Molecular-Assisted Distinctness and Uniformity Testing Using SLAF-Sequencing Approach in Soybean

 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction, SLAF Library Construction and Sequencing
2.3. SSR Detection
2.4. SLAF-seq Data Grouping and Genotyping
2.5. Data Analysis
2.6. Assessment of Purity
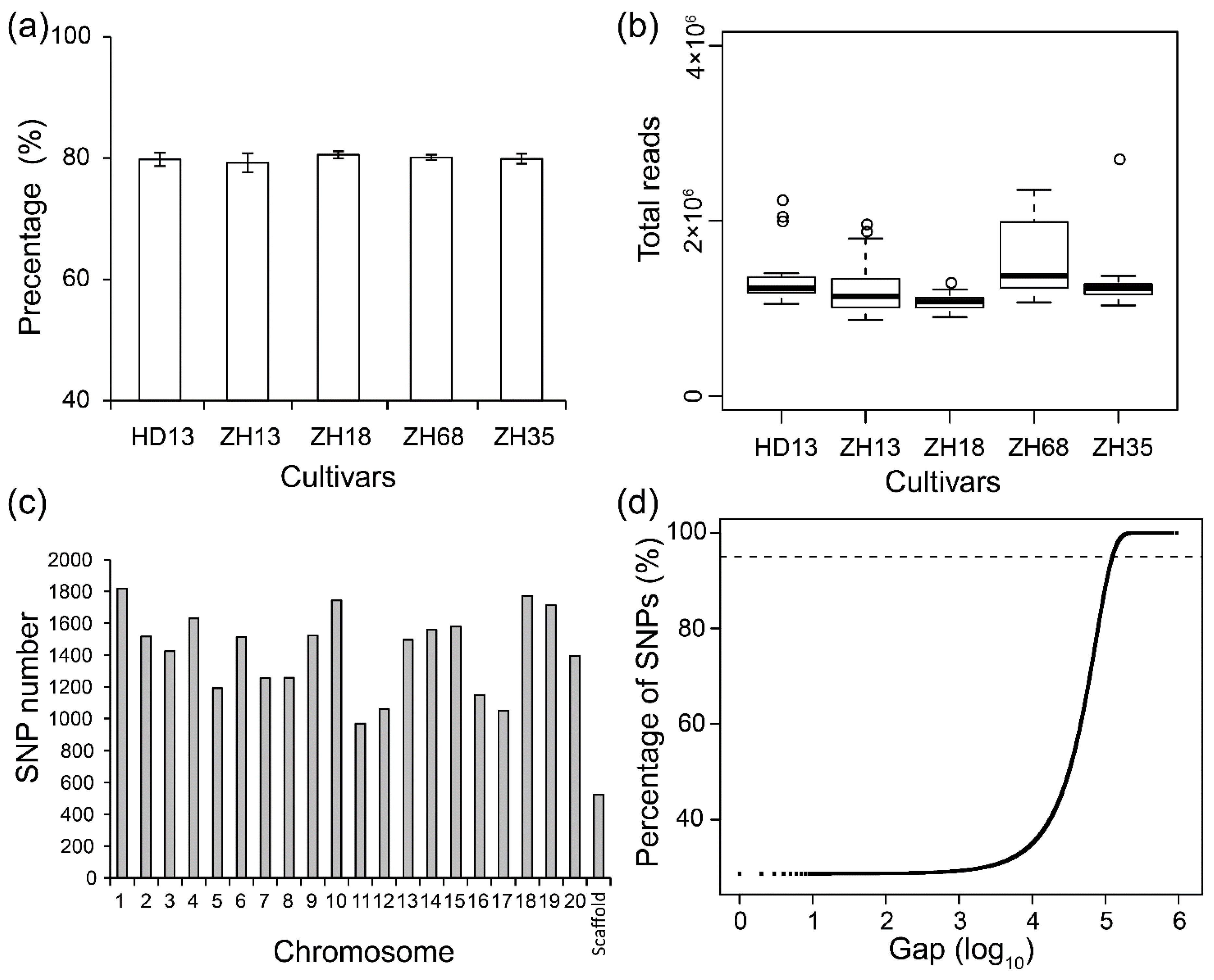
3. Results
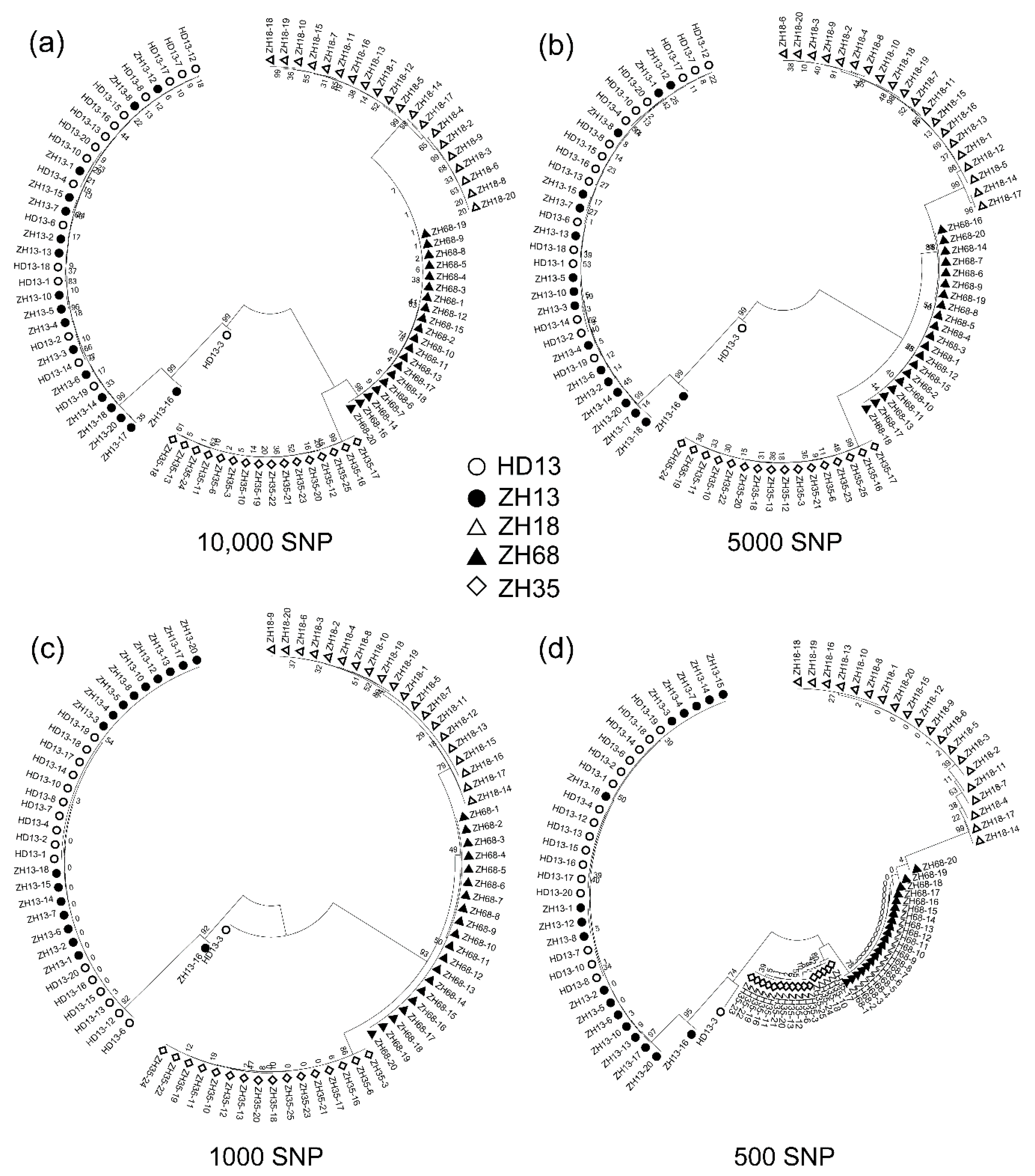
3.1. Assessment of Distinctness and Uniformity of Soybean Cultivars Based on SLAF-seq
3.2. Assessment of Distinctness and Uniformity of Soybean Cultivars based on SSR Markers
3.3. The SNP Number Affects Efficiency of Assessment of Distinctness and Uniformity of Cultivars
3.4. Assessment of Genetic Purity for Soybean Cultivars Using SLAF-seq
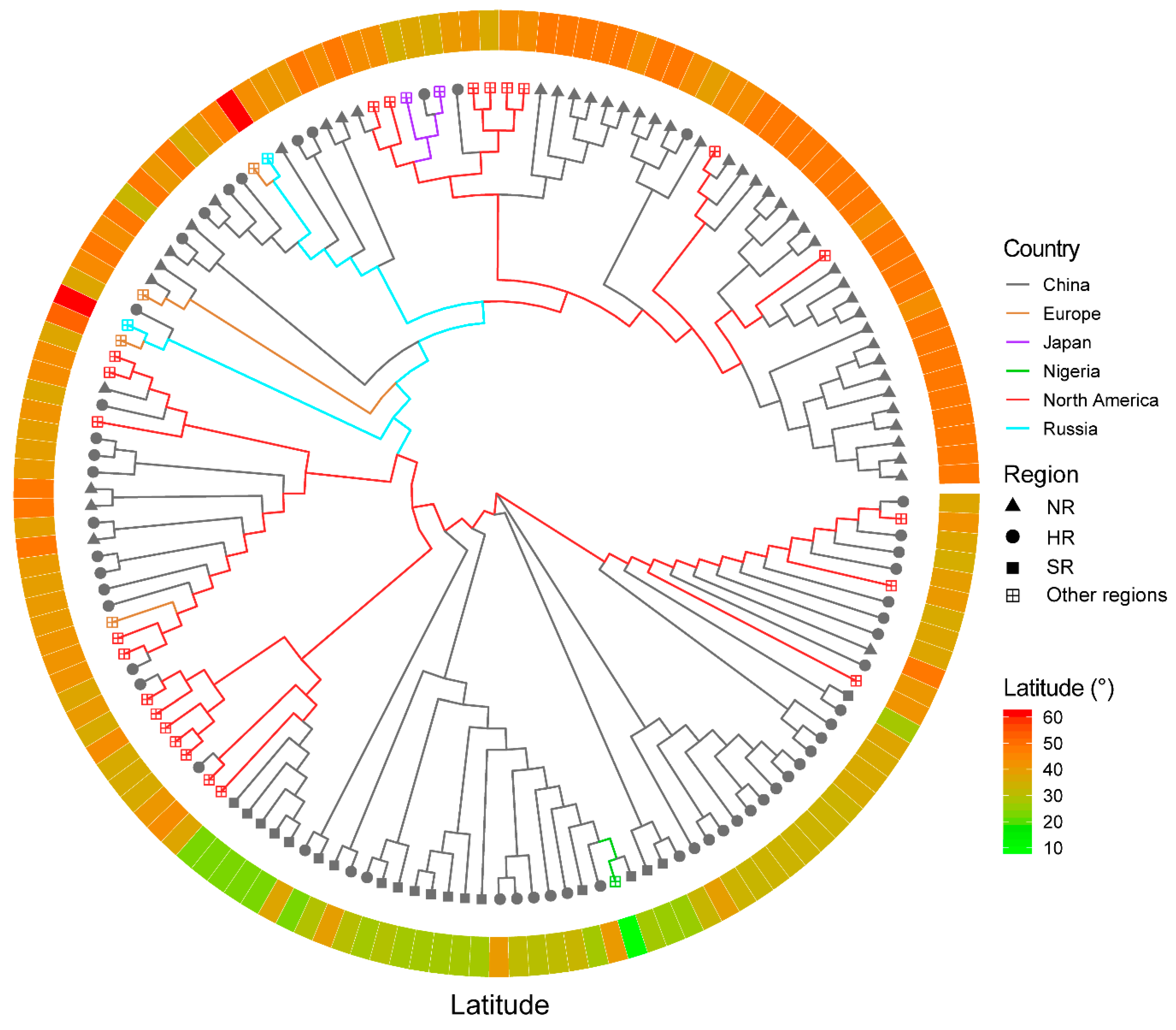
3.5. The Application of SLAF-seq Technology for the Assessment of the Distinctness in 150 Soybean Cultivars
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Specific-locus amplified fragment | SLAF |
| distinctness, uniformity and stability | DUS |
| principal component analysis | PCA |
| International Union for the Protection of New Varieties of Plants | UPOV |
| fragment length polymorphisms | RFLP |
| random amplified polymorphic DNA | RAPD |
| simple sequence repeat | SSR |
| inter-simple sequence repeat | ISSR |
| single nucleotide polymorphism | SNP |
| amplified fragment length polymorphisms | AFLP |
| reduced representation library | RRL |
References
- Sohn, H.B.; Kim, S.J.; Hwang, T.Y.; Park, H.M.; Lee, Y.Y.; Markkandan, K.; Lee, D.; Lee, S.; Hong, S.Y.; Song, Y.H.; et al. Barcode system for genetic identification of soybean [Glycine max (L.) Merrill] cultivars using inDel markers specific to dense variation blocks. Front. Plant Sci. 2017, 8, 520–531. [Google Scholar] [CrossRef]
- Song, Q.; Quigley, C.; Nelson, R.; Carter, T.; Boerma, H.; Strachan, J.; Cregan, P. A selected set of trinucleotide simple sequence repeat markers for soybean cultivar identification. Plant Var. Seeds 1999, 12, 207–220. [Google Scholar]
