Molecular Evolution, Neurodevelopmental Roles and Clinical Significance of HECT-Type UBE3 E3 Ubiquitin Ligases

 ,
, 
Abstract
1. Development of the Neocortical Circuits
2. Neuronal Circuit Assembly and Neurodevelopmental Disorders
3. Protein Ubiquitination as a Potent Regulatory Principle
4. Protein Ubiquitination Is a Reversible Post-Translational Modification
5. Uninterrupted Function of E3 Ligases Is Fundamental for Brain Development
6. UBE3 Ligases Are Able to Assemble Different polyUb Chain Types
7. Functional and Pathophysiological Implications of the HECT Domain Organization of UBE3 Ligases
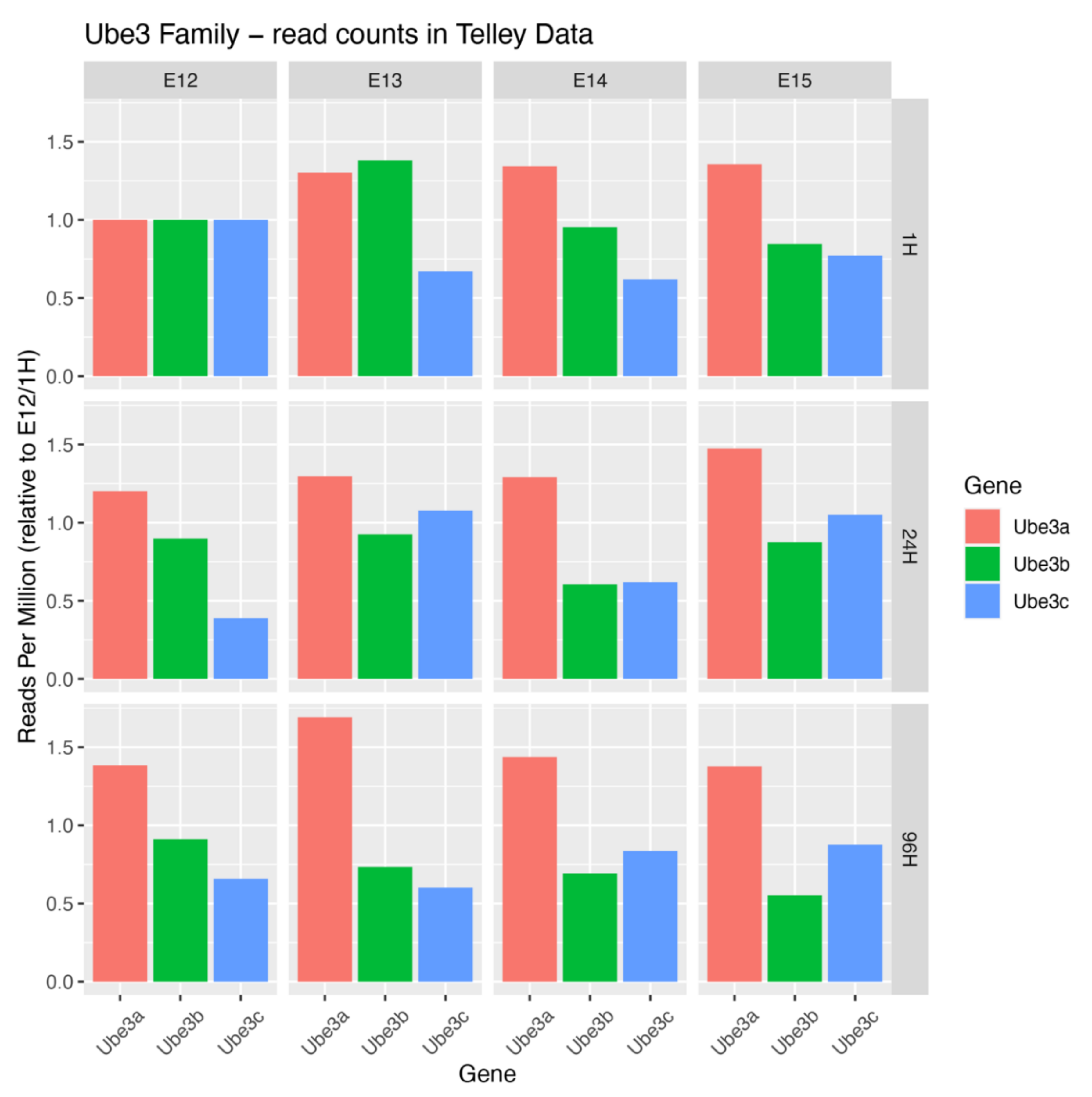
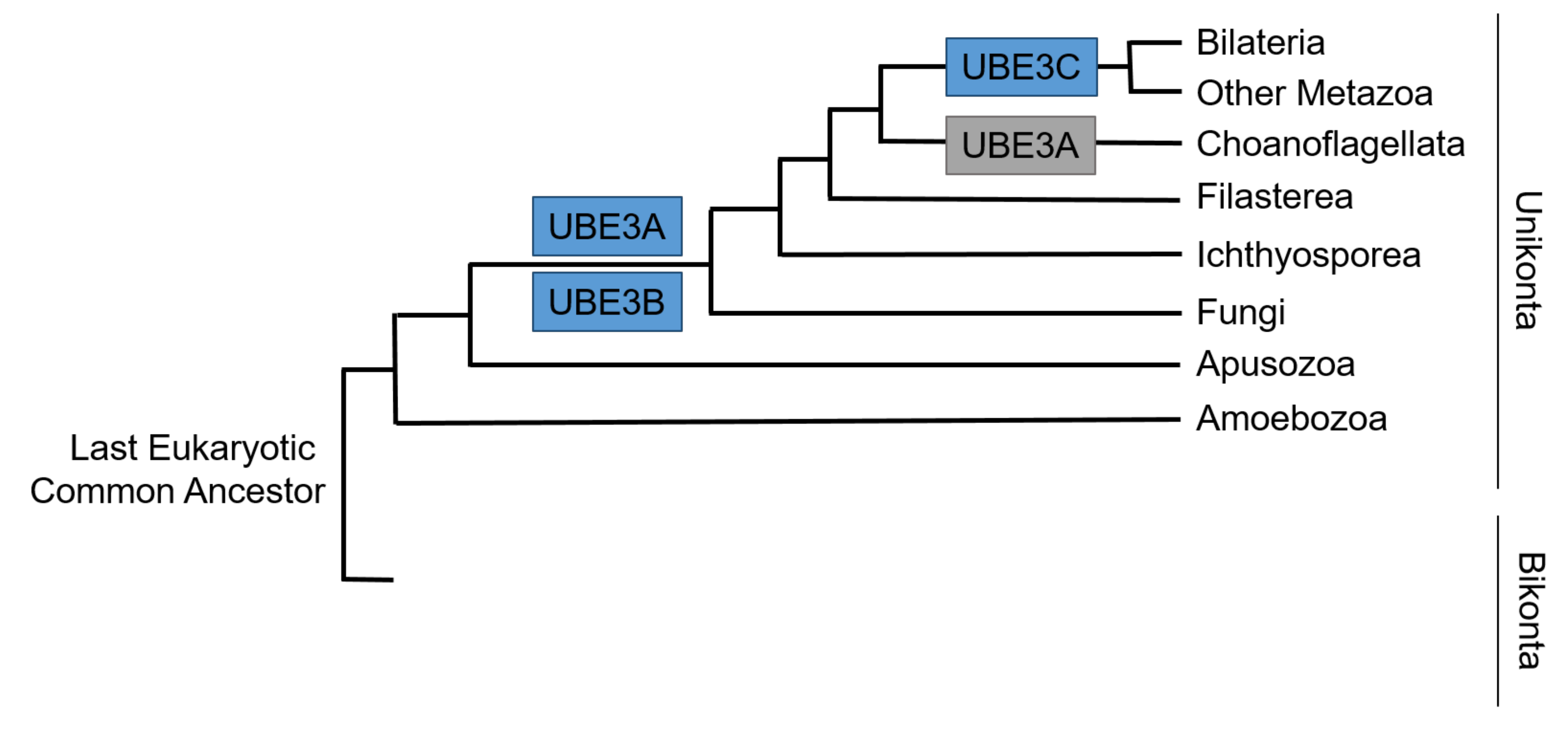
8. Developmental Expression and Molecular Evolution of UBE3 Ligases
9. The Founding Member of HECT Family, E6AP/UBE3A, Angelman Syndrome, and Its Role in the Developing Nervous System
10. Mother Allele-Specific Neuronal Expression of UBE3A
11. Molecular Biology of UBE3A, a Proteasome Regulator and Modulator of Cellular Signaling
12. Isoform-Specific Neuromorphoregulatory Roles of UBE3A
13. UBE3A Acts Locally at the Spine and Globally in the Nucleus to Regulate Synaptic Transmission
14. Critical Developmental Window of UBE3A Expression
15. Neuronal Phenotypes and Physiological Relevance of UBE3A Gain-of-Function
16. The Case of Forward Genetics: Kaufman Oculocerebrofacial Syndrome (KOS) and UBE3B Gene Discovery
17. UBE3B in Non-KOS Diseases
18. Molecular Context of UBE3B Actions in Neurons
19. UBE3B as a Master Regulator of Metabolic Homeostasis
20. Neuronal Roles of UBE3B
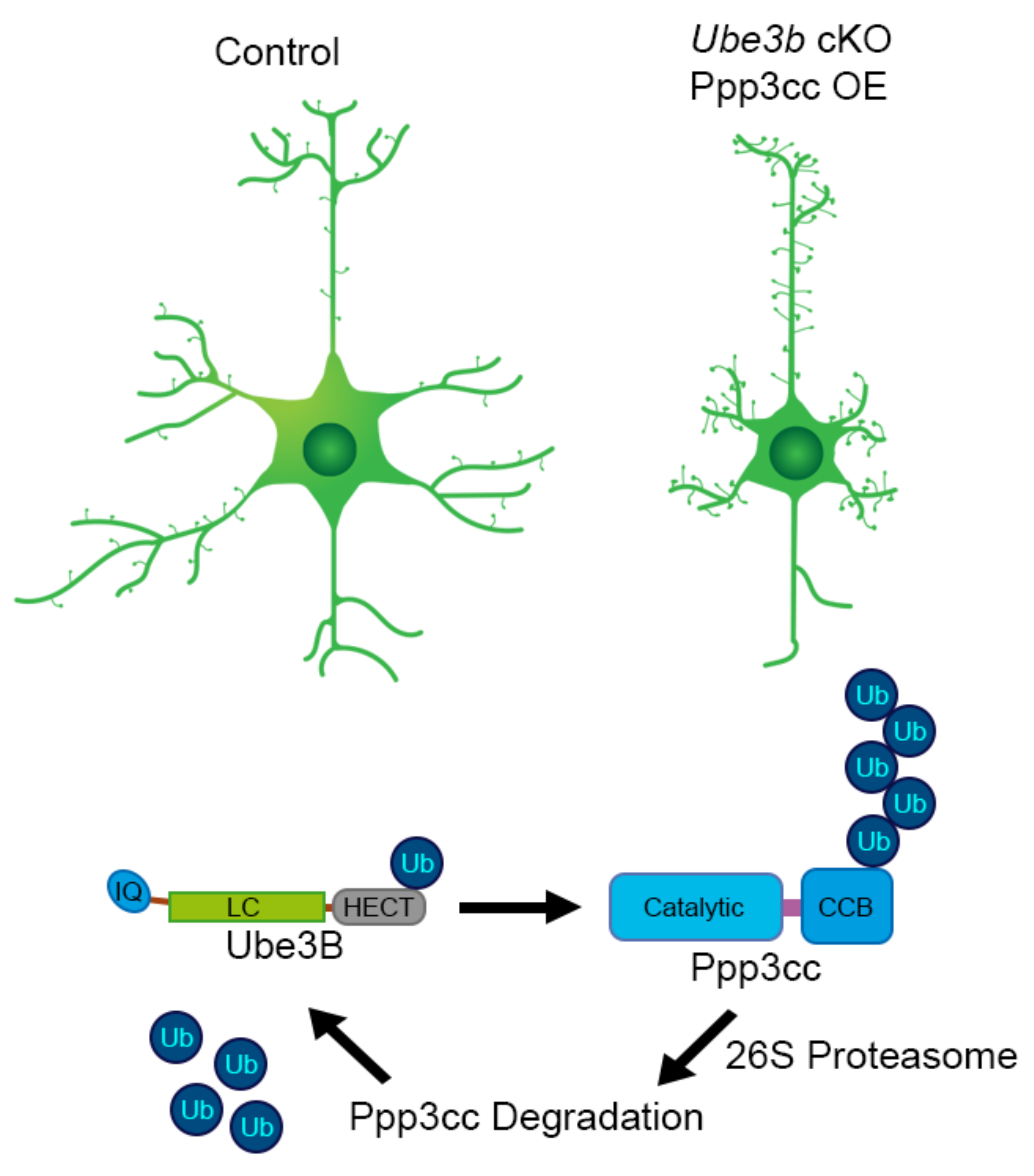
21. Ube3b Conditional and Conventional KO Mouse Models: Lessons Learned
22. Biological Functions and Clinical Importance of UBE3C: More Is Yet to Come
23. UBE3C in Human Disease
24. UBE3C as a Proteasomal Regulator
25. UBE3C Generates K29-Linked Ub Chains
26. Novel Concepts and Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rakic, P. Evolution of the neocortex: A perspective from developmental biology. Nat. Rev. Neurosci. 2009, 10, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, T.J.; Bhaduri, A.; Pollen, A.A.; Alvarado, B.; Mostajo-Radji, M.A.; Di Lullo, E.; Haeussler, M.; Sandoval-Espinosa, C.; Liu, S.J.; Velmeshev, D.; et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 2017, 358, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Telley, L.; Agirman, G.; Prados, J.; Amberg, N.; Fièvre, S.; Oberst, P.; Bartolini, G.; Vitali, I.; Cadilhac, C.; Hippenmeyer, S.; et al. Temporal patterning of apical progenitors and their daughter neurons in the developing neocortex. Science 2019, 364. [Google Scholar] [CrossRef] [PubMed]
- Dierssen, M.; Ramakers, G.J.A. Dendritic pathology in mental retardation: From molecular genetics to neurobiology. Genes Brain Behav. 2006, 5 (Suppl. 2), 48–60. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Lomo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. (Lond.) 1973, 232, 331–356. [Google Scholar] [CrossRef]
- Griffiths, B.J.; Parish, G.; Roux, F.; Michelmann, S.; van der Plas, M.; Kolibius, L.D.; Chelvarajah, R.; Rollings, D.T.; Sawlani, V.; Hamer, H.; et al. Directional coupling of slow and fast hippocampal gamma with neocortical alpha/beta oscillations in human episodic memory. Proc. Natl. Acad. Sci. USA 2019, 116, 21834–21842. [Google Scholar] [CrossRef] [PubMed]
- Buzsáki, G.; Draguhn, A. Neuronal oscillations in cortical networks. Science 2004, 304, 1926–1929. [Google Scholar] [CrossRef]
