Three New Mutations and Mild, Asymmetrical Phenotype in the Highly Distinctive LAMM Syndrome: A Report of Eight Further Cases

 , and
, and
Abstract
1. Introduction
2. Patients and Methods
3. Results
3.1. Clinical Description of Patients
3.1.1. Patients 1 and 2
3.1.2. Patient 3
3.1.3. Patient 4, 5 and 6
3.1.4. Patient 7
3.1.5. Patient 8
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Latchman, K.; Tekin, M. Labyrinthine Aplasia, Microtia, and Microdontia (LAMM) Syndrome and FGF3 Mutations; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2013. [Google Scholar]
- Tekin, M.; Hişmi, B.O.; Fitoz, S.; Ozdag, H.; Cengiz, F.B.; Sırmacı, A.; Aslan, I.; Inceoglu, B.; Yüksel-Konuk, E.B.; Yılmaz, S.T.; et al. Homozygous Mutations in Fibroblast Growth Factor 3 Are Associated with a New Form of Syndromic Deafness Characterized by Inner Ear Agenesis, Microtia, and Microdontia. Am. J. Hum. Genet. 2007, 80, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Sennaroglu, L. Histopathology of inner ear malformations: Do we have enough evidence to explain pathophysiology? Cochlear Implant. Int. 2016, 17, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Wright, T.J.; Mansour, S.L. Fgf3 and Fgf10 are required for mouse otic placode induction. Development 2003, 130, 3379–3390. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, Y.; Alonso, M.T.; Vendrell, V.; Zelarayan, L.C.; Chamero, P.; Theil, T.; Bösl, M.R.; Kato, S.; Maconochie, M.; Riethmacher, D.; et al. Requirements for FGF3 and FGF10 during inner ear formation. Development 2003, 130, 6329–6338. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.P.; Noyes, C.A.; Wang, X.; Wright, T.J.; Mansour, S.L. Fgf3 is required for dorsal patterning and morphogenesis of the inner ear epithelium. Development 2007, 134, 3615–3625. [Google Scholar] [CrossRef] [PubMed]