- Bonow, S.; Von Pinho, E.V.; Vieira, M.G.; Vosman, B. Microsatellite markers in and around rice genes: applications in variety identification and DUS testing. Crop Sci. 2009, 49, 880–886. [Google Scholar] [CrossRef]
- Tian, H.; Wang, F.; Zhao, J.; Yi, H.; Wang, L.; Wang, R.; Yang, Y.; Song, W. Development of maizeSNP3072, a high-throughput compatible SNP array, for DNA fingerprinting identification of Chinese maize varieties. Mol. Breed. 2015, 35, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Tommasini, L.; Batley, J.; Arnold, G.; Cooke, R.; Donini, P.; Lee, D.; Law, J.; Lowe, C.; Moule, C.; Trick, M.; et al. The development of multiplex simple sequence repeat (SSR) markers to complement distinctness, uniformity and stability testing of rape (Brassica napus L.) varieties. Theor. Appl. Genet. 2003, 106, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Jamali, S.H.; Cockram, J.; Hickey, L.T. Insights into deployment of DNA markers in plant variety protection and registration. Theor. Appl. Genet. 2019, 132, 1911–1929. [Google Scholar] [CrossRef] [PubMed]
- Jördens, R. Progress of plant variety protection based on the International Convention for the Protection of New Varieties of Plants (UPOV Convention). World Pat. Inf. 2005, 27, 232–243. [Google Scholar] [CrossRef]
- Morell, M.; Peakall, R.; Appels, R.; Preston, L.; Lloyd, H. DNA profiling techniques for plant variety identification. Aust. J. Exp. Agric. 1995, 35, 807–819. [Google Scholar] [CrossRef]
- Choudhury, P.R.; Kohli, S.; Srinivasan, K.; Mohapatra, T.; Sharma, R.P. Identification and classification of aromatic rices based on DNA fingerprinting. Euphytica 2001, 118, 243–251. [Google Scholar] [CrossRef]
- Geleta, N.; Labuschagne, M.T.; Viljoen, C.D. Genetic diversity analysis in sorghum germplasm as estimated by AFLP, SSR and morpho-agronomical markers. Biodivers. Conserv. 2006, 15, 3251–3265. [Google Scholar] [CrossRef]
- Congiu, L.; Chicca, M.; Cella, R.; Rossi, R.; Bernacchia, G. The use of random amplified polymorphic DNA (RAPD) markers to identify strawberry varieties: a forensic application. Mol. Ecol. 2000, 9, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.; Lorenc, M.T.; Edwards, D. Molecular marker databases. In Plant Genotyping: Methods and Protocols; Batley, J., Ed.; Springer New York: New York, NY, USA, 2015; pp. 49–62. [Google Scholar] [CrossRef]