- Masi, A.; DeMayo, M.M.; Glozier, N.; Guastella, A.J. An Overview of Autism Spectrum Disorder, Heterogeneity and Treatment Options. Neurosci Bull. 2017, 33, 183–193. [Google Scholar] [CrossRef]
- Ambrozkiewicz, M.C.; Kawabe, H. HECT-type E3 ubiquitin ligases in nerve cell development and synapse physiology. Febs Lett. 2015, 589, 1635–1643. [Google Scholar] [CrossRef]
- Kawabe, H.; Brose, N. The role of ubiquitylation in nerve cell development. Nat. Rev. Neurosci. 2011, 12, 251–268. [Google Scholar] [CrossRef]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. THE UBIQUITIN SYSTEM. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, M. Lingering Mysteries of Ubiquitin-Chain Assembly. Cell 2006, 124, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar]
- McDowell, G.S.; Philpott, A. Non-canonical ubiquitylation: Mechanisms and consequences. Int. J. Biochem. Cell Biol. 2013, 45, 1833–1842. [Google Scholar] [CrossRef]
- Kaiser, S.E.; Riley, B.E.; Shaler, T.A.; Trevino, R.S.; Becker, C.H.; Schulman, H.; Kopito, R.R. Protein standard absolute quantification (PSAQ) method for the measurement of cellular ubiquitin pools. Nat. Methods 2011, 8, 691–696. [Google Scholar] [CrossRef]
- McGouran, J.F.; Gaertner, S.R.; Altun, M.; Kramer, H.B.; Kessler, B.M. Deubiquitinating enzyme specificity for ubiquitin chain topology profiled by di-ubiquitin activity probes. Chem. Biol. 2013, 20, 1447–1455. [Google Scholar] [CrossRef]
- Kawabe, H.; Neeb, A.; Dimova, K.; Young, S.M.; Takeda, M.; Katsurabayashi, S.; Mitkovski, M.; Malakhova, O.A.; Zhang, D.-E.; Umikawa, M.; et al. Regulation of Rap2A by the ubiquitin ligase Nedd4-1 controls neurite development. Neuron 2010, 65, 358–372. [Google Scholar] [CrossRef]
- Hsia, H.-E.; Kumar, R.; Luca, R.; Takeda, M.; Courchet, J.; Nakashima, J.; Wu, S.; Goebbels, S.; An, W.; Eickholt, B.J.; et al. Ubiquitin E3 ligase Nedd4-1 acts as a downstream target of PI3K/PTEN-mTORC1 signaling to promote neurite growth. Proc. Natl. Acad. Sci. USA 2014, 111, 13205–13210. [Google Scholar] [CrossRef]
- Ambrozkiewicz, M.C.; Schwark, M.; Kishimoto-Suga, M.; Borisova, E.; Hori, K.; Salazar-Lázaro, A.; Rusanova, A.; Altas, B.; Piepkorn, L.; Bessa, P.; et al. Polarity Acquisition in Cortical Neurons Is Driven by Synergistic Action of Sox9-Regulated Wwp1 and Wwp2 E3 Ubiquitin Ligases and Intronic miR-140. Neuron 2018, 100, 1097–1115.e15. [Google Scholar] [CrossRef]
- George, A.J.; Hoffiz, Y.C.; Charles, A.J.; Zhu, Y.; Mabb, A.M. A Comprehensive Atlas of E3 Ubiquitin Ligase Mutations in Neurological Disorders. Front. Genet. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Hicke, L.; Riezman, H. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell 1996, 84, 277–287. [Google Scholar] [CrossRef]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Reyes-Turcu, F.; Licchesi, J.D.F.; Odenwaelder, P.; Wilkinson, K.D.; Barford, D. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. Embo Rep. 2009, 10, 466–473. [Google Scholar] [CrossRef]
- Kim, H.C.; Huibregtse, J.M. Polyubiquitination by HECT E3s and the Determinants of Chain Type Specificity. Mol. Cell. Biol. 2009, 29, 3307–3318. [Google Scholar] [CrossRef]
