Abstract
Glycosylation is the most common post-translational modification of proteins; it mediates their correct folding and stability, as well as their transport through the secretory transport. Changes in N- and O-linked glycans have been associated with multiple pathological conditions including congenital disorders of glycosylation, inflammatory diseases and cancer. Glycoprotein glycosylation at the Golgi involves the coordinated action of hundreds of glycosyltransferases and glycosidases, which are maintained at the correct location through retrograde vesicle trafficking between Golgi cisternae. In this review, we describe the molecular machinery involved in vesicle trafficking and tethering at the Golgi apparatus and the effects of mutations in the context of glycan biosynthesis and human diseases.
1. Protein Glycosylation and the Golgi
The enzymatic addition of glycans to proteins, dubbed “protein glycosylation”, is a fundamental post-translation and co-translational modification that controls the folding and stability of proteins and their transport through the secretory pathway [1,2,3]. The composite repertoire of glycan structures in glycoproteins impacts on many different cellular and developmental processes such as signal transduction, molecular trafficking, cell-cell communication, immunity and early mammalian development [4,5]. Since one fifth of all proteins in structural databases are classified as glycoproteins, it is not surprising that up to 2% of the eukaryotic proteome is required for glycosylation [3,6]. It has been estimated that 700 proteins are required to generate the glycan structures in human glycome [7]. Glycoprotein glycosylation is a sequential process that involves the coordinated action of hundreds of glycosyltransferases (GTs) and glycosydases, which are trafficked to the specific locations of the endoplasmic reticulum (ER) and Golgi apparatus [8]. Only ten monosaccharides are essential for the synthesis of animal glycans: glucose (Glc), galactose (Gal), N-acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc), fucose (Fuc), mannose (Man), xylose (Xyl), glucuronic acid (GlcA), sialic acid (SA, either as N-acetylneuraminic acid, Neu5Ac or 5-N-glycolylneuraminic acid acid, Neu5Gc). The main types of protein glycoprotein glycans are N- or O-linked. N-glycosylation starts in the ER with the synthesis of a glycan precursor with the composition Glc3Man9GlcNac2 on the carrier dolichol isoprenoid lipid [9]. The precursor glycan is then transferred en bloc onto asparagine residues of nascent proteins by multisubunit oligosaccharyltransferase (OST) complexes within the lumen of the ER [10,11,12]. Distal glucose moieties of the Glc3Man9GlcNac2 glycan are trimmed before reaching the Golgi apparatus by sequential actions of ER glucosidases, representing an important step in the control of folding processes for secretory pathway glycoproteins prior to ER exit [13,14,15]. Glycoproteins that exit the ER reach the Golgi apparatus with eight or nine mannose residues for further processing of the glycan moieties into complex and hybrid glycan forms (Figure 1).
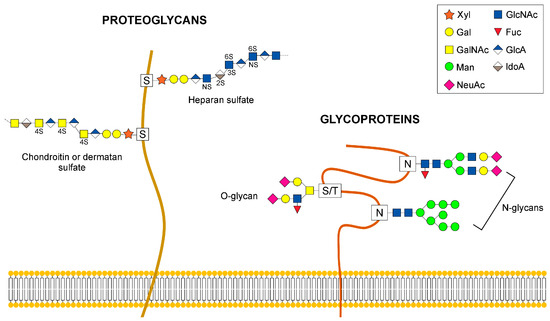
Figure 1.
Examples of glycans that are elaborated in the Golgi. The diagram depicts N- and O-linked glycans attached to glycoproteins and proteoglycans. Abbreviations are: Xyl, xylose; Gal, galactose; GalNAc, N-acetylgalactosamine; Man, mannose; NeuAc, N-Acetylneuraminic acid; GlcNAc, N-acetylglucosamine; Fuc, Fucose; GlcA, glucuronic acid; IdoA, iduronic acid.
The Golgi apparatus is organized into discrete cisternae, each containing a distinct subset of glycosylation machinery proteins which include glycosyltransferases, glycosidases and nucleotide sugar transporters [16,17]. As the secretory proteins arrive at the cis-Golgi, mannose trimming generates the Man5GlcNAc2Asn intermediate followed by the transfer of GlcNAc residue to a terminal Man residue on the α3-arm of the glycan structure by the medial Golgi GT GlcNAcT-1. The addition of GlcNAc to Man5GlcNAc2Asn is an essential step to generate hybrid and complex N-linked glycans [15,17]. Hybrid N-glycans are generated by the extension of the α3-arm that received GlcNAc with the addition of Gal, GalNAc, Fuc and SA. Removal of two terminal of the five Man residues, allows branching with an additional GlcNAc residue and synthesis of biantennary complex N-glycans. Further branching may occur, leading to multiantennary complex glycans in medial-late and trans-Golgi compartments [17,18]. Complex glycans can be decorated with Gal, GalNAc, Fuc and SA.
In contrast to N-linked glycosylation, O-glycosylation does not involve a lipid-linked oligosaccharide precursor for transfer to nascent polypeptides. Instead, it consists of the formation of a single glycosidic linkage between Ser or Thr residues and GalNAc, Man, Glc, Xyl, or Fuc [17]. The most abundant type of O-glycans are the mucins initiated by GalNAc-Ser/Thr and the glycosaminoglycan (GAG) chains on proteoglycans initiated by a conserved tetrasaccharide Glc-A-β1,3-Gal-β1.3-Gal-β1,4Xyl-β (Figure 1). Synthesis of O-glycans starts in the early cis-Golgi or in a translational compartment (ERGIC, with characteristics of the ER), and is completed via the sequential addition of glycans while the secretory protein moves through the cis-, medial- and trans-Golgi cisternae.
Efficient protein N- and O-linked glycosylation is strictly dependent on the precise distribution of glycosyltransferases, glycosidases and nucleotide sugar transporters which should be maintained in the correct Golgi compartment as the secretory cargo protein passes through the Golgi. How Golgi enzymes are retained in the Golgi membranes is strictly regulated by vesicle trafficking across the Golgi [19].
So far, more than 100 monogenic congenital disorders of glycosylation (CDGs) have been identified and associated with a wide variety of symptoms such as neurological deficit, liver disfunction, and skin and bone alterations [20,21]. Traditionally CDGs have been divided into two groups: CDG-I affect the synthesis and/or transfer of the dolichol pyrophosphate oligosaccharide precursor of N-linked glycoproteins, while CDG-II affect the processing of N-linked glycans or the biosynthesis of O-linked glycans [22]. Many pathogenic mutations in intra-Golgi trafficking regulators have been associated with CDG II. This review will highlight the strict relationship between the Golgi trafficking machinery and protein glycosylation.
2. Enzyme Sorting at the Golgi Cisternae
Several models for Golgi trafficking have been postulated, but the most favored model for membrane traffic within the Golgi is the cisternal maturation model of Golgi transport ([19,23]; Figure 2).
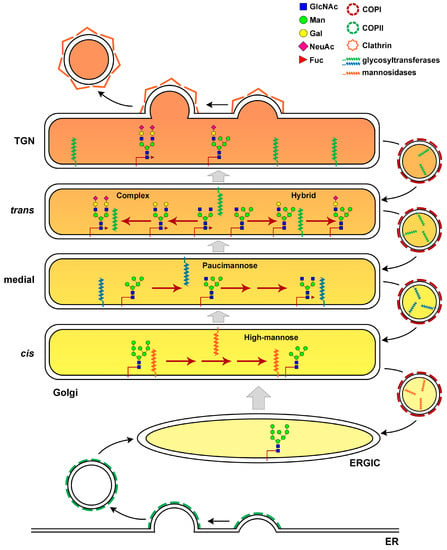
Figure 2.
Elaboration of N-linked Glycans in the Golgi cisternae. N-glycan processing in the Golgi gives rise to three main classes of glycans: high-mannose, hybrid and complex glycans. Glycoproteins exit the ERGIC and reach the Golgi apparatus with eight or nine mannose residues. The Golgi apparatus is organized into discrete cisternae where the enzymes needed for glycan elaboration are specifically compartmentalized. Mannose trimming enzymes are located in the cis and medial-Golgi. The addition of GlcNAc is an essential step to generate hybrid and complex N-linked glycans in the trans-Golgi. Hybrid N-glycans are generated by the extension of the α3-arm that received GlcNAc with the addition of Gal, Fuc and SA. Removal of two terminal of the five Man residues, allows branching with an additional GlcNAc residue and synthesis of biantennary complex N-glycans with Gal, Fuc and SA. The polarized distribution of glycosylation enzymes is maintained through retrograde transport of COPI coated vesicles. A proper cisternal pH (different shades of orange) is needed to control retrograde sorting of enzymes and the association of glycoenzymes with the trafficking cargo. GlcNAc, N-acetylglucosamine; Man, mannose; Gal, galactose; NeuAc, N-Acetylneuraminic acid; Fuc, Fucose.