- Gene, Cards. FGF3 Gene. Available online: https://www.genecards.org/cgibin/carddisp.pl?gene=FGF3 (accessed on 8 July 2019).
- Tekin, M.; Öztürkmen Akay, H.; Fitoz, S.; Birnbaum, S.; Cengiz, F.; Sennaroğlu, L.; Incesulu, A.; Konuk, E.Y.; Bayrak, A.H.; Sentürk, S.; et al. Homozygous FGF3 mutations result in congenital deafness with inner ear agenesis, microtia, and microdontia. Clin. Genet. 2008, 73, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Ramsebner, R.; Ludwig, M.; Parzefall, T.; Ludwig, M.; Parzefall, T.; Lucas, T.; Baumgartner, W.D.; Bodamer, O.; Cengiz, F.B.; Schoefer, C.; et al. A FGF3 mutation associated with differential inner ear malformation, microtia, and microdontia. Laryngoscope 2010, 120, 359–364. [Google Scholar] [PubMed]
- Alsmadi, O.; Meyer, B.F.; Alkuraya, F.; Wakil, S.; Alkayal, F.; Al-Saud, H.; Ramzan, K.; Al-Sayed, M. Syndromic congenital sensorineural deafness, microtia and microdontia resulting from a novel homoallelic mutation in fibroblast growth factor 3 (FGF3). Eur. J. Hum. Genet. EJHG. 2009, 17, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Riazuddin, S.; Ahmed, Z.M.; Hegde, R.S.; Khan, S.N.; Nasir, I.; Shaukat, U.; Riazuddin, S.; Butman, J.A.; Griffith, A.J.; Friedman, T.B.; et al. Variable expressivity of FGF3 mutations associated with deafness and LAMM syndrome. BMC Med. Genet. 2011, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Sensi, A.; Ceruti, S.; Trevisi, P.; Gualandi, F.; Busi, M.; Donati, I.; Neri, M.; Ferlini, A.; Martini, A. LAMM syndrome with middle ear dysplasia associated with compound heterozygosity for FGF3 mutations. Am. J. Med. Genet. Part. A 2011, 155, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.; Minoux, M.; Lauer, J.; Pelletier, V.; Schmittbuhl, M.; Manière, M.C.; Clauss, F.; Veillon, F.; Riehm, S.; Stoetzel, C.; et al. A novel mutation involving the initiation codon of FGF3 in a family described with complete inner ear agenesis, microtia and major microdontia (LAMM syndrome). J. Genet. Syndr. Gene Ther. 2014, 5, 1–5. [Google Scholar]