- Ambrozkiewicz, M.C.; Borisova, E.; Schwark, M.; Ripamonti, S.; Schaub, T.; Smorodchenko, A.; Weber, A.I.; Rhee, H.J.; Altas, B.; Yilmaz, R.; et al. The murine ortholog of Kaufman oculocerebrofacial syndrome protein Ube3b regulates synapse number by ubiquitinating Ppp3cc. Mol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Michel, M.A.; Elliott, P.R.; Swatek, K.N.; Simicek, M.; Pruneda, J.N.; Wagstaff, J.L.; Freund, S.M.V.; Komander, D. Assembly and specific recognition of k29- and k33-linked polyubiquitin. Mol. Cell 2015, 58, 95–109. [Google Scholar] [CrossRef]
- Kristariyanto, Y.A.; Abdul Rehman, S.A.; Campbell, D.G.; Morrice, N.A.; Johnson, C.; Toth, R.; Kulathu, Y. K29-Selective Ubiquitin Binding Domain Reveals Structural Basis of Specificity and Heterotypic Nature of K29 Polyubiquitin. Mol. Cell 2015, 58, 83–94. [Google Scholar] [CrossRef]
- Huang, L.; Kinnucan, E.; Wang, G.; Beaudenon, S.; Howley, P.M.; Huibregtse, J.M.; Pavletich, N.P. Structure of an E6AP-UbcH7 complex: Insights into ubiquitination by the E2-E3 enzyme cascade. Science (N. Y.) 1999, 286, 1321–1326. [Google Scholar] [CrossRef]
- Kim, H.C.; Steffen, A.M.; Oldham, M.L.; Chen, J.; Huibregtse, J.M. Structure and function of a HECT domain ubiquitin-binding site. Embo Rep. 2011, 12, 334–341. [Google Scholar] [CrossRef]
- Ries, L.K.; Liess, A.K.L.; Feiler, C.G.; Spratt, D.E.; Lowe, E.D.; Lorenz, S. Crystal structure of the catalytic C-lobe of the HECT-type ubiquitin ligase E6AP. Protein Sci. A Publ. Protein Soc. 2020, 29, 1550–1554. [Google Scholar] [CrossRef]
- Ronchi, V.P.; Klein, J.M.; Haas, A.L. E6AP/UBE3A ubiquitin ligase harbors two E2~ubiquitin binding sites. J. Biol. Chem. 2013, 288, 10349–10360. [Google Scholar] [CrossRef]
- Ronchi, V.P.; Klein, J.M.; Edwards, D.J.; Haas, A.L. The active form of E6-associated protein (E6AP)/UBE3A ubiquitin ligase is an oligomer. J. Biol. Chem. 2014, 289, 1033–1048. [Google Scholar] [CrossRef]
- Beasley, S.A.; Kellum, C.E.; Orlomoski, R.J.; Idrizi, F.; Spratt, D.E. An Angelman syndrome substitution in the HECT E3 ubiquitin ligase C-terminal Lobe of E6AP affects protein stability and activity. PLoS ONE 2020, 15, e0235925. [Google Scholar] [CrossRef]
- Chan, A.-L.; Grossman, T.; Zuckerman, V.; Campigli Di Giammartino, D.; Moshel, O.; Scheffner, M.; Monahan, B.; Pilling, P.; Jiang, Y.-H.; Haupt, S.; et al. c-Abl phosphorylates E6AP and regulates its E3 ubiquitin ligase activity. Biochemistry 2013, 52, 3119–3129. [Google Scholar] [CrossRef]
- Basel-Vanagaite, L.; Dallapiccola, B.; Ramirez-Solis, R.; Segref, A.; Thiele, H.; Edwards, A.; Arends, M.J.; Miró, X.; White, J.K.; Désir, J.; et al. Deficiency for the Ubiquitin Ligase UBE3B in a Blepharophimosis-Ptosis-Intellectual-Disability Syndrome. Am. J. Hum. Genet. 2012, 91, 998–1010. [Google Scholar] [CrossRef]
- Zhang, C.; Milunsky, J.M.; Newton, S.; Ko, J.; Zhao, G.; Maher, T.A.; Tager-Flusberg, H.; Bolliger, M.F.; Carter, A.S.; Boucard, A.A.; et al. A Neuroligin-4 Missense Mutation Associated with Autism Impairs Neuroligin-4 Folding and Endoplasmic Reticulum Export. J. Neurosci. 2009, 29, 10843–10854. [Google Scholar] [CrossRef]
- Singh, S.; Sivaraman, J. Crystal Structure of HECT Domain of UBE3C E3 Ligase and its Ubiquitination Activity. Biochem. J. 2020, 477, 905–923. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krummm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef]
- Yuen, R.K.C.; Merico, D.; Cao, H.; Pellecchia, G.; Alipanahi, B.; Thiruvahindrapuram, B.; Tong, X.; Sun, Y.; Cao, D.; Zhang, T.; et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genom. Med. 2016, 1. [Google Scholar] [CrossRef]
- Haynes, C.; Oldfield, C.J.; Ji, F.; Klitgord, N.; Cusick, M.E.; Radivojac, P.; Uversky, V.N.; Vidal, M.; Iakoucheva, L.M. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2006, 2, e100. [Google Scholar] [CrossRef]