According to the cisternal maturation model, as a new cis-Golgi cisterna is formed, it is gradually converted by the accumulation of medial and then trans glycosylation enzymes that travel from more mature Golgi compartments in vesicles moving in retrograde direction. Although there is strong support for this model, recent observations of the transport of soluble cargoes in mammalian cells have led researchers to postulate the existence of intercisternal tubules that offer specialized Golgi traffic routes [24]. Retrograde transport and sorting of glycosylation enzymes depends mostly on COPI-coated vesicles, composed of seven different protein subunits αCOP, βCOP, β’COP, γCOP, δCOP, εCOP and ζCOP [25]. The COPI coat is recruited from the cytosol to the membrane primarily through binding to activated Arf1 GTPase. As coat proteins are recruited, they assemble a cage-like structure that enhances membrane curvature to promote vesicle formation [26,27]. A recent study showed that COPI carriers can traffic in anterograde as well as retrograde directions, with the small GTPase Cdc42 regulating the bidirectional COPI transport specificity [28]. Golgi glycosylation also requires maintenance of a stringent cisternal pH, which influences retrograde sorting of enzymes and the association of a glycosylation enzyme with a trafficking cargo [29]. Indeed, disruption of the cisternal pH leads to mislocalization of glycosyltransferases [30,31]. Consistent with the role of the pH in Golgi glycosylation, loss-of-function mutations in ATP6V0A2, the gene encoding the membrane-bound V0a2 subunit of the V-ATPase, disrupt retrograde intra-Golgi trafficking, resulting in glycosylation defects [32]. Patients carrying pathogenic variants in ATP6V0A2 present with wrinkly skin syndrome, autosomal recessive cutis laxa type II and neurological impairment [32]. Recent work showed that Golgi localization of glycosylation enzymes and their function also depend on lipid composition of the Golgi compartments [33,34]. Disruption of sphingomyelin homeostasis at the trans-Golgi network of mammalian cells, by using short-chain ceramide, affects the organization of Golgi membranes, leading to separation of Golgi-resident proteins from each other [33]. As a consequence, the enzyme sialyltransferase is unable to interact with its substrate TGN46, that subsequently fails to be glycosylated. Moreover, the biosynthesis of complex sphingolipids is required for the retention of a Golgi mannosyltransferase in yeast [34].
3. Molecular Actors in Vesicle Targeting and Fusion at the Golgi
The fusion of transported COPI vesicles at the target Golgi membrane requires the orchestrated action of Rab GTPases, tethering complexes and SNARE proteins [20,35,36,37]. Small Rab GTPases are important molecular regulators of multiple vesicle trafficking steps [38]. There are about 20 different Rab proteins that associate with the Golgi membranes [36]. In the GTP-bound active state (membrane-associated), Rab proteins recruit molecular motors and tethering factors to specific membrane domains to promote vesicle fusion [39]. Prior to fusion, transport vesicles and their cognate target membranes must be properly aligned, a process known as tethering. The tethering of vesicles at the cognate target membrane is regulated by large coiled-coil tethers (CCTs, [40,41,42,43,44]) or multisubunit tethering complexes (MTC; [35,45]). CCTs that localize at the Golgi apparatus are named ˝golgins˝ [43]. In humans, there are 11 golgins including GM130, Golgin-45, Golgin-97. Golgins are anchored to the Golgi membranes by the carboxy-terminus, and protrude into the cytoplasm to capture vesicles, usually through the amino-terminus of the protein [40]. These tethers are large dimeric proteins displaying two globular heads connected by a long coiled-coil structure, which greatly facilitate the first contact with the vesicles over distances of more than 200 nm [35]. Golgins can occupy several locations within the Golgi, residing at the cis-Golgi, the rims of the Golgi stacks and the trans-Golgi network. Due to their ability to selectively tether vesicles of specific cargo content, these proteins play an essential role in directing vesicle trafficking within the Golgi. Most golgins have been found to cooperate with several trafficking players including Rab proteins and other GTPases, MTCs and SNAREs [46]. Rabs bind the coiled-coil regions within the golgin protein, whereas small Arf GTPases interact with GRAB/GRIP domains, located in the terminus of many golgins [36].
The multisubunit tethering complexes (MTCs), consisting of three to ten subunits and an overall molecular weight of 250–800 kDa, can associate with vesicles over much shorter distances compared to CCTs [45]. Recent structural studies and sequence comparisons have led researchers to group MTCs into CATCHRs and non-CATCHRs [47]. The family of complexes associated with tethering containing helical rods (CATCHR) include DSL1/ZW10 complex, Conserved Oligomeric Golgi (COG), GARP (Golgi associated retrograde complex) and the exocyst [45]. The non-CATCHR group includes two complexes involved in regulating vesicle transport from late Golgi to endosomal/lysosomal compartments named Homotypic fusion and protein sorting (HOPS) and class C core vacuole/endosome tethering (CORVET) [45]. The multisubunit Transport Particle (TRAPP) complexes (I, II and III) have been classified as MTCs with a unique structure [48]. TRAPPI function is involved in ER-Golgi transport [49].
Although MTCs and CCTs function as tethers, they are implicated in different steps of vesicular tethering [50]. The long CCTs are required for the initial capturing of distant vesicles through transient and low affinity interactions. Conversely MTCs, which can bind vesicles at short distances from the target membrane, act in later events of tethering and facilitate SNAREpin in assembly. Thanks to their multiple subunit structures, MTCs can simultaneously interact with various vesicle trafficking regulators (i.e., CCTs, Rab1 and SNAREs), playing a key role in coordinating vesicular tethering with docking and fusion [35].
The fusion of the uncoated vesicle with the target membrane at the Golgi requires the assembly of functional SNAREs (soluble N-ethylmaleimide-sensitive factor activating protein receptors) complexes [50]. SNARE proteins share a characteristic coiled-coil motif of approximately 70 amino acids comprising heptad repeats, and can localize on either the target (t-SNARE) or the vesicle membrane (v-SNARE). The interaction of cognate v-SNAREs and t-SNAREs leads to the formation of a fusogenic trans-SNARE complex or SNAREpin, in which four SNARE motifs form a twisted parallel four helix-bundle, bringing the membranes together and leading to vesicle fusion [51,52]. Sec1/Munc18 (SM) proteins, evolutionarily conserved peripheral membrane proteins of 60–90 kDa, act in conjunction with tethering factors and SNAREs [50,53]. SM proteins can bind to the syntaxin family of SNAREs [54] or to the whole SNARE complex [55,56,57]. In both cases SM proteins have been shown to promote SNAREpin assembly and membrane fusion at the Golgi membranes.
4. GOLPH3 Interacts with COPI to Recruit Glycosyltransferases to Golgi Compartments
Golgi phosphoprotein 3 (GOLPH3), a highly conserved protein from yeast to humans, has been characterized as a Phosphatidylinositol 4-phosphate [PI(4)P] effector that regulates Golgi trafficking [58]. Identified in proteomic-based studies of the Golgi [59,60], GOLPH3 localizes to the trans-Golgi via the direct interaction with PI(4)P, mediated by its unique C-terminal GPP34 domain [61,62]. Localization of GOLPH3 protein to the Golgi is required to maintain Golgi architecture in human cells and Drosophila melanogaster [61,63].
Much evidence indicates that GOLPH3 is an oncogene, that is overexpressed in several solid tumors such as lung cancer, breast cancer, colon cancer and melanoma [58,64]. However, the molecular mechanisms underlying the oncogenic properties of GOLPH3 are poorly understood. It has been proposed that GOLPH3 might contribute to cellular transformation by affecting the glycosylation of key cancer relevant glycoproteins [58]. Experimental data from human cells and in model organisms indicate that GOLPH3 plays a fundamental role for retrograde intra-Golgi trafficking of protein glycosyltransferases ([58]; Figure 3).
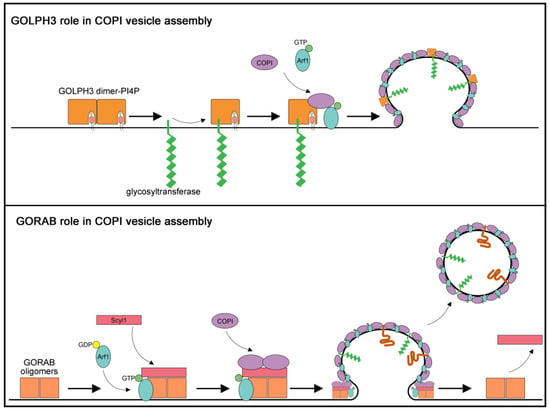
Figure 3.
Proposed models for roles of GORAB and GOLPH3 in COPI-mediated trafficking. (Upper panel) The diagram depicts a model for GOLPH3 role in N-glycosylation based on the results of Eckert and coauthors [65]. GOLPH3 protein binds to the coatomer as well as to a set of glycosyltransferases, such as C2GnT and SiaT as well as the coatomer, thereby mediating incorporation of these enzymes into COPI coated vesicles. (Lower panel) The diagram depicts a model for GORAB role in COPI vesicle release based on the results of Witkos and coauthors [66] GORAB self-associates in oligomers that form discrete domains at the trans-Golgi membrane. GORAB oligomers recruit Scyl1 protein driving Arf1-GTP accumulation in the GORAB-enriched domains. High levels of Scyl1 and Arf1-GTP proteins lead to the selective COPI loading. In turn, coatomer assembly leads to vesicle budding and cargo incorporation while GORAB/Scyl1 complex stabilizes the bud neck during vesicle formation. After releasing of the COPI vesicle from the Golgi membrane, GORAB and Scyl1 detach from the complex to start a new cycle.