- Singh, A.; Tekin, M.; Falcone, M.; Kapoor, S. Delayed presentation of rickets in a child with labyrinthine aplasia, microtia and microdontia (LAMM) syndrome. Indian. Pediatr. 2014, 51, 919–920. [Google Scholar] [CrossRef] [PubMed]
- Basdemirci, M.; Zamani, A.G.; Sener, S.; Tassoker, M.; Cetmili, H.; Zamani, A.; Aydogdu, D.; Basdemirci, A.; Yildirim, M.S. LAMM syndrome: Two new patients with a novel mutation in FGF3 gene and additional clinical findings. Clin. Dysmorphol. 2019, 28, 81–85. [Google Scholar] [CrossRef] [PubMed]
- GnomAD Database. Available online: http://gnomad.broadinstitute.org/ (accessed on 8 July 2019).
- Exome Aggregation Consortium (ExAC). Available online: http://exac.broadinstitute.org/ (accessed on 8 July 2019).
- Collaborative Spanish Variant Server-CSVS Databases. Available online: http://csvs.babelomics.org/ (accessed on 8 July 2019).
- Gregory-Evans, C.Y.; Moosajee, M.; Hodges, M.D.J.; Mackay, D.S.; Game, L.; Vargesson, N.; Bloch-Zupan, A.; Rüschendorf, F.; Santos-Pinto, L.; Wackens, G. SNP genome scanning localizes oto-dental syndrome to chromosome 11q13 and microdeletions at this locus implicate FGF3 in dental and inner-ear disease and FADD in ocular coloboma. Hum. Mol. Genet. 2007, 16, 2482–2493. [Google Scholar] [CrossRef] [PubMed]
- Sennaroglu, L.; Saatci, I. A New Classification for Cochleovestibular Malformations. Laryngoscope 2002, 112, 2230–2241. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
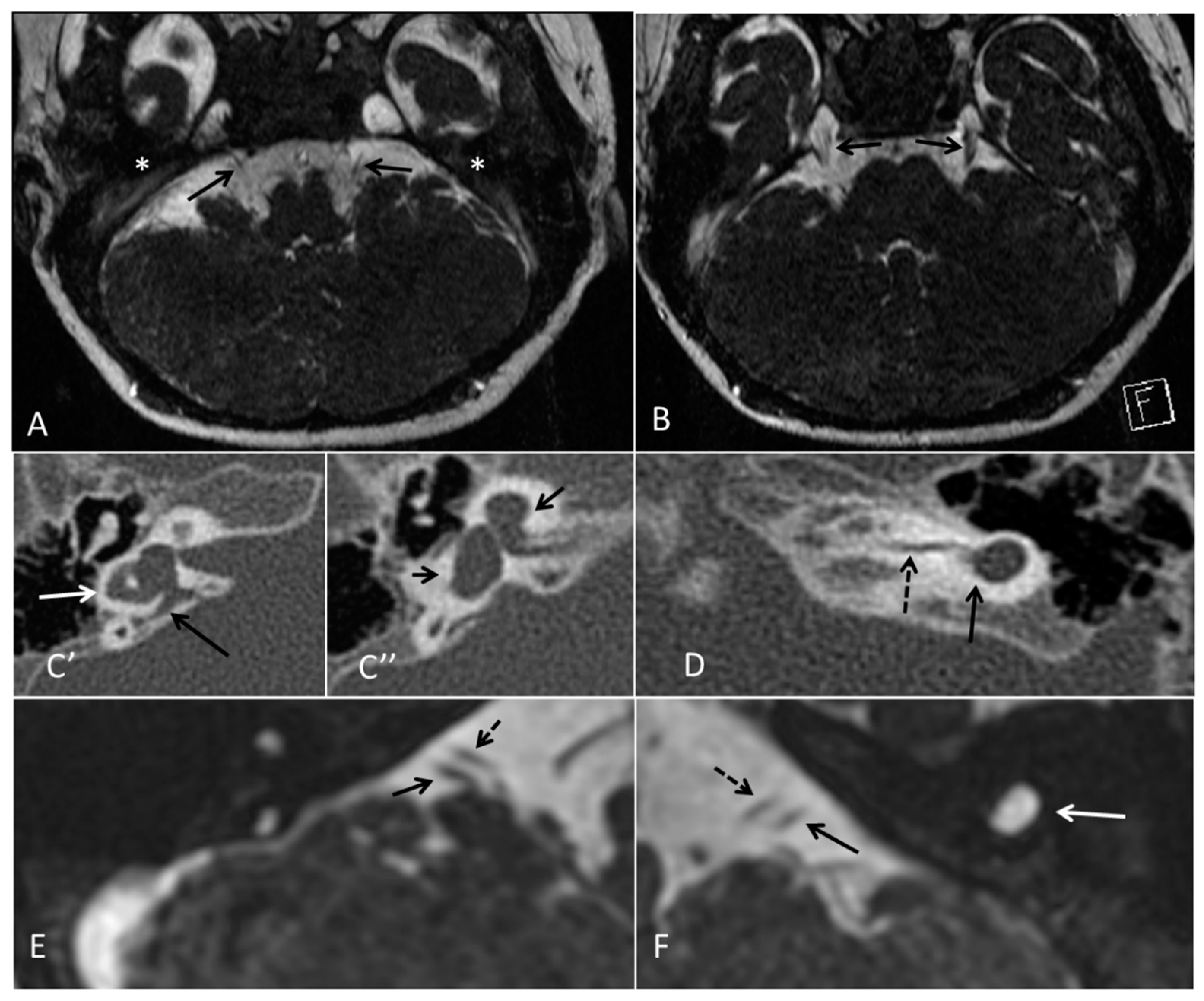
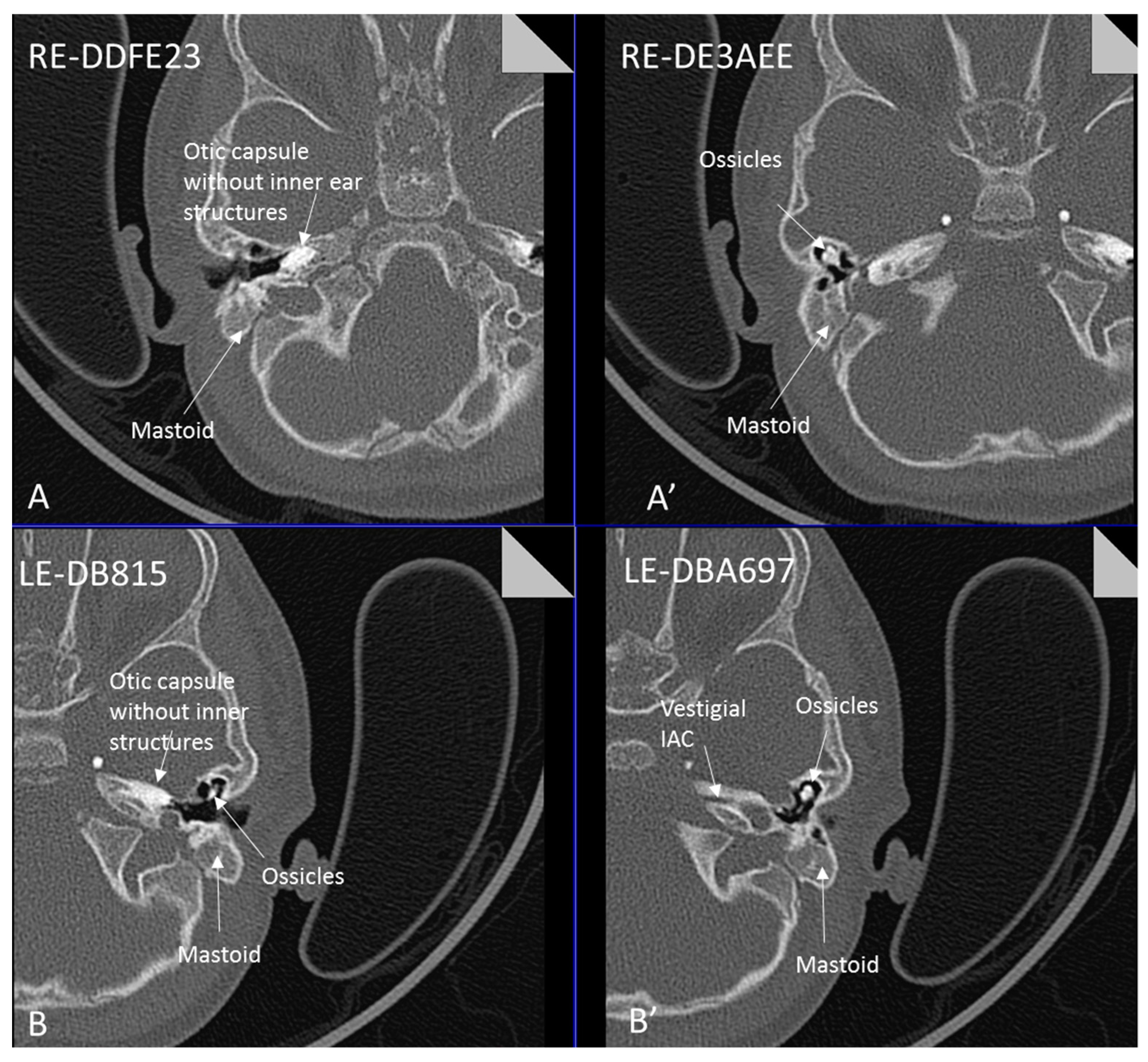
| ID | Mutation | Right Ear | Left Ear | Teeth | Other Images | MRI Results | Interesting or Unusual Features |
|---|---|---|---|---|---|---|---|