- Tanksley, S.D.; Young, N.D.; Paterson, A.H.; Bonierbale, M.W. RFLP mapping in plant breeding: new tools for an old science. Bio/Technology 1989, 7, 257–264. [Google Scholar] [CrossRef]
- Welsh, J.; McClelland, M. Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res. 1990, 18, 7213–7218. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.G.K.; Kubelik, A.R.; Livak, K.J.; Rafalski, J.A.; Tingey, S.V. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990, 18, 6531–6535. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Milligan, B.G. Analysis of population genetic structure with RAPD markers. Mol. Ecol. 1994, 3, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, A.J. Hypervariable minisatellite regions in human DNA. Nature 1985, 314, 67–73. [Google Scholar] [CrossRef]
- Akkaya, M.S.; Bhagwat, A.A.; Cregan, P.B. Length polymorphisms of simple sequence repeat DNA in soybean. Genetics 1992, 132, 1131–1139. [Google Scholar]
- Pradeep Reddy, M.; Sarla, N.; Siddiq, E.A. Inter simple sequence repeat (ISSR) polymorphism and its application in plant breeding. Euphytica 2002, 128, 9–17. [Google Scholar] [CrossRef]
- Jorde, L.B. Linkage disequilibrium and the search for complex disease genes. Genome Res. 2000, 10, 1435–1444. [Google Scholar] [CrossRef]
- Goldstein, D.B. Islands of linkage disequilibrium. Nat. Genet. 2001, 29, 109–111. [Google Scholar] [CrossRef]
- Sachidanandam, R.; Weissman, D.; Schmidt, S.C.; Kakol, J.M.; Stein, L.D.; Marth, G.; Sherry, S.; Mullikin, J.C.; Mortimore, B.J.; Willey, D.L.; et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 2001, 409, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 2011, 6, e19379. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.; Heger, A.; Ponting, C. Sequencing depth and coverage: key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tian, L.; Zhang, J.; Huang, L.; Han, F.; Yan, S.; Wang, L.; Zheng, H.; Sun, J. Construction of a high-density genetic map based on large-scale markers developed by specific length amplified fragment sequencing (SLAF-seq) and its application to QTL analysis for isoflavone content in Glycine max. BMC Genom. 2014, 15, 1086–1101. [Google Scholar] [CrossRef]