Glycosyltransferases are all type II integral membrane proteins containing of a short cytosolically exposed N-terminal region, a single membrane-spanning domain and a luminal enzymatic domain [5]. The cytoplasmic tails of Golgi glycosyltransferases lack the canonical COPI-binding sorting signals. The role of GOLPH3 in enzyme recycling was initially demonstrated for the yeast homologue Vps74p which was shown to bind the N-terminal tails of a set of mannosyltransferases as well as the COPI coatomer, thereby anchoring these enzymes at the Golgi [67,68]. Mutations in the PI(4)P-binding pocket of Vps74p caused loss of Vps74p-membrane association in yeast cells and mislocalization of mannosyltransferases [69]. Both GOLPH3 and Vps74p form oligomers and tetramerization of Vps74p was essential to bind to a pentameric sequence motif at the cytoplasmic tail of a subset of Golgi glycosyltransferases in in vitro assays [70]. A cluster of evolutionarily conserved arginine residues at the N-terminus of GOLPH3 proteins was shown to mediate coatomer binding [70]. Vps74p-coatomer binding was required for retention of glycosyltransferases to the Golgi, but not for Vps74p association with the Golgi membrane which was instead dependent on the oligomerization status and the PI(4)P binding capabilities [70]. Vps74p recognizes a (F/L)(L/I/V)XX(R/K) motif in the cytoplasmic tail of numerous yeast mannosyltransferases raising the possibility that it might function as an adaptor for incorporation of glycosyltransferases in the COPI vesicles. A sequence similar to the (F/L)(L/I/V)XX(R/K) motif, was also found in the cytoplasmic tail (CT) of Core 2 N-acetylglucosaminyltransferase 1 (C2GnT1; [71]). C2GnT1 plays a crucial role in the synthesis of core 2-associated sialyl Lewis x (C2-O-sLex), a ligand involved in selectin-mediated leukocyte trafficking and tumorigenesis [72]. GOLPH3 protein interacts with the CT of C2GnT1 via a LLRRR sequence and controls Golgi localization of C2GnT1 in KG1a and K562 cells [71]. Moreover, depletion of GOLPH3 affects synthesis of C2-O-sLex associated with P-selectin glycoprotein ligand-1 with effects on cell tethering, rolling and adhesion [71].
The involvement of GOLPH3 in O-glycosylation was further demonstrated by its interaction with the CT of POMGnT1, the O-mannosyl-β-1,2-N- acetylglucosaminyltransferase 1 required for O-mannosylation of α-dystroglycan [73]. α-dystroglycan, a key component of the α-dystroglycan glycoprotein complex mediates the association with laminin and other components of the extracellular matrix [74]. Perturbation of the glycosylation status of α-dystroglycan has been linked to various forms of congenital muscular dystrophies [75]. Loss of GOLPH3/ POMGnT1 interaction impairs Golgi localization of POMGnT1 and results in a reduction in IIH6 immunoreactivity indicating a defective glycosylation status of α-dystroglycan.
Chang and coauthors [76] showed that GOLPH3 controls the ability of Golgi to retain exostosins, a class of glycosyltransferases implicated in O-glycosylation of heparan sulfate proteoglycans. Exostosins catalyze the addition of alternating β1-4-linked glucuronic acid (GlcA) and α1-4-linked N-acetylglucosamine (GlcNAc) disaccharides in the biosynthesis of glycosaminoglycan chains [77]. In humans, mutations affecting the exosostins EXT1 and EXT2 cause multiple osteochondrosoma (MO), an autosomal dominant skeletal disease characterized by multiple cartilaginous tumors [78,79,80]. Drosophila GOLPH3 physically interacts with the exosostin EXT1and EXT2. Moreover, knockdown or overexpression of GOLPH3 affects the distribution of EX1 and EX2 proteins within the Golgi cisternae, resulting in defective HSPGs and Hedgehog signaling [76]. Consistent with the results in Drosophila, Golgi localization of EXT1 and EXT2 is sensitive to GOLPH3 protein levels in cells cultivated from bone cells such as osteosarcoma cells (U2OS, MG63), chondrosarcoma cells (SW1353), and rhabdomyosarcoma (RD) cells [76].
Several studies have supported the role of the GOLPH3 protein in N-glycosylation ([65,81]; Figure 3). Isaji and coworkers [81] showed that GOLPH3 knockdown (KD) affects integrin-mediated cell migration which correlates with defective sialylation of N-glycans, especially α2,6-sialylation on β1-integrin. They further demonstrated that GOLPH3 associates with α2,6-sialyltransferase-I (ST6GAL1) in HeLa cells and that expression of α2,6-sialyltransferase-I (ST6GAL1) rescues integrin-dependent cell migration and cellular signaling defects in GOLPH3 KD cells. Eckert and coauthors [65] reported a role of human GOLPH3 in spatial regulation of ST6GAL1. They showed that GOLPH3 protein directly interacts with Core 2N-acetylglucosaminyltransferase (C2GnT) and ST6GAL1, and directs incorporation of these enzymes into COPI coated vesicles. Conversely, galactosyltransferase, an enzyme that does not bind to GOLPH3, is not incorporated into COPI vesicles, and does not depend on GOLPH3 for its Golgi localization. Remarkably, previous studies reported that increased α2-6 sialylation of β1-integrins affect the adhesive and migratory capacity of tumor cells, and may contribute to colon tumor progression [82,83]. Collectively, these data suggest a strong correlation between GOLPH3-dependent glycosylation defects and tumor progression.
5. GORAB, a Scaffolding Protein for COPI, Is Involved in Gerodermia Osteodysplastica
The RAB6-interacting protein GORAB, also known as NTKL-binding protein 1, was also dubbed “Scyl1-binding protein 1” after its interaction with Scyl1/NTKL protein [84]. Scyl1, a component of the Scy1-like family of catalytically inactive protein kinases, was involved in COPI-mediated retrograde trafficking because of its association with isoform 2 of γCOP subunit, through a C-terminal RKXX-COO- dibasic motif [85,86]. GORAB loss-of-function mutations have been linked to Gerodermia osteodysplastica GO, OMIM 231070; [87], an autosomal recessive inherited disorder characterized by lax and wrinkly skin, which gives patients a prematurely aged appearance [87,88]. Moreover, individuals suffering from GO exhibit a severe osteoporosis with reduced bone mass, susceptibility to fractures and malar and mandibular hypoplasia [87,88]. To date, the molecular mechanisms that link loss of GORAB with the onset of GO disease remain to be clarified. Evidence indicates that GORAB is a key regulator of the E3 ligase Mdm2, promoting both its transcription, by binding Mdm2 promoter [89], and Mdm2 protein self-ubiquitylation [90]. A recent work implicated the Drosophila homologue of GORAB in centriole structure and duplication [91]. Although the above studies proved a subcellular localization of GORAB in the cytoplasm and in the nucleus, the GO disease has been mostly associated with Golgi-associated functions [87,92]. Indeed, in dermal fibroblasts and osteoblasts, which represent the mostly affected cell types in GO, GORAB protein was detected almost exclusively at the Golgi apparatus [87,92].
Originally classified as a member of the Golgin family, GORAB localizes to the trans-Golgi through the association with the active, GTP-bound, form of the small RAB6 and ARF5 GTPases through the “internal Golgi-targeting RAB6 and ARF5 binding domain” (IGRAB) [87,92]. However, a recent study has suggested that GORAB functions as a scaffolding protein for COPI assembly at the trans-Golgi, rather than behaving as a member of the Golgin family [66]. Witkos and coauthors [66] demonstrated that GORAB undergoes oligomerization and localizes to discrete puncta throughout the trans-Golgi where Scyl1 and GORAB directly interact via their respective N-terminal domains. Moreover, a mutational analysis indicated that the ability of GORAB to self-associate in oligomers is involved in the assembly of the coatomer. Taken together, these data indicate that the GORAB/Scyl1 complex is necessary and sufficient to recruit the COPI complex to the Golgi membranes [66]. GORAB and Scyl1 were also shown to directly bind to the active GTP-bound form Arf1, indicating that they might function as Arf1 effector proteins. Of note, the predominant Arf1 binding coat proteins at the trans-Golgi network (TGN) are AP1 and the GGAs [93]. Witkos and coauthors propose that GORAB and Scyl1 oligomers provide discrete domains at the TGN, where high levels of Scyl1 and GTP loaded Arf1 favor the selective COPI coat assembly at the expense of AP1 ([66]; Figure 3).
The roles of GORAB protein in the COPI-mediated retrograde trafficking also involve maintenance of the proper set of glycosyltransferases in the Golgi trans-cisterna. Indeed, in HeLa cells depleted of GORAB, the β-galactoside α-2,6-sialyltransferase 1(ST6GAL1) undergoes a shift towards the later compartments of Golgi [66]. Moreover, N-glycome analysis by mass spectrometry from fibroblasts of GO patients and from skin samples of GORAB-knockout mice revealed a significant decrease of sialylated complex glycans [66]. Recent work indicated the requirement for GORAB protein for O-glycosylation. An analysis of a Gorab null full knock out in a mouse model revealed a substantial reduction of the dermatan sulfate glycosaminoglycan (GAG) attached to proteoglycans decorin and biglycan through O-glycosylation [94]. According to these data, GO can be considered as a type II congenital disorder of glycosylation (CDGs-II), affecting the tissues that are mostly dependent on protein glycosylation and glycanation, such as skin and bone. Indeed, these tissues present a large component of extracellular matrix; for this reason, they could be very sensitive to GORAB mutations [66,87,94].
6. The Vesicle Tethering COG Complex and Other Tethering Factors Are Required for Normal Glycosylation
Recent work involved mutations in CCTs in glycosylation defects and CDGs. GMAP-210 functions as a tether for both ER-to-Golgi and intra-Golgi vesicles [95]. Loss-of-function mutations in the gene encoding GMAP-210 are associated with neonatal lethal skeletal dysplasia in mice and achondrogenesis type 1A in humans [96]. Mice carrying null mutations in Trip11, which encodes GMAP-210, displayed swollen ER and alterations of Golgi architecture in multiple tissues including cartilage [96]. Glycan processing defects in the Golgi were found in fibroblasts and chondrocytes of mice lacking GMAP-210, resulting in intracellular accumulation of perlecan, but not of type II collagen or aggrecan in chondrocytes. Similarly, fibroblasts from patients carrying either a heterozygous or homozygous nonsense mutant variant of GMAP-210 displayed incomplete glycosylation of the model secretory protein vesicular stomatitis virus G protein, indicating a link between altered glycan processing and GMAP-201 mediated vesicle tethering [96].