- Sato, M. Early origin and evolution of the angelman syndrome ubiquitin ligase gene Ube3a. Front. Cell. Neurosci. 2017, 11, 1–8. [Google Scholar] [CrossRef]
- Marín, I. Animal HECT ubiquitin ligases: Evolution and functional implications. BMC Evol. Biol. 2010, 10, 56. [Google Scholar] [CrossRef]
- Grau-Bové, X.; Sebé-Pedrós, A.; Ruiz-Trillo, I. A genomic survey of HECT ubiquitin ligases in eukaryotes reveals independent expansions of the HECT system in several lineages. Genome Biol. Evol. 2013, 5, 833–847. [Google Scholar] [CrossRef]
- Sadikovic, B.; Fernandes, P.; Zhang, V.W.; Ward, P.A.; Miloslavskaya, I.; Rhead, W.; Rosenbaum, R.; Gin, R.; Roa, B.; Fang, P. Mutation Update for UBE3A variants in Angelman syndrome. Hum. Mutat. 2014, 35, 1407–1417. [Google Scholar] [CrossRef]
- LaSalle, J.M.; Reiter, L.T.; Chamberlain, S.J. Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics 2015, 7, 1213–1228. [Google Scholar] [CrossRef]
- Burette, A.C.; Judson, M.C.; Li, A.N.; Chang, E.F.; Seeley, W.W.; Philpot, B.D.; Weinberg, R.J. Subcellular organization of UBE3A in human cerebral cortex. Mol. Autism 2018, 9, 54. [Google Scholar] [CrossRef]
- Greer, P.L.; Hanayama, R.; Bloodgood, B.L.; Mardinly, A.R.; Lipton, D.M.; Flavell, S.W.; Kim, T.-K.; Griffith, E.C.; Waldon, Z.; Maehr, R.; et al. The Angelman Syndrome Protein Ube3A Regulates Synapse Development by Ubiquitinating Arc. Cell 2010, 140, 704–716. [Google Scholar] [CrossRef]
- Rougeulle, C.; Glatt, H.; Lalande, M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet. 1997, 17, 14–15. [Google Scholar] [CrossRef]
- Vu, T.H.; Hoffman, A.R. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat. Genet. 1997, 17, 12–13. [Google Scholar] [CrossRef]
- Meng, L.; Person, R.E.; Beaudet, A.L. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum. Mol. Genet. 2012, 21, 3001–3012. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Joh, K.; Ohta, T.; Masuzaki, H.; Ishimaru, T.; Mukai, T.; Niikawa, N.; Ogawa, M.; Wagstaff, J.; Kishino, T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum. Mol. Genet. 2003, 12, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, J.S.; Germain, N.D.; Wilderman, A.; Stoddard, C.; Wojenski, L.A.; Villafano, G.J.; Core, L.; Cotney, J.; Chamberlain, S.J. A bipartite boundary element restricts UBE3A imprinting to mature neurons. Proc. Natl. Acad. Sci. USA 2019, 116, 2181–2186. [Google Scholar] [CrossRef]
- Huang, H.-S.; Allen, J.A.; Mabb, A.M.; King, I.F.; Miriyala, J.; Taylor-Blake, B.; Sciaky, N.; Dutton, J.W.; Lee, H.-M.; Chen, X.; et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 2011, 481, 185–189. [Google Scholar] [CrossRef]
- Meng, L.; Ward, A.J.; Chun, S.; Bennett, C.F.; Beaudet, A.L.; Rigo, F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 2015, 518, 409–412. [Google Scholar] [CrossRef]
- Lopez, S.J.; Segal, D.J.; LaSalle, J.M. UBE3A: An E3 Ubiquitin Ligase With Genome-Wide Impact in Neurodevelopmental Disease. Front. Mol. Neurosci. 2019, 11. [Google Scholar] [CrossRef]
- Ramirez, J.; Lectez, B.; Osinalde, N.; Sivá, M.; Elu, N.; Aloria, K.; Procházková, M.; Perez, C.; Martínez-Hernández, J.; Barrio, R.; et al. Quantitative proteomics reveals neuronal ubiquitination of Rngo/Ddi1 and several proteasomal subunits by Ube3a, accounting for the complexity of Angelman syndrome. Hum. Mol. Genet. 2018, 27, 1955–1971. [Google Scholar] [CrossRef]