- Han, Y.; Zhao, X.; Cao, G.; Wang, Y.; Li, Y.; Liu, D.; Teng, W.; Zhang, Z.; Li, D.; Qiu, L.; et al. Genetic characteristics of soybean resistance to HG type 0 and HG type 1.2.3.5.7 of the cyst nematode analyzed by genome-wide association mapping. BMC Genom. 2015, 16, 598–608. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, G.; Chen, B.; Du, H.; Zhang, F.; Zhang, H.; Wang, Q.; Geng, S. Candidate genes for first flower node identified in pepper using combined SLAF-seq and BSA. PLoS ONE 2018, 13, e0194071. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, H.; Wang, Z.; Ren, Y.; Niu, L.; Liu, J.; Liu, B. Photoperiodism dynamics during the domestication and improvement of soybean. Sci. China Life Sci. 2017, 60, 1416–1427. [Google Scholar] [CrossRef]
- International Union for the Protection of New Varieties of Plants. Guidance on the Use of Biochemical and Molecular Markers in the Examination of Distinctness, Uniformity and Stability (DUS). Available online: http://www.upov.int/edocs/tgpdocs/en/tgp_15.pdf (accessed on 1 November 2019).
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: an efficient method of large-scale De Novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef]
- Guan, R.; Fang, H.; He, Y.; Chang, R.; Qiu, L. Molecular homozygosity of soybean varieties (lines) in regional test of China by using SSR markers. Acta Agron. Sin. 2012, 38, 1760–1765. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics (Oxford, England) 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: molecular evolutionary genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.; Zhu, H.; Guan, Y.; Lam, T. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Yu, G.; Lam, T.; Zhu, H.; Guan, Y. Two methods for mapping and visualizing associated data on phylogeny using ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef]