The requirement for the conserved oligomeric Golgi (COG) complex for normal Golgi-glycosylation has been demonstrated by a large amount of research data [20]. The eight-subunit COG complex, a member of the CATCHR family of tethers, regulates retrograde intra-Golgi vesicle trafficking [97,98,99,100]. The COG complex consists of eight subunits and can exist as a hetero-octameric complex or two distinct subcomplexes: Lobe A (COG1-4) and Lobe B (COG5-8) [101,102,103]. COGI-COG8 interaction, through the formation of alpha-helical bundles, is required to bridge the two lobes together [101,102,104]. Lobe A predominantly associates with the Golgi membranes of every cisterna whereas Lobe B localizes to COPI vesicles [105]. Depletion of COG subunits in yeast and mammalian cells results in a massive accumulation of Golgi-derived vesicles of approximately 60 nm [106,107,108]. These vesicles called COG complex dependent (CCD) vesicles, are enriched in medial/trans-Golgi enzymes and intra-Golgi v-SNAREs and can be tethered in vitro by purified COG, demonstrating the role of COG as a tethering complex [109]. The COG complex has a central role in intra-Golgi trafficking due to its ability to associate with several vesicle trafficking players. Indeed, COG subunits were shown to interact with intra-Golgi SNAREs, SM proteins, COPI coat proteins, small Rab GTPases and CCTs [50,110,111]. Several studies indicate that the COG complex may coordinate tethering and vesicle fusion events [50]. Importantly the interaction of the COG complex subunit COG4 with SM proteins and SNAREs is required for the assembly of two different SNAREpin complexes, the intra-Golgi Stx5-GS28-Ykt6-GS15 and the trans-Golgi STX16/STX6/VT1A/VAMP4 [112,113].
In humans, mutations in COG1, COG2, COG4-COG8 cause inherited, autosomal recessive, congenital disorders of glycosylation CDGs-II, [114,115,116,117,118,119,120,121,122,123,124,125]. Deficiencies in COG complex subunits (COG-CDG) have been linked to developmental impairments including microcephaly, mental retardation, hypothonia accompanied with dysmorphic features and feeding problems [114,115,116,117,118,119,120,121,122,123,124,125]. Mutations in COG7 gene, cause the highest mortality within the first year of life [123,124]. Glycan analysis by mass-spectrometry revealed hyposialylation of serum proteins, abnormal synthesis of N- and O-linked glycans and altered glycolipids in most COG-CDG patients [114,119,120,122,123,124,125]. COG complex is evolutionarily conserved with COG subunits present in all the eukaryotic kingdom [126,127]. The requirement for COG complex for proper glycosylation was demonstrated in model organisms and humans [106,109,128,129,130,131,132,133,134]. Several groups studied the effects of siRNA-mediated knockdown (KD) of individual COG subunits in HeLa cells in studies that complemented the analysis of patients’ fibroblasts [106,111,133,135,136]. In response to depletion of COG complex subunits, the entire COG complex is non-functional and several glycosylation enzymes including medial- (MGAT1 and MAN2A1), and trans-Golgi (B4GALT1 and ST6GAL1)- glycosyltransferases were severely mislocalized [106,133]. N-glycan profiling revealed decreased levels of sialylation in COG3 and COG4 KDs [133]. Another study reported defects of terminal sialylation in COG3 and COG7 KD [136]. More recently the CRISPR/Cas9 approach was used to generate HEK293T knockout cell lines missing individual COG subunits. KO cell lines displayed retrograde trafficking defects accompanied by sialylation and fucosylation alterations, albeit with effects that varied with the affected subunit [110].
The effects of COG mutations have been studied in many model organisms including yeast, worms and fruit flies, and several phenotypic effects at the cellular and organismal levels have been reported [103]. The loss of COG1 (cog1/LDLB cells) or COG2 (cog2/LDLC cells) in Chinese hamster ovary (CHO) cells disrupts Golgi structure, resulting in defects of protein sorting and secretion and immature of N-, O-, and glycolipid glycosylation with reduced sialic acid in glycans [97,106,128,137,138]. Glycosylation defects attributed to Golgi trafficking also characterized the phenotypes of COG mutants in S. cerevisiae at the restrictive temperature [130,139,140,141,142]. In C. elegans COG1 and COG3 proteins are required for normal glycosylation and gonadal localization of MIG-17 protease, a member of the ADAM family proteins, which controls migration of gonadal distal tip cells [143]. Structural analysis of N-glycans by MS showed that cog1 mutant failed to form the fucose-rich N-glycans and to add fucose to residues on N-glycans. MS analysis also revealed an increase in high mannose and paucimmanose glycans in C. elegans cog1 mutants [129]. The Drosophila homologues of human COG5, and COG7 are essential for male fertility, normal cytokinesis and spermatid differentiation [144,145,146]. Loss-of-function Cog7 mutations reduce life span and cause psychomotor defects in Drosophila similar to COG7-CDG patients [134]. Analysis of N-linked glycoprotein glycans by multidimensional ion trap mass spectrometry in heads of Drosophila Cog7 mutants, revealed altered N-linked glycome profiles with an increased abundance of high mannose and paucimannose type glycans and of a family of neural-specific, difucosylated N-glycans. Additionally, Cog7 mutants displayed hyposialylation. Thus, the phenotypic traits of the Drosophila COG7-CDG disease model parallel the pathological characteristics of COG7-CDG patients, including neuromotor defects and defective N-glycome profiles. Additionally, Drosophila Cog7 mutants revealed defects in the larval neuromuscular junctions leading to a reduction in bouton numbers. Different defects in larval NMJs were observed in Drosophila Cog1 null mutants, which increased synaptic branch length without affecting bouton density [147]. Comparison of N-glycome alterations in Cog1 null mutants with those of Cog7 mutants should elucidate on the requirements for specific glycans in the larval NMJ architecture. The analysis of COG complex interactome in Drosophila melanogaster confirmed the interaction with Rab1 and allowed to identify GOLPH3 as a novel molecular partner of Cog5 and Cog7 [134,148]. Cog 7 protein is required for Golgi localization of GOLPH3 and facilitates GOLPH3 binding with the coatomer and Rab1 GTPase. These results suggest that the COG complex, besides its role in vesicle tethering, might act together with GOLPH3 to coordinate the incorporation of glycosyltransferases into COPI coated vesicles. Overexpression of Rab1 can rescue the locomotor defects caused by loss of Drosophila Cog7. Taken together, these results suggest that the Drosophila COG7-CDG model can be used to test novel potential therapeutic strategies by modulating trafficking pathways.
7. Concluding Remarks
Glycan processing at the Golgi plays an essential role in cellular physiology and differentiation. Defects of N-and O-linked glycans have been linked to multiple human diseases including CDGs, inflammatory diseases and cancer [149]. Cancer cells often display characteristic glycosylation patterns such as abnormal core fucosylation, high mannose N-linked glycans and truncated O-glycans Tn (GalNAcα1-Ser/Thr) and sialyl-Tn (STn) (Neu5Acα2,6GalNAcα1-Ser/Thr) [83,150]. Tumor-associated changes in glycosylation drive cancer progression and metastasis, providing novel diagnostic biomarkers and therapeutic targets [83]. Current applications of sophisticated mass spectrometry approaches in clinical diagnosis coupled with the advances in exome sequencing in patients have contributed to boosting the number of new glycosylation disorders [151].
In this review, we have illustrated how the steps of glycan modifications are intimately connected to secretory trafficking with perturbation in Golgi glycosylation resulting from mutations in proteins required for COPI coat scaffolding, the incorporation of glycosyltransferases in vesicles and vesicle tethering. In the context of N-glycosylation GORAB, GOLPH3 and the COG complex are all required for the normal synthesis of complex glycans, with effects on sialylation of N-glycans. However, these proteins act at different steps of this process, with GORAB acting as a scaffold for the COPI coat complex, GOLPH3 regulating the incorporation of a subset group of glycosyltransferases into COPI vesicles, and the COG complex mostly being involved in vesicle tethering. However further studies should clarify the reciprocal dependence between these vesicle trafficking players in N- and O-glycosylation, and whether they display a specificity towards defined groups of glycosyltransferases and a cell-type limited function. Although the discovery of novel glycosylation disorders is increasing rapidly, genetic variants of important trafficking regulators have not been studied within the context of CDGs to date. For example, GOLPH3 and COG3 have been shown to have a role in glycosylation in model organisms and mammalian cell cultures, but they have not yet been associated with a CDG [65,76,81,106]. We may expect that the potential impact of mutations in some secretory trafficking players on glycosylation is so severe that it is not compatible with viability. Studies on model organisms (e.g., zebrafish, flies, mice) provide a valuable resource with which to dissect the phenotypic consequences of glycosylation disfunction in the whole organism and to identify potential therapeutic strategies for CDGs or cancer aimed at modulating secretory trafficking pathways [152].