- Elu, N.; Osinalde, N.; Beaskoetxea, J.; Ramirez, J.; Lectez, B.; Aloria, K.; Rodriguez, J.A.; Arizmendi, J.M.; Mayor, U. Detailed Dissection of UBE3A-Mediated DDI1 Ubiquitination. Front. Physiol. 2019, 10, 534. [Google Scholar] [CrossRef]
- Tomaic, V.; Pim, D.; Thomas, M.; Massimi, P.; Myers, M.P.; Banks, L. Regulation of the Human Papillomavirus Type 18 E6/E6AP Ubiquitin Ligase Complex by the HECT Domain-Containing Protein EDD. J. Virol. 2011, 85, 3120–3127. [Google Scholar] [CrossRef]
- Kühnle, S.; Martínez-Noël, G.; Leclere, F.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Angelman syndrome-associated point mutations in the Zn2+-binding N-terminal (AZUL) domain of UBE3A ubiquitin ligase inhibit binding to the proteasome. J. Biol. Chem. 2018, 293, 18387–18399. [Google Scholar] [CrossRef]
- Yi, J.J.; Paranjape, S.R.; Walker, M.P.; Choudhury, R.; Wolter, J.M.; Fragola, G.; Emanuele, M.J.; Major, M.B.; Zylka, M.J. The autism-linked UBE3A T485A mutant E3 ubiquitin ligase activates the Wnt/β-catenin pathway by inhibiting the proteasome. J. Biol. Chem. 2017, 292, 12503–12515. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.J.; Berrios, J.; Newbern, J.M.; Snider, W.D.; Philpot, B.D.; Hahn, K.M.; Zylka, M.J. An Autism-Linked Mutation Disables Phosphorylation Control of UBE3A. Cell 2015, 162, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Ramirez, J.; Franco, M.; Lectez, B.; Gonzalez, M.; Barrio, R.; Mayor, U. Ube3a, the E3 ubiquitin ligase causing Angelman syndrome and linked to autism, regulates protein homeostasis through the proteasomal shuttle Rpn10. Cell Mol. Life Sci. 2014, 71, 2747–2758. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, K.J.; Nalbandian, A.; Gomez, A.; Wei, D.; Walker, N.; Kimonis, V.E. Administration of CoQ10 analogue ameliorates dysfunction of the mitochondrial respiratory chain in a mouse model of Angelman syndrome. Neurobiol. Dis. 2015, 76, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Noël, G.; Luck, K.; Kühnle, S.; Desbuleux, A.; Szajner, P.; Galligan, J.T.; Rodriguez, D.; Zheng, L.; Boyland, K.; Leclere, F.; et al. Network Analysis of UBE3A/E6AP-Associated Proteins Provides Connections to Several Distinct Cellular Processes. J. Mol. Biol. 2018, 430, 1024–1050. [Google Scholar] [CrossRef]
- Kühnle, S.; Kogel, U.; Glockzin, S.; Marquardt, A.; Ciechanover, A.; Matentzoglu, K.; Scheffner, M. Physical and functional interaction of the HECT ubiquitin-protein ligases E6AP and HERC2. J. Biol. Chem. 2011, 286, 19410–19416. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Huibregtse, J.M.; Howley, P.M. The human E6-AP gene (UBE3A) encodes three potential protein isoforms generated by differential splicing. Genomics 1997, 41, 263–266. [Google Scholar] [CrossRef]
- Miao, S.; Chen, R.; Ye, J.; Tan, G.-H.; Li, S.; Zhang, J.; Jiang, Y.; Xiong, Z.-Q. The Angelman syndrome protein Ube3a is required for polarized dendrite morphogenesis in pyramidal neurons. J. Neurosci. 2013, 33, 327–333. [Google Scholar] [CrossRef]
- Tonazzini, I.; Van Woerden, G.M.; Masciullo, C.; Mientjes, E.J.; Elgersma, Y.; Cecchini, M. The role of ubiquitin ligase E3A in polarized contact guidance and rescue strategies in UBE3A-deficient hippocampal neurons. Mol. Autism 2019, 10, 41. [Google Scholar] [CrossRef]
- Sirois, C.L.; Bloom, J.E.; Fink, J.J.; Gorka, D.; Keller, S.; Germain, N.D.; Levine, E.S.; Chamberlain, S.J. Abundance and localization of human UBE3A protein isoforms. Hum. Mol. Genet. 2020. [Google Scholar] [CrossRef]