- Lipka, A.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.; Gore, M.; Buckler, E.; Zhang, Z. GAPIT: genome association and prediction integrated tool. Bioinformatics (Oxford, England) 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Jia, J.; Kong, X. A SNP-based molecular barcode for characterization of common wheat. PloS ONE 2016, 11, e0150947. [Google Scholar] [CrossRef] [PubMed]
- Cockram, J.; Jones, H.; Norris, C.; O’Sullivan, D. Evaluation of diagnostic molecular markers for DUS phenotypic assessment in the cereal crop, barley (Hordeum vulgare ssp. vulgare L.). Theor. Appl. Genet. 2012, 125, 1735–1749. [Google Scholar] [CrossRef] [PubMed]
- Annicchiarico, P.; Nazzicari, N.; Ananta, A.; Carelli, M.; Wei, Y.; Brummer, C. Assessment of cultivar distinctness in Alfalfa: A comparison of genotyping-by-sequencing, simple-sequence repeat marker, and morphophysiological observations. Plant Genome 2016, 9, 1–12. [Google Scholar] [CrossRef]
- Zhao, X.; Teng, W.; Li, Y.; Liu, D.; Cao, G.; Li, D.; Qiu, L.; Zheng, H.; Han, Y.; Li, W. Loci and candidate genes conferring resistance to soybean cyst nematode HG type 2.5.7. BMC Genom. 2017, 18, 462. [Google Scholar] [CrossRef]
- Bomblies, K.; Yant, L.; Laitinen, R.A.; Kim, S.-T.; Hollister, J.D.; Warthmann, N.; Fitz, J.; Weigel, D. Local-scale patterns of genetic variability, outcrossing, and spatial structure in natural stands of Arabidopsis thaliana. PLoS Gemet. 2010, 6, e1000890. [Google Scholar] [CrossRef]
- Li, Y.H.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.G.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cultivar | Breeder | Release Year | Parents | Features |
|---|---|---|---|---|
| Zhonghuang 13 | ICS, CAAS 1 | 2001 | Yudou8 × Zhongzuo90052-76 | High yield, wide adaptability, disease resistance, lodging resistance, high protein |
| Zhonghuang 35 | ICS, CAAS 1 | 2007 | Zhongzuo9943 × Zheng76062 | High yield, lodging resistance, high oil |
| Zhonghuang 18 | ICS, CAAS 1 | 2001 | Zhongpin661 × Century-2 | High yield, disease resistance, lodging resistance, high protein |