Author Contributions
Conceptualization, A.F., A.K.-G., S.S., M.G.G.; writing-original draft preparation; M.G.G. and A.F.; writing-review and editing, M.G.G. and A.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant from Fondazione AIRC per la Ricerca sul Cancro (IG2017, Id.20779) to M.G.G., A.F. was supported by a fellowship from the Fondazione Italiana per la Ricerca sul Cancro-Fondazione AIRC per la Ricerca sul Cancro (Id. 19686).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dennis, J.W.; Lau, K.S.; Demetriou, M.; Nabi, I.R. Adaptive regulation at the cell surface by N-glycosylation. Traffic 2009, 10, 1569–1578. [Google Scholar] [CrossRef]
- Haltiwanger, R.S. Regulation of signal transduction by glycosylation. Int. J. Exp. Pathol. 2004, 85, A49–A50. [Google Scholar] [CrossRef]
- Freeze, H.H.; Ng, B.G. Golgi glycosylation and human inherited diseases. Cold Spring Harb. Perspect. Biol. 2011, 3, a005371. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post- translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef]
- Freeze, H.H. Genetic defects in the human glycome. Nat. Rev. Genet. 2006, 7, 537–551. [Google Scholar] [CrossRef]
- Cummings, R.D. The repertoire of glycan determinants in the human glycome. Mol. Biosyst. 2009, 5, 1087–1104. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Burda, P.; Aebi, M. The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta 1999, 1426, 239–257. [Google Scholar] [CrossRef]
- Li, H.; Chavan, M.; Schindelin, H.; Lennarz, W.J.; Li, H. Structure of the oligosaccharyl transferase complex at 12 A resolution. Structure 2008, 16, 432–440. [Google Scholar] [CrossRef]
- Zielinska, D.F.; Gnad, F.; Wi’sniewski, J.R.; Mann, M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 2010, 141, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Shwartz, F.; Aebi, M. Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef] [PubMed]
- Lederkremer, G.Z. Glycoprotein folding, quality control and ER-associated degradation. Curr. Opin. Struct. Biol. 2009, 19, 515–523. [Google Scholar] [CrossRef]
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [Google Scholar] [CrossRef]
- Rabouille, C.; Hui, N.; Hunte, F.; Kieckbusch, R.; Berger, E.G.; Warren, G.; Nilsson, T. Mapping the distribution of Golgi enzymes involved in the construction of complex oligosaccharides C. J. Cell Sci. 1995, 108, 1617–1627. [Google Scholar] [PubMed]
- Stanley, P. Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 2011, 3, a005199. [Google Scholar] [CrossRef]
- Stanley, P.; Schachter, H.; Taniguchi, N. N-Glycans. In Essentials of Glycobiology; Varki, A., Ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009; pp. 101–114. [Google Scholar]
- Glick, B.S.; Luini, A. Models for Golgi traffic: A critical assessment. Cold Spring Harb. Perspect. Biol. 2011, 3, a005215. [Google Scholar] [CrossRef] [PubMed]
- Fisher, P.; Ungar, D. Bridging the Gap between Glycosylation and Vesicle Traffic. Front. Cell Dev. Biol. 2016, 4, 15. [Google Scholar] [CrossRef]
- Linders, P.; Peters, E.; Ter Beest, M.; Lefeber, D.J.; Van den Bogaart, G. Sugary Logistics Gone Wrong: Membrane Trafficking and Congenital Disorders of Glycosylation. Int. J. Mol. Sci. 2020, 21, 4654. [Google Scholar] [CrossRef]
- Goreta, S.S.; Dabelic, S.; Dumic, J. Insights into complexity of congenital disorders of glycosylation. Biochem. Med. (Zagreb) 2012, 22, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Pantazopoulou, A.; Glick, B.S. A Kinetic View of Membrane Traffic Pathways Can Transcend the Classical View of Golgi Compartments. Front. Cell Dev. Biol. 2019, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Beznoussenko, G.V.; Parashuraman, S.; Rizzo, R.; Polishchuk, R.; Martella, O.; Di Giandomenico, D.; Fusella, A.; Spaar, A.; Sallese, M.; Capestrano, M.G.; et al. Transport of soluble proteins through the Golgi occurs by diffusion via continuities across cisternae. Elife 2014, 3, e02009. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.; Rawet, M.; Wieland, F.T.; Cassel, D. The COPI system: Molecular mechanisms and function. FEBS Lett. 2009, 583, 2701–2709. [Google Scholar] [CrossRef]
- D’Souza-Schorey, C.; Chavrier, P. ARF proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Pucadyil, T.J.; Schmid, S.L. Conserved functions of membrane active GTPases in coated vesicle formation. Science 2009, 325, 1217–1220. [Google Scholar] [CrossRef]
- Park, S.Y.; Yang, J.S.; Schmider, A.B.; Soberman, R.J.; Hsu, V.W. Coordinated regulation of bidirectional COPI transport at the Golgi by cdc42. Nature 2015, 521, 529–532. [Google Scholar] [CrossRef]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Rivinoja, A.; Hassinen, A.; Kokkonen, N.; Kauppila, A.; Kellokumpu, S. Elevated Golgi pH impairs terminal N-glycosylation by inducing mislocalization of Golgi glycosyltransferases. J. Cell Physiol. 2009, 220, 144–154. [Google Scholar] [CrossRef]
- Maeda, Y.; Kinoshita, T. The acidic environment of the Golgi is critical for glycosylation and transport. Methods Enzymol. 2010, 480, 495–510. [Google Scholar] [CrossRef]
- Kornak, U.; Reynders, E.; Dimopoulou, A.; Reeuwijk, J.; Fischer, B.; Rajab, A.; Budde, B.; Nürnberg, P.; Foulquier, F.; Mundlos, A.S. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat. Genet. 2008, 40, 32–34. [Google Scholar] [CrossRef]
- Van Galen, J.; Campelo, F.; Martínez-Alonso, E.; Scarpa, M.; Martínez-Menárguez, J.A.; Malhotra, V. Sphingomyelin homeostasis is required to form functional enzymatic domains at the trans-Golgi network. J. Cell Biol. 2014, 206, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.S.; Hung, C.S.; Huoh, Y.S.; Mousley, C.J.; Stefan, C.J.; Bankaitis, V.; Ferguson, K.M.; Burd, C.G. Local control of phosphatidylinositol 4-phosphate signaling in the Golgi apparatus by Vps74 and Sac1 phosphoinositide phosphatase. Mol. Biol. Cell. 2012, 23, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Bröcker, C.; Engelbrecht-Vandré, S.; Ungermann, C. Multisubunit tethering complexes and their role in membrane fusion. Curr. Biol. 2010, 20, R943–R952. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef]
- Barr, F.; Lambright, D.G. Rab GEFs and GAPs. Curr. Opin. Cell Biol. 2010, 22, 461–470. [Google Scholar] [CrossRef]
- Pfeffer, S.R. Rab GTPase regulation of membrane identity. Curr. Opin. Cell Biol. 2013, 25, 414–419. [Google Scholar] [CrossRef]
- Gillingham, A.K. At the ends of their tethers! How coiled-coil proteins capture vesicles at the Golgi. Biochem. Soc. Trans. 2017, 46, 43–50. [Google Scholar] [CrossRef]
- Munro, S. The Golgin coiled-coil proteins of the Golgi apparatus. Cold Spring Harb. Perspect Biol. 2011, 3, a005256. [Google Scholar] [CrossRef]
- Pfeffer, S.R. Rab GTPase localization and Rab cascades in Golgi transport. Biochem. Soc. Trans. 2012, 40, 1373–1377. [Google Scholar] [CrossRef]
- Lowe, M. The Physiological Functions of the Golgin Vesicle Tethering Proteins. Front. Cell Dev. Biol. 2019, 7, 94. [Google Scholar] [CrossRef]
- Witkos, T.M.; Lowe, M. The golgin family of coiled-coil tethering proteins. Front. Cell Dev. Biol. 2015, 3, 86. [Google Scholar] [CrossRef]
- Yu, I.-M.; Hughson, F.M. Tethering factors as organizers of intracellular vesicular traffic. Annu. Rev. Cell Dev. Biol. 2010, 26, 137–156. [Google Scholar] [CrossRef]
- Wong, M.; Munro, S. Membrane trafficking. The specificity of vesicle traffic to the Golgi is encoded in the golgin coiled-coil proteins. Science 2014, 346, 1256898. [Google Scholar] [CrossRef]
- Ungermann, C.; Kümmel, D. Structure of membrane tethers and their role in fusion. Traffic 2019, 20, 479–490. [Google Scholar] [CrossRef]
- Kim, J.J.; Lipatova, Z.; Segev, N. TRAPP Complexes in Secretion and Autophagy. Cell Dev. Biol. 2016, 4, 20. [Google Scholar] [CrossRef]
- Cai, H.; Yu, S.; Menon, S.; Cai, Y.; Lazarova, D.; Fu, C.; Reinisch, K.; Hay, J.C.; Ferro-Novick, S. TRAPPI tethers COPII vesicles by binding the coat subunit Sec23. Nature 2007, 445, 941–944. [Google Scholar] [CrossRef]