- Valluy, J.; Bicker, S.; Aksoy-Aksel, A.; Lackinger, M.; Sumer, S.; Fiore, R.; Wüst, T.; Seffer, D.; Metge, F.; Dieterich, C.; et al. A coding-independent function of an alternative Ube3a transcript during neuronal development. Nat. Neurosci. 2015, 18, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Sadhwani, A.; Sanjana, N.E.; Willen, J.M.; Calculator, S.N.; Black, E.D.; Bean, L.J.H.; Li, H.; Tan, W.-H. Two Angelman families with unusually advanced neurodevelopment carry a start codon variant in the most highly expressed UBE3A isoform. Am. J. Med Genet. A 2018, 176, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kunz, P.A.; Mooney, R.; Philpot, B.D.; Smith, S.L. Maternal Loss of Ube3a Impairs Experience-Driven Dendritic Spine Maintenance in the Developing Visual Cortex. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 4888–4894. [Google Scholar] [CrossRef] [PubMed]
- Margolis, S.S.; Salogiannis, J.; Lipton, D.M.; Mandel-Brehm, C.; Wills, Z.P.; Mardinly, A.R.; Hu, L.; Greer, P.L.; Bikoff, J.B.; Ho, H.-Y.H.; et al. EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell 2010, 143, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lou, S.-S.; Wang, T.; Wu, R.-J.; Li, G.; Zhao, M.; Lu, B.; Li, Y.-Y.; Zhang, J.; Cheng, X.; et al. UBE3A-mediated PTPA ubiquitination and degradation regulate PP2A activity and dendritic spine morphology. Proc. Natl. Acad. Sci. USA 2019, 116, 12500–12505. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, Y.; Jia, Y.; Hao, X.; Lin, W.J.; Tran, J.; Lynch, G.; Baudry, M.; Bi, X. UBE3A-mediated p18/LAMTOR1 ubiquitination and degradation regulate mTORC1 activity and synaptic plasticity. eLife 2018, 7. [Google Scholar] [CrossRef]
- Li, W.; Yao, A.; Zhi, H.; Kaur, K.; Zhu, Y.-C.; Jia, M.; Zhao, H.; Wang, Q.; Jin, S.; Zhao, G.; et al. Angelman Syndrome Protein Ube3a Regulates Synaptic Growth and Endocytosis by Inhibiting BMP Signaling in Drosophila. PLoS Genet. 2016, 12, e1006062. [Google Scholar] [CrossRef]
- Daniel, J.A.; Cooper, B.H.; Palvimo, J.J.; Zhang, F.-P.; Brose, N.; Tirard, M. Analysis of SUMO1-conjugation at synapses. eLife 2017, 6. [Google Scholar] [CrossRef]
- Avagliano Trezza, R.; Sonzogni, M.; Bossuyt, S.N.V.; Zampeta, F.I.; Punt, A.M.; van den Berg, M.; Rotaru, D.C.; Koene, L.M.C.; Munshi, S.T.; Stedehouder, J.; et al. Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat. Neurosci. 2019, 22, 1235–1247. [Google Scholar] [CrossRef]
- Krishnan, V.; Stoppel, D.C.; Nong, Y.; Johnson, M.A.; Nadler, M.J.S.; Ozkaynak, E.; Teng, B.L.; Nagakura, I.; Mohammad, F.; Silva, M.A.; et al. Autism gene Ube3a and seizures impair sociability by repressing VTA Cbln1. Nature 2017, 543, 507–512. [Google Scholar] [CrossRef]
- Sun, A.X.; Yuan, Q.; Fukuda, M.; Yu, W.; Yan, H.; Lim, G.G.Y.; Nai, M.H.; D’Agostino, G.A.; Tran, H.-D.; Itahana, Y.; et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science (N. Y.) 2019, 366, 1486–1492. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhu, G.; Liu, Y.; Standley, S.; Ji, A.; Tunuguntla, R.; Wang, Y.; Claus, C.; Luo, Y.; Baudry, M.; et al. UBE3A Regulates Synaptic Plasticity and Learning and Memory by Controlling SK2 Channel Endocytosis. Cell Rep. 2015, 12, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Sonzogni, M.; Hakonen, J.; Bernabé Kleijn, M.; Silva-Santos, S.; Judson, M.C.; Philpot, B.D.; van Woerden, G.M.; Elgersma, Y. Delayed loss of UBE3A reduces the expression of Angelman syndrome-associated phenotypes. Mol. Autism 2019, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Silva-Santos, S.; van Woerden, G.M.; Bruinsma, C.F.; Mientjes, E.; Jolfaei, M.A.; Distel, B.; Kushner, S.A.; Elgersma, Y. