene expression interferes with the normal epithelial differentiation program. Am. J. Pathol. 2009, 174, 71–81. [Google Scholar] [CrossRef]
- Wisniewski, T.; Newman, K.; Javitt, N.B. Alzheimer’s disease: Brain desmosterol levels. J. Alzheimers Dis. 2013, 33, 881–888. [Google Scholar] [CrossRef]
- Chesnokova, V.; Zonis, S.; Ben-Shlomo, A.; Wawrowsky, K.; Melmed, S. Molecular mechanisms of pituitary adenoma senescence. Front. Horm. Res. 2010, 38, 7–14. [Google Scholar]
- Liu, T.; Krysiak, K.; Shirai, C.L.; Kim, S.; Shao, J.; Ndonwi, M.; Walter, M.J. Knockdown of HSPA9 induces TP53-dependent apoptosis in human hematopoietic progenitor cells. PLoS ONE 2017, 12, e0170470. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Myers, L.; Ravussin, E.; Cherry, K.E.; Jazwinski, S.M. Single nucleotide polymorphisms linked to mitochondrial uncoupling protein genes UCP2 and UCP3 affect mitochondrial metabolism and healthy aging in female nonagenarians. Biogerontology 2016, 17, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Dato, S.; Soerensen, M.; Montesanto, A.; Lagani, V.; Passarino, G.; Christensen, K.; Christiansen, L. UCP3 polymorphisms, hand grip performance and survival at old age: Association analysis in two Danish middle aged and elderly cohorts. Mech. Ageing. Dev. 2012, 133, 530–537. [Google Scholar] [CrossRef]
- Rose, G.; Crocco, P.; De Rango, F.; Montesanto, A.; Passarino, G. Further support to the uncoupling-to-survive theory: The genetic variation of human UCP genes is associated with longevity. PLoS ONE 2011, 6, e29650. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Nguyen, K.C.; Nguyen, C.T.; Jang, I.S.; Han, J.M.; Fabian, C.; Lee, S.E.; Rhee, J.H.; Cho, K.A. Flagellin-dependent TLR5/caveolin-1 as a promising immune activator in immunosenescence. Aging Cell 2015, 14, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Khurts, S.; Masutomi, K.; Delgermaa, L.; Arai, K.; Oishi, N.; Mizuno, H.; Hayashi, N.; Hahn, W.C.; Murakami, S. Nucleolin interacts with telomerase. J. Biol. Chem. 2004, 279, 51508–51515. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, Z.; Tong, T. Cloning and characterization of cellular senescence-associated genes in human fibroblasts by suppression subtractive hybridization. Exp. Cell Res. 2004, 298, 465–472. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data | Molecules in Networks | Score | P-Value | Top Functions |
|---|---|---|---|---|
| Msc_2vs.Msc_1 | COL4A2, COL4A1, COL3A1, ITGB1, COL6A3, TEK, RAC1, COL1A2, COL6A2, GYS1, JAK1, COL6A1, COL1A1, LAMC1, LAMB1, PIK3R1, GHR, THBS4 | 33 | 0.001 | PI3K-Akt signaling pathway |
| MYLK2, ACTN2, MYL12A, ACTN3, PPP1CC, ITGB1, PPP1CB, PXN, ACTG1, PPP1CA, PFN2, RAC1, PAK1, PIK3R1 | 0.016 | Regulation of actin cytoskeleton | ||
| PFN2, MYH1, MYH2, MYH7, ACTN2, ACTN3, TMOD1 | 0.001 | actin binding | ||
| Msc_3vs.Msc_2 | ACTG1, PFN2, ACTN4, CFL2, TMSB4X, ACTN2, ACTN3, ITGB1, PPP1CB, PIK3R1 | 18 | 0.034 | Regulation of actin cytoskeleton |
| GNAI3, CAMK2D, PPP1CB, CAMK2A, PIK3R1, CALM1 | 0.050 | cAMP signaling pathway | ||
| CAV2, ACTN4, COL3A1, ACTN2, ACTN3, ITGB1, PPP1CB, FLNA, ACTG1, COL1A2, PIK3R1 | 0.005 | Focal adhesion | ||
| Msc_4vs.Msc_3 | ACTN4, ACTN2, STAT3, JAK1, PIK3R1 | 20 | 0.005 | Viral carcinogenesis |
| MAP2K1, RAF1, PPP1CC, PPP1CB, CALM, CAMK2D, PPP1R12A, PAK1, CAMK2A, PIK3R1 | 0.005 | cAMP signaling pathway | ||
| CALM, PHKG1, PHKB, PHKA1, CAMK2D, MYLK2, NOS3, PPP3CA, CAMK2A | 0.050 | Calcium signaling pathway |
| miRNA ID | Muscle Development | miRNA ID | Muscle Aging |
|---|---|---|---|
| miR-17-5p | skeletal muscle proliferation and differentiation | miR-7 | ageing-related inflammation |
| miR-22-3p | skeletal muscle proliferation and differentiation | miR-17-5p | restored the osteogenic capacity of old mesenchymal stem cells and aging-associated pathologies |
| miR-26a | skeletal muscle differentiation and regeneration | miR-196b | regulating the muscle protein synthesis pathway |
| miR-133a | skeletal muscle proliferation, differentiation, and hypertrophy | miR-199a-5p | regulating the MPS pathway |
| miR-133b | miR-378b | myogenic differentiation of myogenic progenitors from adult and old individual | |
| miR-133c | |||
| miR-151-3p | skeletal muscle proliferation and differentiation | ||
| miR-186 | skeletal muscle differentiation | ||
| miR-192 | skeletal muscle proliferation and differentiation | ||
| miR-199a-5p | skeletal muscle proliferation and differentiation | ||
| miR-378b | skeletal muscle autophagy and apoptosis |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, B.; Liu, Y.; Li, Q.; Zhang, J.; Ge, C.; Wang, G.; Chen, G.; Liu, D.; Yang, F. Altered miRNA and mRNA Expression in Sika Deer Skeletal Muscle with Age. Genes 2020, 11, 172. https://doi.org/10.3390/genes11020172
Jia B, Liu Y, Li Q, Zhang J, Ge C, Wang G, Chen G, Liu D, Yang F. Altered miRNA and mRNA Expression in Sika Deer Skeletal Muscle with Age. Genes. 2020; 11(2):172. https://doi.org/10.3390/genes11020172
Chicago/Turabian StyleJia, Boyin, Yuan Liu, Qining Li, Jiali Zhang, Chenxia Ge, Guiwu Wang, Guang Chen, Dongdong Liu, and Fuhe Yang. 2020. "Altered miRNA and mRNA Expression in Sika Deer Skeletal Muscle with Age" Genes 11, no. 2: 172. https://doi.org/10.3390/genes11020172
APA StyleJia, B., Liu, Y., Li, Q., Zhang, J., Ge, C., Wang, G., Chen, G., Liu, D., & Yang, F. (2020). Altered miRNA and mRNA Expression in Sika Deer Skeletal Muscle with Age. Genes, 11(2), 172. https://doi.org/10.3390/genes11020172