- Hong, W.; Lev, S. Tethering the assembly of SNARE complexes. Trends Cell Biol. 2014, 24, 35–43. [Google Scholar] [CrossRef]
- Weber, T.; Zemelman, B.V.; McNew, J.A.; Westermann, B.; Gmachl, M.; Parlati, F.; Söllner, T.H.; Rothman, J.E. SNAREpins: Minimal machinery for membrane fusion. Cell 1998, 92, 759–772. [Google Scholar] [CrossRef]
- Hua, Y.; Scheller, R.H. Three SNARE complexes cooperate to mediate membrane fusion. Proc. Natl. Acad. Sci. USA 2001, 98, 8065–8070. [Google Scholar] [CrossRef] [PubMed]
- Rizo, J.; Südhof, T.C. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices--guilty as charged? Annu. Rev. Cell Dev. Biol. 2012, 28, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Dulubova, I.; Min, S.W.; Chen, X.; Rizo, J.; Südhof, T.C. Sly1 binds to Golgi and ER syntaxins via a conserved N-terminal peptide motif. Dev. Cell 2002, 2, 295–305. [Google Scholar] [CrossRef]
- Carr, C.M.; Grote, E.; Munson, M.; Hughson, F.M.; Novick, P.J. Sec1p binds to SNARE complexes and concentrates at sites of secretion. J. Cell Biol. 1999, 146, 333–344. [Google Scholar] [CrossRef]
- Togneri, J.; Cheng, Y.-S.; Munson, M.; Hughson, F.M.; Carr, C.M. Specific SNARE complex binding mode of the Sec1/Munc-18 protein, Sec1p. Proc. Natl. Acad. Sci. USA 2006, 103, 17730–17735. [Google Scholar] [CrossRef]
- Lobingier, B.T.; Merz, A.J. Sec1/Munc18 protein Vps33 binds to SNARE domains and the quaternary SNARE complex. Biol. Cell 2012, 23, 4611–4622. [Google Scholar] [CrossRef] [PubMed]
- Sechi, S.; Frappaolo, A.; Karimpour-Ghahnavieh, A.; Piergentili, R.; Giansanti, M.G. Oncogenic Roles of GOLPH3 in the Physiopathology of Cancer. Int. J. Mol. Sci. 2020, 21, 933. [Google Scholar] [CrossRef]
- Wu, C.C.; Taylor, R.S.; Lane, D.R.; Ladinsky, M.S.; Weisz, J.A.; Howell, K.E. GMx33: A novel family of trans-Golgi proteins identified by proteomics. Traffic 2000, 1, 963–975. [Google Scholar] [CrossRef]
- Bell, A.W.; Ward, M.A.; Blackstock, W.P.; Freeman, H.N.; Choudhary, J.S.; Lewis, A.P.; Chotai, D.; Fazel, A.; Gushue, J.N.; Paiement, J.; et al. Proteomics characterization of abundant Golgi membrane proteins. J. Biol. Chem. 2001, 276, 5152–5165. [Google Scholar] [CrossRef]
- Dippold, H.C.; Ng, M.M.; Farber-Katz, S.E.; Lee, S.K.; Kerr, M.L.; Peterman, M.C.; Sim, R.; Wiharto, P.A.; Galbraith, K.A.; Madhavarapu, S.; et al. GOLPH3 bridges phosphatidylinositol-4-phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 2009, 139, 337–351. [Google Scholar] [CrossRef]
- Snyder, C.M.; Mardones, G.A.; Ladinsky, M.S.; Howell, K.E. GMx33 associates with the trans-Golgi matrix in a dynamic manner and sorts within tubules exiting the Golgi. Mol. Biol. Cell. 2005, 17, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Sechi, S.; Colotti, G.; Belloni, G.; Mattei, V.; Frappaolo, A.; Raffa, G.D.; Fuller, M.T.; Giansanti, M.G. GOLPH3 Is Essential for Contractile Ring Formation and Rab11 Localization to the Cleavage Site during Cytokinesis in Drosophila melanogaster. PLoS Genet. 2014, 10, e1004305. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.L.; Kabbarah, O.; Liang, M.C.; Ivanova, E.; Anagnostou, V.; Wu, J.; Dhakal, S.; Wu, M.; Chen, S.; Feinberg, T.; et al. GOLPH3 modulates mTOR signaling and rapamycin sensitivity in cancer. Nature 2009, 459, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Eckert, E.; Reckmann, I.; Hellwig, A.; Röhling, S.; El-Battari, A.; Wieland, F.T.; Popoff, V. Golgi phosphoprotein 3 triggers signal-mediated incorporation of glycosyltransferases into coatomer-coated (COPI) vesicles. J. Biol. Chem. 2014, 289, 31319–33129. [Google Scholar] [CrossRef]
- Witkos, T.M.; Chan, W.L.; Joensuu, M.; Rhiel, M.; Pallister, E.; Thomas-Oates, J.; Mould, A.P.; Mironov, A.A.; Lowe, M. GORAB scaffolds COPI at the trans-Golgi for efficient enzyme recycling and correct protein glycosylation. Nat. Commun. 2019, 10, 127. [Google Scholar] [CrossRef]
- Schmitz, K.R.; Liu, J.; Li, S.; Setty, T.G.; Wood, C.S.; Burd, C.G.; Ferguson, K.M. Golgi localization of glycosyltransferases requires a Vps74p oligomer. Dev. Cell 2008, 14, 523–534. [Google Scholar] [CrossRef]
- Tu, L.; Tai, W.C.; Chen, L.; Banfield, D.K. Signal-mediated dynamic retention of glycosyltransferases in the Golgi. Science 2008, 321, 404–407. [Google Scholar] [CrossRef]
- Wood, C.S.; Schmitz, K.R.; Bessman, N.J.; Setty, T.G.; Ferguson, K.M.; Burd, C.G. PtdIns4P recognition by Vps74/GOLPH3 links PtdIns 4-kinase signaling to retrograde Golgi trafficking. J. Cell Biol. 2009, 187, 967–975. [Google Scholar] [CrossRef]
- Tu, L.; Chen, L.; Banfield, D.K. A conserved N-terminal arginine-motif in GOLPH3-family proteins mediates binding to coatomer. Traffic 2012, 13, 1496–1507. [Google Scholar] [CrossRef]
- Ali, M.F.; Chachadi, V.B.; Petrosyan, A.; Pi-Wan, C. Golgi phosphoprotein 3 determines cell binding properties under dynamic flow by controlling Golgi localization of core 2 N-acetylglucosaminyltransferase 1. Biol. Chem. 2012, 287, 39564–39577. [Google Scholar] [CrossRef]
- Ochs, H.D.; Wedgwood, R.J.; Heller, S.R.; Beatty, P.G. Complement, membrane glycoproteins, and complement receptors. Their role in regulation of the immune response. Clin. Immunol. Immunopathol. 1986, 40, 94–104. [Google Scholar] [CrossRef]
- Pereira, N.A.; Pu, H.X.; Goh, H.; Song, Z. Golgi phosphoprotein 3 mediates the Golgi localization and function of protein O-linked mannose β-1,2-N-acetlyglucosaminyltransferase 1. Biol. Chem. 2014, 289, 14762–14770. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.D.; Campbell, K.P. Dystroglycan inside and out. Curr. Opin. Cell Biol. 1999, 11, 602–607. [Google Scholar] [CrossRef]
- Muntoni, F.; Brockington, M.; Godfrey, C.; Ackroyd, M.; Robb, S.; Manzur, A.; Kinali, M.; Mercuri, E.; Kaluarachchi, M.; Feng, L.; et al. Muscular dystrophies due to defective glycosylation of dystroglycan. Acta Myol. 2007, 26, 129–135. [Google Scholar] [PubMed]
- Chang, W.L.; Chang, C.W.; Chang, Y.Y.; Sung, H.H.; Lin, M.D.; Chang, S.C.; Chen, C.H.; Huang, C.W.; Tung, K.S.; Chou, T.B. The Drosophila GOLPH3 homolog regulates the biosynthesis of heparan sulfate proteoglycans by modulating the retrograde trafficking of exostosins. Development 2013, 140, 2798–2807. [Google Scholar] [CrossRef]
- Busse-Wichera, M.; Wicherb, K.B.; Kusche-Gullberg, M. The extostosin family: Proteins with many functions. Matrix Biol. 2014, 35, 25–33. [Google Scholar] [CrossRef]
- Wuyts, W.; Van Hul, W.; De Boulle, K.; Hendrickx, J.; Bakker, E.; Vanhoenacker, F.; Mollica, F.; Lüdecke, H.J.; Sayli, B.S.; Pazzaglia, U.E.; et al. Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am. J. Hum. Genet. 1998, 62, 346–354. [Google Scholar] [CrossRef]
- Bovée, J.V. EXTra hit for mouse osteochondroma. Proc. Natl. Acad. Sci. USA 2010, 107, 1813–1814. [Google Scholar] [CrossRef]
- Jennes, I.; Pedrini, E.; Zuntini, M.; Mordenti, M.; Balkassmi, S.; Asteggiano, C.G.; Casey, B.; Bakker, B.; Sangiorgi, L.; Wuyts, W. Multiple osteochondromas: Mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum. Mutat. 2009, 30, 1620–1627. [Google Scholar] [CrossRef]
- Isaji, T.; Im, S.; Gu, W.; Wang, Y.; Hangm, Q.; Lu, J.; Fukuda, T.; Hashii, N.; Takakura, D.; Kawasaki, N.; et al. An oncogenic protein Golgi phosphoprotein 3 up-regulates cell migration via sialylation. J. Biol. Chem. 2014, 289, 20694–20705. [Google Scholar] [CrossRef]
- Seales, E.C.; Jurado, G.A.; Brunson, B.A.; Wakefield, J.K.; Frost, A.R.; Bellis, S.L. Hypersialylation of beta1 integrins, observed in colon adenocarcinoma, may contribute to cancer progression by up-regulating cell motility. Cancer Res. 2005, 65, 4645–4652. [Google Scholar] [CrossRef] [PubMed]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein Glycosylation in Cancer. Annu. Rev. Pathol. 2015, 10, 473–510. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.; Li, J.; Fang, J.; Xu, Z.; He, X.; Zhang, F.; Ling, J.; Li, X.; Xu, D.; Li, L.; et al. Cloning and characterization of a novel gene which encodes a protein interacting with the mitosis-associated kinase-like protein NTKL. J. Hum. Genet. 2003, 48, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Burman, J.L.; Bourbonniere, L.; Philie, J.; Stroh, T.; Dejgaard, S.Y.; Presley, J.F.; McPherson, P.S. Scyl1, mutated in a recessive form of spinocerebellar neurodegeneration, regulates COPI-mediated retrograde traffic. J. Biol. Chem. 2008, 283, 22774–22786. [Google Scholar] [CrossRef]