| 1 | p.Glu109Thrfs*18 c.325_327delinsA homozygous |  |  |  |  (Mother’s teeth; heterozygous) | Michel’s aplasia | Novel mutation; sacral pit; asymmetrical kidney size |
| 2 | p.Glu109Thrfs*18 c.325_327delinsA homozygous |  |  |  | Michel’s aplasia | Novel mutation; possible social communication disorder | |
| 3 | p.Arg95Trp c.283C>T homozygous | Normal |  |  | CT: Michel’s aplasia | Mild external ear and dental phenotype | |
| 4 | p.Arg95Trp c.283C>T homozygous |  |  |  | Michel’s aplasia (R); Minimal cochlear rest (L) | Large arachnoid cyst (R); asymmetrical kidney size; type 1 diabetes | |
| 5 | p.Arg95Trp c.283C>T homozygous |  |  |  | Bilateral Michel’s aplasia | ||
| 6 | p.Arg95Trp c.283C>T homozygous | Not available | Not available | Not available | Michel’s aplasia and rudimentary otocyst (R) | Rudimentary otocyst with possible inner ear development (previously reported) | |
| 7 | p.Arg95Gln p.Phe178Leu |  |  |  | Parents teeth (both heterozygous): | Cystic cochleo-vestibular malformation (R); cochlear aplasia with a rudimentary vestibule (L). | Novel mutation; compound heterozygote; milder phenotype of external ears without microtia |
| 8 | p.Leu58Pro p.Arg95Trp |  |  |  | Michel’s aplasia | Novel mutation; compound heterozygote; |
| Current Report | Tekin 2007 | Tekin 2008 | Ramsebner 2010 | Alsmadi 2009 | Riazuddin 2011 | Sensi 2011 | Schaefer 2014 | Singh 2014 | Basdemirci 2019 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Fam A n = 5 | Fam B n = 1 | Fam C n = 3 | Fam1 n = 1 | Fam2 n = 3 | Fam1 n = 4 | Fam1 n = 22 | Fam1 n = 4 | Fam2 n = 7 | Fam3 n = 3 | Fam1 n = 2 | Patient B | Fam1 n = 2 | 1 | Fam1 n = 2 | |
| Ethnicity | Indian | Indian | Yemeni | Somalian | Somalian | Somalian | Indian | Spanish | Turkish | Turkish | Somali | Saudi | Pakistani | Pakistani | Pakistani | Albanian | Italian | French | Indian | Turkish | |||