| Zhonghuang 68 | ICS, CAAS 1 | 2013 | Zhonghuang18 × Karikoi-434 | High yield, disease resistance, no beany flavour |
| Hedou 13 | HAAS 2 | 2005 | Yudou8 × He95-1 | High yield, wide adaptability, disease resistance, lodging resistance, high protein |
| ZH13 | HD13 | ZH18 | ZH68 | ZH35 | ZH13/HD13 1 | ZH13-16/ZH13 2 | HD13-3/HD13 3 | |
|---|---|---|---|---|---|---|---|---|
| Maximum | 0.036 | 0.028 | 0.052 | 0.011 | 0.014 | 0.035 | 0.140 | 0.233 |
| Minimum | 0.010 | 0.006 | 0.009 | 0.002 | 0.006 | 0.006 | 0.123 | 0.221 |
| Average | 0.020 | 0.019 | 0.024 | 0.007 | 0.011 | 0.021 | 0.130 | 0.229 |
| Cultivar | Purity (%) | Min (%) | Max (%) | Std. * (%) |
|---|---|---|---|---|
| HD13 | 92.44 | 91.32 | 94.58 | 0.96 |
| ZH13 | 92.55 | 91.24 | 94.41 | 1.02 |
| ZH18 | 91.89 | 91.38 | 93.19 | 0.44 |
| ZH68 | 93.36 | 91.36 | 96.25 | 1.09 |
| ZH35 | 92.53 | 92.32 | 95.50 | 1.12 |
| Cultivar | HD13 | ZH13 | ZH18 | ZH68 | ZH35 |
|---|---|---|---|---|---|
| HD13 | 38 | ||||
| ZH13 | 21 | 48 | |||
| ZH18 | 7 | 8 | 50 | ||
| ZH68 | 3 | 10 | 24 | 36 | |
| ZH35 | 7 | 5 | 12 | 16 | 46 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Li, B.; Chen, Y.; Shaibu, A.S.; Zheng, H.; Sun, J. Molecular-Assisted Distinctness and Uniformity Testing Using SLAF-Sequencing Approach in Soybean. Genes 2020, 11, 175. https://doi.org/10.3390/genes11020175
Zhang S, Li B, Chen Y, Shaibu AS, Zheng H, Sun J. Molecular-Assisted Distinctness and Uniformity Testing Using SLAF-Sequencing Approach in Soybean. Genes. 2020; 11(2):175. https://doi.org/10.3390/genes11020175
Chicago/Turabian StyleZhang, Shengrui, Bin Li, Ying Chen, Abdulwahab S. Shaibu, Hongkun Zheng, and Junming Sun. 2020. "Molecular-Assisted Distinctness and Uniformity Testing Using SLAF-Sequencing Approach in Soybean" Genes 11, no. 2: 175. https://doi.org/10.3390/genes11020175
APA StyleZhang, S., Li, B., Chen, Y., Shaibu, A. S., Zheng, H., & Sun, J. (2020). Molecular-Assisted Distinctness and Uniformity Testing Using SLAF-Sequencing Approach in Soybean. Genes, 11(2), 175. https://doi.org/10.3390/genes11020175