- Hamlin, J.N.; Schroeder, L.K.; Fotouhi, M.; Dokainish, H.; Ioannou, M.S.; Girard, M.; Summerfeldt, N.; Melançon, P.; McPherson, P.S. Scyl1 scaffolds class II Arfs to specific subcomplexes of coatomer through the gamma-COP appendage domain. J. Cell. Sci. 2014, 127, 1454–1463. [Google Scholar] [CrossRef]
- Hennies, H.C.; Kornak, U.; Zhang, H.; Egerer, J.; Zhang, X.; Seifert, W.; Kühnisch, J.; Budde, B.; Nätebus, M.; Brancati, F.; et al. Gerodermia osteodysplastica is caused by mutations in SCYL1BP1, a Rab-6 interacting golgin. Nat. Genet. 2008, 40, 1410–1412. [Google Scholar] [CrossRef]
- Hunter, A.G. Is geroderma osteodysplastica underdiagnosed? J. Med. Genet. 1988, 25, 854–857. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Y.; Lu, X.; Wang, Y.; Duan, Y.; Cheng, C.; Shen, A. SCYL1BP1 modulates neurite outgrowth and regeneration by regulating the Mdm2/p53 pathway. Mol. Biol. Cell 2012, 23, 4506–4514. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, D.; Di, Y.; Shi, H.; Rao, H.; Huo, K. A newly identified Pirh2 substrate SCYL1-BP1 can bind to MDM2 and accelerate MDM2 self-ubiquitination. FEBS Lett. 2010, 584, 3275–3278. [Google Scholar] [CrossRef]
- Kovacs, L.; Chao-Chu, J.; Schneider, S.; Gottardo, M.; Tzolovsky, G.; Dzhindzhev, N.S.; Riparbelli, M.G.; Callaini, G.; Glover, D.M. Gorab is a Golgi protein required for structure and duplication of Drosophila centrioles. Nat. Genet. 2018, 50, 1021–1031. [Google Scholar] [CrossRef]
- Egerer, J.; Emmerich, D.; Fischer-Zirnsak, B.; Meierhofer, D.; Tuysuz, B.; Marschner, K.; Sauer, S.; Barr, F.A.; Mundlos, S.; Kornak, U. GORAB Missense Mutations Disrupt RAB6 and ARF5 Binding and Golgi Targeting. J. Investig. Dermatol. 2015, 135, 2368–2376. [Google Scholar] [CrossRef]
- Gleeson, P.A.; Lock, J.G.; Luke, M.R.; Stow, J.L. Domains of the TGN: Coats, tethers and G proteins. Traffic 2004, 5, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.L.; Steiner, M.; Witkos, T.; Egerer, J.; Busse, B.; Mizumoto, S.; Pestka, J.M.; Zhang, H.; Hausser, I.; Khayal, L.A.; et al. Impaired proteoglycan glycosylation, elevated TGF-beta signaling, and abnormal osteoblast differentiation as the basis for bone fragility in a mouse model for gerodermia osteodysplastica. PLoS Genet. 2018, 14, e1007242. [Google Scholar] [CrossRef] [PubMed]
- Roboti, P.; Sato, K.; Lowe, M. The golgin GMAP-210 is required for efficient membrane trafficking in the early secretory pathway. J. Cell Sci. 2015, 128, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Smits, P.; Bolton, A.D.; Funari, V.; Hong, M.; Boyden, E.D.; Lu, L.; Manning, D.K.; Dwyer, N.D.; Moran, J.L.; Prysak, M.; et al. Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210. N. Engl. J. Med. 2010, 362, 206–216. [Google Scholar] [CrossRef]
- Ungar, D.; Oka, T.; Brittle, E.E.; Vasile, E.; Lupashin, V.V.; Chatterton, J.E.; Heuser, E.E.; Krieger, M.; Waters, M.G. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 2002, 157, 405–415. [Google Scholar] [CrossRef]
- Climer, L.K.; Dobretsov, M.; Lupashin, V. Defects in the COG complex and COG-related trafficking regulators affect neuronal Golgi function. Front. Neurosci. 2015, 9, 405. [Google Scholar] [CrossRef]
- Miller, V.J.; Ungar, D. Re’COG’nition at the Golgi. Traffic 2012, 13, 891–897. [Google Scholar] [CrossRef]
- Smith, R.D.; Lupashin, V.V. Role of the conserved oligomeric Golgi (COG) complex in protein glycosylation. Carbohydr. Res. 2008, 343, 2024–2431. [Google Scholar] [CrossRef]
- Fotso, P.; Koryakina, Y.; Pavliv, O.; Tsiomenko, A.B.; Lupashin, V.V. Cog1p plays a central role in the organization of the yeast conserved oligomeric Golgi complex. J. Biol. Chem. 2005, 280, 27613–27623. [Google Scholar] [CrossRef]
- Ungar, D.; Oka, T.; Vasile, E.; Krieger, M.; Hughson, F.M. Subunit architecture of the conserved oligomeric Golgi complex. J. Biol. Chem. 2005, 280, 32729–32735. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, J.B.; D’Souza, Z.; Lupashin, V.V. Maintaining order: COG complex controls Golgi trafficking, processing, and sorting. FEBS Lett. 2019, 593, 2466–2487. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.A.; Yip, C.K.; Walz, T.; Hughson, F.M. Molecular organization of the COG vesicle tethering complex. Nat. Struct. Mol. Biol. 2010, 17, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Willett, R.; Blackburn, J.B.; Climer, L.; Pokrovskaya, I.; Kudlyk, T.; Wang, W.; Lupashin, V. COG lobe B sub-complex engages v-SNARE GS15 and functions via regulated interaction with lobe A sub-complex. Sci. Rep. 2016, 6, 29139. [Google Scholar] [CrossRef]
- Shestakova, A.; Zolov, S.; Lupashin, V. COG complex-mediated recycling of Golgi glycosyltransferases is essential for normal protein glycosylation. Traffic 2006, 7, 191–204. [Google Scholar] [CrossRef]
- Wuestehube, L.J.; Duden, R.; Eun, A.; Hamamoto, S.; Korn, P.; Ram, R.; Schekman, R. New mutants of Saccharomyces cerevisiae affected in the transport of proteins from the endoplasmic reticulum to the Golgi complex. Genetics 1996, 142, 393–406. [Google Scholar]
- Zolov, S.N.; Lupashin, V.V. Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J. Cell Biol. 2005, 168, 747–759. [Google Scholar] [CrossRef]
- Cottam, N.P.; Wilson, K.M.; Ng, B.G.; Korner, C.; Freeze, H.H.; Ungar, D. Dissecting functions of the conserved oligomeric Golgi tethering complex using a cell-free assay. Traffic 2014, 15, 12–21. [Google Scholar] [CrossRef]
- Bailey-Blackburn, J.; Pokrovskaya, I.; Fisher, P.; Ungar, D.; Lupashin, V.V. COG complex complexities: Detailed characterization of a complete set of HEK293T cells lacking individual COG subunits. Front. Cell Dev. Biol. 2016, 4, 23. [Google Scholar] [CrossRef]
- Willett, R.; Ungar, D.; Lupashin, V. The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochem. Cell Biol. 2013, 140, 271–283. [Google Scholar] [CrossRef]
- Laufman, O.; Kedan, A.; Hong, W.; Lev, S. Direct interaction between the COG complex and the SM protein, Sly1, is required for Golgi SNARE pairing. EMBO J. 2009, 28, 2006–2017. [Google Scholar] [CrossRef]
- Laufman, O.; Hong, W.; Lev, S. The COG complex interacts directly with Syntaxin 6 and positively regulates endosome-to-TGN retrograde transport. J. Cell Biol. 2011, 194, 459–472. [Google Scholar] [CrossRef]
- Foulquier, F.; Vasile, E.; Schollen, E.; Callewaert, N.; Raemaekers, T.; Quelhas, D.; Jaeken, J.; Mills, P.; Winchester, B.; Krieger, M.; et al. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc. Natl. Acad. Sci. USA 2006, 103, 3764–3769. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.W.; Matthijs, G.; Sturiale, L.; Garozzo, D.; Wong, K.Y.; Wong, R. COG5-CDG with a mild neurohepatic presentation. JIMD Rep. 2012, 3, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Kranz, C.; Ng, B.G.; Sun, L.; Sharma, V.; Eklund, E.A.; Miura, Y.; Ungar, D.; Lupashin, V.; Winkel, R.D.; Cipollo, J.F.; et al. COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum. Mol. Genet. 2007, 16, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Kodera, H.; Ando, N.; Yuasa, I.; Wada, Y.; Tsurusaki, Y.; Nakashima, M.; Miyake, N.; Saitoh, S.; Matsumoto, N.; Saitsu, H. Mutations in COG2 encoding a subunit of the conserved oligomeric golgi complex cause a congenital disorder of glycosylation. Clin. Genet. 2015, 87, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Lübbehusen, J.; Thiel, C.; Rind, N.; Ungar, D.; Prinsen, B.H.; De Koning, T.J.; Peter, M.; Hasselt, V.; Körner, C. Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Hum. Mol. Genet. 2010, 19, 3623–3633. [Google Scholar] [CrossRef]