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J. Clin. Investig. 2015, 125, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Rotaru, D.C.; van Woerden, G.M.; Wallaard, I.; Elgersma, Y. Adult Ube3a Gene Reinstatement Restores the Electrophysiological Deficits of Prefrontal Cortex Layer 5 Neurons in a Mouse Model of Angelman Syndrome. J. Neurosci. 2018, 38, 8011–8030. [Google Scholar] [CrossRef]
- Judson, M.C.; Wallace, M.L.; Sidorov, M.S.; Burette, A.C.; Gu, B.; van Woerden, G.M.; King, I.F.; Han, J.E.; Zylka, M.J.; Elgersma, Y.; et al. GABAergic Neuron-Specific Loss of Ube3a Causes Angelman Syndrome-Like EEG Abnormalities and Enhances Seizure Susceptibility. Neuron 2016, 90, 56–69. [Google Scholar] [CrossRef]
- Noor, A.; Dupuis, L.; Mittal, K.; Lionel, A.C.; Marshall, C.R.; Scherer, S.W.; Stockley, T.; Vincent, J.B.; Mendoza-Londono, R.; Stavropoulos, D.J. 15q11.2 Duplication Encompassing Only the UBE3A Gene Is Associated with Developmental Delay and Neuropsychiatric Phenotypes. Hum. Mutat 2015, 36, 689–693. [Google Scholar] [CrossRef]
- Copping, N.A.; Christian, S.G.B.; Ritter, D.J.; Islam, M.S.; Buscher, N.; Zolkowska, D.; Pride, M.C.; Berg, E.L.; LaSalle, J.M.; Ellegood, J.; et al. Neuronal overexpression of Ube3a isoform 2 causes behavioral impairments and neuroanatomical pathology relevant to 15q11.2-q13.3 duplication syndrome. Hum. Mol. Genet. 2017, 26, 3995–4010. [Google Scholar] [CrossRef]
- Khatri, N.; Gilbert, J.P.; Huo, Y.; Sharaflari, R.; Nee, M.; Qiao, H.; Man, H.-Y. The Autism Protein Ube3A/E6AP Remodels Neuronal Dendritic Arborization via Caspase-Dependent Microtubule Destabilization. J. Neurosci. 2018, 38, 363–378. [Google Scholar] [CrossRef]
- Xu, X.; Li, C.; Gao, X.; Xia, K.; Guo, H.; Li, Y.; Hao, Z.; Zhang, L.; Gao, D.; Xu, C.; et al. Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res. 2018, 28, 48–68. [Google Scholar] [CrossRef]
- Kaufman, R.L.; Rimoin, D.L.; Prensky, A.L.; Sly, W.S. An oculocerebrofacial syndrome. Birth Defects Orig. Artic. Ser. 1971, 7, 135–138. [Google Scholar] [PubMed]
- Basel-Vanagaite, L.; Yilmaz, R.; Tang, S.; Reuter, M.S.; Rahner, N.; Grange, D.K.; Mortenson, M.; Koty, P.; Feenstra, H.; Farwell Gonzalez, K.D.; et al. Expanding the clinical and mutational spectrum of Kaufman oculocerebrofacial syndrome with biallelic UBE3B mutations. Hum. Genet. 2014, 133, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Flex, E.; Ciolfi, A.; Caputo, V.; Fodale, V.; Leoni, C.; Melis, D.; Bedeschi, M.F.; Mazzanti, L.; Pizzuti, A.; Tartaglia, M.; et al. Loss of function of the E3 ubiquitin-protein ligase UBE3B causes Kaufman oculocerebrofacial syndrome. J. Med. Genet. 2013, 50, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.S.; Otaify, G.A.; Ismail, S.; Issa, M.Y.; El-Ruby, M.O.; Sadek, A.A.; Ashaat, E.A.; El Saeidi, S.A.; Aglan, M.S.; Temtamy, S.; et al. Blepharophimosis-ptosis-intellectual disability syndrome: A report of nine Egyptian patients with further expansion of phenotypic and mutational spectrum. Am. J. Med. Genet. A 2020. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.I.; Gong, T.-W.; Cho, Y.; Huang, L.; Oh, S.-H.; Adler, H.J.; Raphael, Y.; Altschuler, R.A. Differential Gene Expression Following Noise Trauma in Birds and Mammals. Noise Health 2001, 3, 19–35. [Google Scholar] [PubMed]