| Consanguinity | + | + | + | + | + | + | - | - | + | + | + | + | + | Unknown | ++ | ++ | ++ | + | - | - | + | - | ++ |
| Profound congenital SNHL | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | +/- | + | + | + | + |
| Vestibular function | - | - | - | ? | ? | ? | - | ? | - | - | - | ? | - | ? | ? | ? | ? | ? | - | - | ? | ? | - |
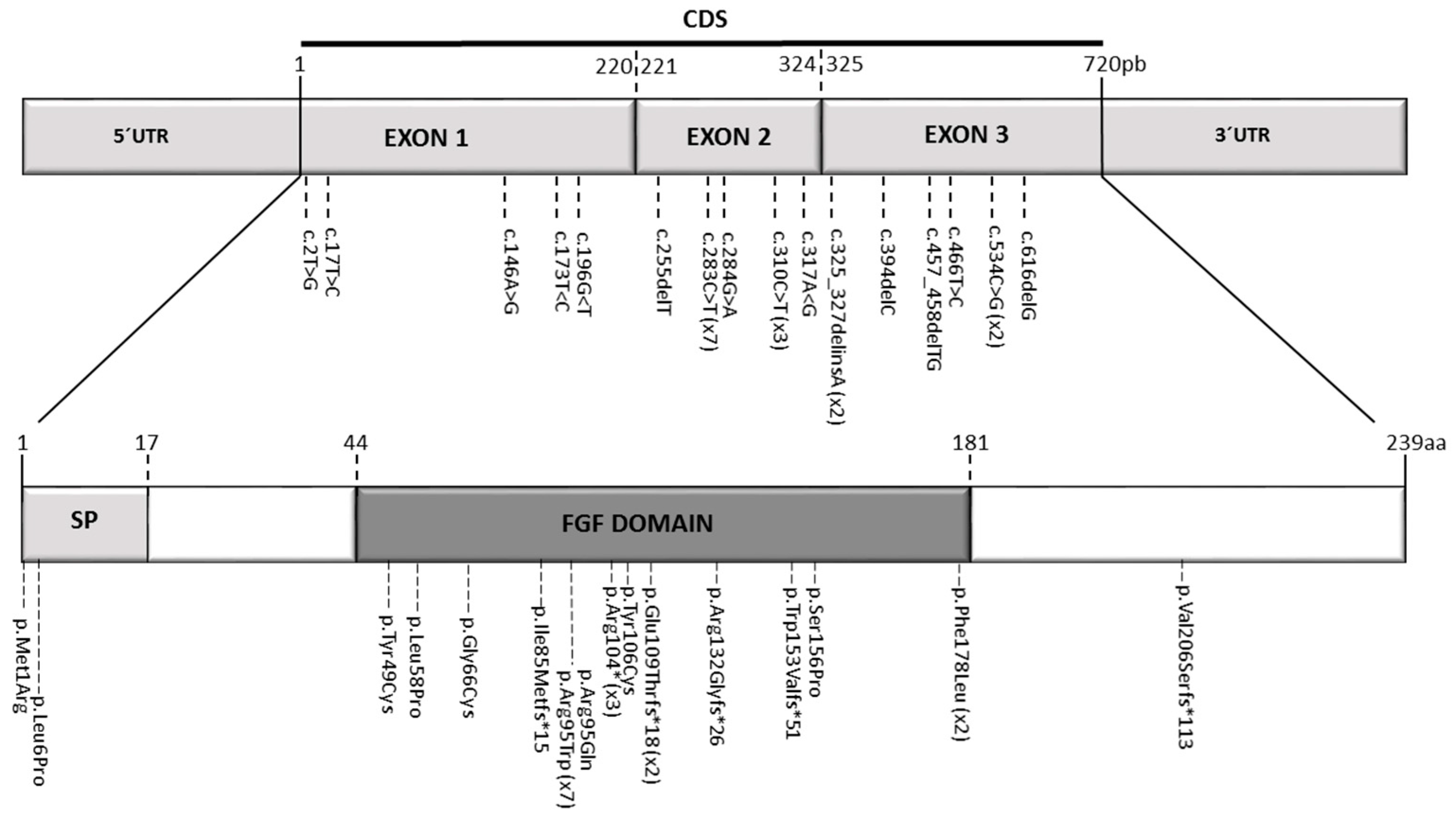
| Mutation | c.325_327delinsA | c.325_327delinsA | c.283C>T | c.283C>T | c.283C>T | c.283C>T | c.284G>A c.534C>G | c.173T>C c.283C>T | c.466T>C | c.616delG | c.310C>T | c.255delT | c.17T>C | c.283C>T | c.196G >T | c.310C>T | c.283C>T | c.394delC | c.317A>G c.457_458 delTG | c.146A>G c.310C>T | c.2T>G | c.534C>G | c.196G>A |
| Protein effect | p.Glu109Thrfs*18 | p.Glu109Thrfs*18 | p.Arg95Trp | p.Arg95Trp | p.Arg95Trp | p.Arg95Trp | p.Arg95Gln p.Phe178Leu | p.Leu58Pro p.Arg95Trp | p.Ser156Pro | p.Val206Serfs*113 | p.Arg104* | p. Ile85Metfs*15 | p.Leu6Pro | p.Arg95Trp | p.Gly66Cys | p.Arg104* | p.Arg95Trp | p.Agr132Glyfs*26 | p.Tyr106Cys p.Trp153Valfs *51 | p.Tyr49Cys p.Arg104* | p.Met1Arg | p.Phe178Leu | p.Gly66Ser |
| External ear anomaly | + | + | + | + | + | + | + | + | + | + | + | + | + | +/- | + | + | + (Mild) | + | + | + | + | + | + |
| External ear Symmetry | S | S | A | A | S | A | A | S | S | S | S | S | S | A | A | S | S | S | S | S | S | S | A |
| External ear details | T1-MO Bilat preauricular skin tag, cupped pinna | Cup shape, simple, protruding | Tiny preauricular skin hillock on one ear, no microtia | Cup shaped ears | Lop ear, T1M0 | Lop ears | Large ears, skin tag on R. | Microtia+ Bilat. cupshaped ears, a tiny preauricular skin hillock on the left ear | T1-MO | T1-MO | T1-MO | T1-MO | T1-MO | Varied phenotype from normal auricles to T1-MO | T1-MO and anteverted ears | T1-MO | Some with near normal auricles; mild | T1-MO | T1-MO, anteverted ears, creased ear lobe. | T1-M0 | T1-M0 | T1-MO | Skin tag left auricule (n = 1) Shortened upper part auricules (n = 2) |