- Morava, E.; Zeevaert, R.; Korsch, E.; Huijben, K.; Wopereis, S.; Matthijs, G.; Keymolen, K.; Lefeber, D.J.; De Meirleir, L.; Wevers, R.A. A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur. J. Hum. Genet. 2007, 15, 638–645. [Google Scholar] [CrossRef]
- Ng, B.G.; Kranz, C.; Hagebeuk, E.E.; Duran, M.; Abeling, N.G.; Wuyts, B.; Ungar, D.; Lupashin, V.V.; Hartdorff, C.M.; Poll-The, B.T.; et al. Molecular and clinical characterization of a Moroccan Cog7 deficient patient. Mol. Genet. Metab. 2007, 91, 201–214. [Google Scholar] [CrossRef]
- Paesold-Burda, P.; Maag, C.; Troxler, H.; Foulquier, F.; Kleinert, P.; Schnabel, S.; Baumgartner, M.; Hennet, T. Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Hum. Mol. Genet. 2009, 18, 4350–4356. [Google Scholar] [CrossRef]
- Reynders, E.; Foulquier, F.; Leão Teles, E.; Quelhas, D.; Morelle, W.; Rabouille, C.; Wim, A.; Gert, M. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Hum. Mol. Genet. 2009, 18, 3244–3256. [Google Scholar] [CrossRef] [PubMed]
- Spaapen, L.J.; Bakker, J.A.; Van der Meer, S.B.; Sijstermans, H.J.; Steet, R.A.; Wevers, R.A.; Jaeken, J. Clinical and biochemical presentation of siblings with COG-7 deficiency, a lethal multiple O- and N-glycosylation disorder. J. Inherit. Metab. Dis. 2005, 28, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Steet, R.A.; Bohorov, O.; Bakker, J.; Newell, J.; Krieger, M.; Spaapen, L.; Kornfeld, S.; Freeze, H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004, 10, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Zeevaert, R.; Foulquier, F.; Cheillan, D.; Cloix, I.; Guffon, N.; Sturiale, L.; Garozzo, D.; Matthijsc, M.; Jaeken, J. A new mutation in COG7 extends the spectrum of COG subunit deficiencies. Eur. J. Med. Genet. 2009, 52, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Quental, R.; Azevedo, L.; Matthiesen, R.; Amorim, A. Comparative analyses of the Conserved Oligomeric Golgi (COG) complex in vertebrates. BMC Evol. Biol. 2010, 10, 212. [Google Scholar] [CrossRef] [PubMed]
- Koumandou, V.L.; Dacks, J.B.; Coulson, M.R.; Field, M.C. Control systems for membrane fusion in the ancestral eukaryote; evolution of tethering complexes and SM proteins. MC Evol. Biol. 2007, 7, 29. [Google Scholar] [CrossRef]
- Kingsley, D.M.; Kozarsky, K.F.; Segal, M.; Krieger, M. Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J. Cell Biol. 1986, 102, 1576–1585. [Google Scholar] [CrossRef]
- Struwe, W.B.; Reinhold, V.N. The conserved oligomeric Golgi complex is required for fucosylation of N-glycans in Caenorhabditis elegans. Glycobiology 2012, 22, 863–875. [Google Scholar] [CrossRef]
- Suvorova, E.S.; Duden, R.; Lupashin, V.V. The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. J. Cell Biol. 2002, 157, 631–643. [Google Scholar] [CrossRef]
- Podos, S.D.; Reddy, P.; Ashkenas, J.; Krieger, M. LDLC encodes a brefeldin A-sensitive, peripheral Golgi protein required for normal Golgi function. J. Cell. Biol. 1994, 127, 679–691. [Google Scholar] [CrossRef]
- Steet, R.; Kornfeld, S. COG-7-deficient human fibroblasts exhibit altered recycling of Golgi proteins. Mol. Biol. Cell 2006, 17, 2312–2321. [Google Scholar] [CrossRef] [PubMed]
- Pokrovskaya, I.D.; Willett, R.; Smith, R.D.; Morelle, W.; Kudlyk, T.; Lupashin, V.V. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology 2011, 21, 1554–1569. [Google Scholar] [CrossRef] [PubMed]
- Frappaolo, A.; Sechi, S.; Kumagai, T.; Robinson, S.; Fraschini, R.; Karimpour- Ghahnavieh, A.; Belloni, G.; Piergentili, R.; Tiemeyer, K.T.; Tiemeyer, M.; et al. COG7 deficiency in Drosophila generates multifaceted developmental, behavioral and protein glycosylation phenotypes. J. Cell Sci. 2017, 130, 3637–3649. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Vasile, E.; Penman, M.; Novina, C.D.; Dykxhoorn, D.M.; Ungar, D.; Hughson, F.M.; Krieger, M. Genetic analysis of the subunit organization and function of the conserved oligomeric golgi (COG) complex: Studies of COG5- and COG7-deficient mammalian cells. J. Biol. Chem. 2005, 280, 32736–32745. [Google Scholar] [CrossRef] [PubMed]
- Peanne, R.; Legrand, D.; Duvet, S.; Mir, A.M.; Matthijs, G.; Rohrer, J.; Foulquier, F. Differential effects of lobe A and lobe B of the Conserved Oligomeric Golgi complex on the stability of {beta}1,4-galactosyltransferase 1 and {alpha}2,6-sialyltransferase 1. Glycobiology 2011, 21, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, D.M.; Krieger, M. Receptor-mediated endocytosis of low density lipoprotein: Somatic cell mutants define multiple genes required for expression of surface-receptor activity. Proc. Natl. Acad. Sci. USA 1984, 81, 5454–5458. [Google Scholar] [CrossRef]
- Oka, T.; Ungar, D.; Hughson, F.M.; Krieger, M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol. Biol. Cell 2004, 15, 2423–2435. [Google Scholar] [CrossRef]
- Whyte, J.R.; Munro, S. A yeast homolog of the mammalian mannose 6-phosphate receptors contributes to the sorting of vacuolar hydrolases. Curr. Biol. 2001, 11, 1074–1078. [Google Scholar] [CrossRef]
- Ram, R.J.; Li, B.; Kaiser, C.A. Identification of Sec36p, Sec37p, and Sec38p: Components of yeast complex that contains Sec34p and Sec35p. Mol. Biol. Cell 2002, 13, 1484–1500. [Google Scholar] [CrossRef]
- Conde, R.; Guadalupe, P.; Cueva, R.; Larriba, G. Screening for new yeast mutants affected in mannosylphosphorylation of cell wall mannoproteins. Yeast 2003, 20, 1189–1211. [Google Scholar] [CrossRef]
- Corbacho, I.; Olivero, I.; Hernández, L.M. Identification of low-dye-binding (ldb) mutants of Saccharomyces cerevisiae. S. Yeast Res. 2004, 4, 437–444. [Google Scholar] [CrossRef][Green Version]
- Kubota, Y.; Sano, M.; Goda, S.; Suzuki, N.; Nishiwaki, K. The conserved oligomeric Golgi complex acts in organ morphogenesis via glycosylation of an ADAM protease in C. elegans. Development 2006, 133, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Farkas, R.M.; Giansanti, M.G.; Gatti, M.; Fuller, M.T. The Drosophila Cog5 homologue is required for cytokinesis, cell elongation, and assembly of specialized Golgi architecture during spermatogenesis. Mol. Biol. Cell 2003, 14, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Fári, K.; Takács, S.; Ungár, D.; Sinka, R. The role of acroblast formation during Drosophila spermatogenesis. Biol. Open 2016, 5, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Belloni, G.; Sechi, S.; Riparbelli, M.G.; Fuller, M.T.; Callaini, G.; Giansanti, M.G. Mutations in Cog7 affect Golgi structure, meiotic cytokinesis and sperm development during Drosophila spermatogenesis. J. Cell Sci. 2012, 125, 5441–5452. [Google Scholar] [CrossRef] [PubMed]
- Comstra, H.S.; McArthy, J.; Rudin-Rush, S.; Hartwig, C.; Gokhale, A.; Zlatic, S.A.; Blackburn, J.B.; Werner, E.; Petris, M.; D’Souza, P.; et al. The interactome of the copper transporter ATP7A belongs to a network of neurodevelopmental and neurodegeneration factors. Elife 2017, 6, e24722. [Google Scholar] [CrossRef]
- Sechi, S.; Frappaolo, A.; Fraschini, R.; Capalbo, L.; Gottardo, M.; Belloni, G.; Glover, D.M.; Wainman, A.; Giansanti, M.G. Rab1 interacts with GOLPH3 and controls Golgi structure and contractile ring constriction during cytokinesis in Drosophila melanogaster. Open Biol. 2017, 7, 160257. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Ju, T.; Wang, Y.; Aryal, R.P.; Lehoux, S.D.; Ding, X.; Kudelka, M.R.; Cutler, C.; Zeng, J.; Wang, J.; Sun, X.; et al. Tn and sialyl-Tn antigens, aberrant O-glycomics as human disease markers. Proteomics Clin. Appl. 2013, 7, 618–631. [Google Scholar] [CrossRef]
- Freeze, H.H.; Chong, J.X.; Bamshad, M.J.; Ng, B.G. Solving Glycosylation Disorders: Fundamental Approaches Reveal Complicated Pathways. Am. J. Hum. Genet. 2014, 94, 161–175. [Google Scholar] [CrossRef]
- Frappaolo, A.; Sechi, S.; Kumagai, T.; Karimpour-Ghahnavieh, A.; Tiemeyer, M.; Giansanti, M.G. Modeling Congenital Disorders of N-Linked Glycoprotein Glycosylation in Drosophila melanogaster. Front. Genet. 2018, 9, 436. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).