| Inner ear MRI features | Michels | Michels | Michels | L- otocyst rudimentary, R- Michel aplasia | Michels | L- Michel aplasia R- otocyst rudimentary | L- cystic vestibule R- cystic cochleovestibular malf | Michels, normal middle ear development | Michels | Michels | Michels | Michels | L- Michels R- rudimentarl cystic vestibule | Two subjects with rudimentary inner ear formation unilaterally | L- rudimentary vestibular structures R- Michels | Michels, normal middle ear development | Two subjects with cochlear basal turn, vestibule and posterior semi-circular canal | Michels, normal middle ear development | Middle ear involved. Hypoplasia petrous pyramids, bilateral labyrinth and IAC dysplasia. Stapes present in female. | CT Michels aplasia bilaterally Otic vesicle remnant ectopic in left mastoid | Michels; Middle ear structures involved | Michels | Michels |
| Inner ear (a)symmetry | S | S | S | A | S | A | A | S | S | S | S | S | A | A | A index | S | A n = 2 | S | S | S | S | S | A |
| Dental anomalies | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + (mild) | + | + | + | + | + | + |
| Dental details SWS = Small, widely spaced | SWS | SWS | Mild | SWS gum hypertrophy | SWS | Microdontia | SWS | Microdontia | SWS | SWS | SWS | Microdontia, conical teeth | SWS | Mild | SWS | SWS | SWS | Persistent deciduous molars, dyschromia, enamel pits, taurodontism, oligodontia | Microdontia, thinned enamel, enlarged pulp | Hypodontia or oligodontia | |||
| Additional features | Renal size disparity | Autism | Sinus surgery | Gum HT, renal size disparity, long sighted | Long sighted | OSA, Squint | Motor delay | Motor delay | Unilateral stenosis of ureteropelvic junction (n = 1) | Strabismus and hypermetropia (n = 1) | Prominent nose tip, anteverted ears | Some perception loud sounds (n = 1); Occipital bone flattening, hypoplasic alae nasi | Carrier mother had unilateral ear defect surgically corrected. | Dextrorotation of heart (n = 1) | Hypophosphatemic rickets, language delay | Myopia, astigmatism, dilated azygos vein | |||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Yassin, A.; D’Arco, F.; Morín, M.; Pagarkar, W.; Harrop-Griffiths, K.; Shaida, A.; Fernández, E.; Cullup, T.; De-Souza, B.; Moreno-Pelayo, M.A.; et al. Three New Mutations and Mild, Asymmetrical Phenotype in the Highly Distinctive LAMM Syndrome: A Report of Eight Further Cases. Genes 2019, 10, 529. https://doi.org/10.3390/genes10070529
Al Yassin A, D’Arco F, Morín M, Pagarkar W, Harrop-Griffiths K, Shaida A, Fernández E, Cullup T, De-Souza B, Moreno-Pelayo MA, et al. Three New Mutations and Mild, Asymmetrical Phenotype in the Highly Distinctive LAMM Syndrome: A Report of Eight Further Cases. Genes. 2019; 10(7):529. https://doi.org/10.3390/genes10070529
Chicago/Turabian StyleAl Yassin, Amina, Felice D’Arco, Matías Morín, Waheeda Pagarkar, Katherine Harrop-Griffiths, Azhar Shaida, Elena Fernández, Tom Cullup, Bianca De-Souza, Miguel Angel Moreno-Pelayo, and et al. 2019. "Three New Mutations and Mild, Asymmetrical Phenotype in the Highly Distinctive LAMM Syndrome: A Report of Eight Further Cases" Genes 10, no. 7: 529. https://doi.org/10.3390/genes10070529
APA StyleAl Yassin, A., D’Arco, F., Morín, M., Pagarkar, W., Harrop-Griffiths, K., Shaida, A., Fernández, E., Cullup, T., De-Souza, B., Moreno-Pelayo, M. A., & Bitner-Glindzicz, M. (2019). Three New Mutations and Mild, Asymmetrical Phenotype in the Highly Distinctive LAMM Syndrome: A Report of Eight Further Cases. Genes, 10(7), 529. https://doi.org/10.3390/genes10070529