- Gong, T.-W.L.; Huang, L.; Warner, S.J.; Lomax, M.I. Characterization of the human UBE3B gene: Structure, expression, evolution, and alternative splicing. Genomics 2003, 82, 143–152. [Google Scholar] [CrossRef]
- Greene, C.C.; McMillan, P.M.; Barker, S.E.; Kurnool, P.; Lomax, M.I.; Burmeister, M.; Lesperance, M.M. DFNA25, a novel locus for dominant nonsyndromic hereditary hearing impairment, maps to 12q21-24. Am. J. Hum. Genet. 2001, 68, 254–260. [Google Scholar] [CrossRef]
- Venhoranta, H.; Pausch, H.; Flisikowski, K.; Wurmser, C.; Taponen, J.; Rautala, H.; Kind, A.; Schnieke, A.; Fries, R.; Lohi, H.; et al. In frame exon skipping in UBE3B is associated with developmental disorders and increased mortality in cattle. BMC Genom. 2014, 15, 890. [Google Scholar] [CrossRef]
- Chahrour, M.H.; Yu, T.W.; Lim, E.T.; Ataman, B.; Coulter, M.E.; Hill, R.S.; Stevens, C.R.; Schubert, C.R.; Greenberg, M.E.; ARRA Autism Sequencing Collaboration; et al. Whole-Exome Sequencing and Homozygosity Analysis Implicate Depolarization-Regulated Neuronal Genes in Autism. PLoS Genet. 2012, 8, e1002635. [Google Scholar] [CrossRef]
- Kohlbrenner, E.A.; Shaskan, N.; Pietersen, C.Y.; Sonntag, K.-C.; Woo, T.-U.W. Gene expression profile associated with postnatal development of pyramidal neurons in the human prefrontal cortex implicates ubiquitin ligase E3 in the pathophysiology of schizophrenia onset. J. Psychiatr Res. 2018, 102, 110–117. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Z.; Chen, X.; Li, J.; Yao, W.; Huang, S.; Gu, A.; Lei, Q.-Y.; Mao, Y.; Wen, W. A multi-lock inhibitory mechanism for fine-tuning enzyme activities of the HECT family E3 ligases. Nat. Commun. 2019, 10, 3162. [Google Scholar] [CrossRef] [PubMed]
- Alexander, K.A.; Cimler, B.M.; Meier, K.E.; Storm, D.R. Regulation of calmodulin binding to P-57. A neurospecific calmodulin binding protein. J. Biol. Chem. 1987, 262, 6108–6113. [Google Scholar] [PubMed]
- Braganza, A.; Li, J.; Zeng, X.; Yates, N.A.; Dey, N.B.; Andrews, J.; Clark, J.; Zamani, L.; Wang, X.-H.; St Croix, C.; et al. UBE3B Is a Calmodulin-regulated, Mitochondrion-associated E3 Ubiquitin Ligase. J. Biol. Chem. 2017, 292, 2470–2484. [Google Scholar] [CrossRef] [PubMed]
- Svilar, D.; Dyavaiah, M.; Brown, A.R.; Tang, J.; Li, J.; McDonald, P.R.; Shun, T.Y.; Braganza, A.; Wang, X.; Maniar, S.; et al. Alkylation sensitivity screens reveal a conserved cross-species functionome. Mol. Cancer Res. 2012, 10, 1580–1596. [Google Scholar] [CrossRef] [PubMed]
- Parkes, J.E.; Rothwell, S.; Day, P.J.; McHugh, N.J.; Betteridge, Z.E.; Cooper, R.G.; Ollier, W.E.; Chinoy, H.; Lamb, J.A. Myositis Genetics Consortium (MYOGEN) Systematic protein-protein interaction and pathway analyses in the idiopathic inflammatory myopathies. Arthritis Res. Ther. 2016, 18, 156. [Google Scholar] [CrossRef]
- Cheon, S.; Kaur, K.; Nijem, N.; Tuncay, I.O.; Kumar, P.; Dean, M.; Juusola, J.; Guillen-Sacoto, M.J.; Bedoukian, E.; Ierardi-Curto, L.; et al. The ubiquitin ligase UBE3B, disrupted in intellectual disability and absent speech, regulates metabolic pathways by targeting BCKDK. Proc. Natl. Acad. Sci. USA 2019, 116, 3662–3667. [Google Scholar] [CrossRef]
- Mazumder, M.K.; Paul, R.; Bhattacharya, P.; Borah, A. Neurological sequel of chronic kidney disease: From diminished Acetylcholinesterase activity to mitochondrial dysfunctions, oxidative stress and inflammation in mice brain. Sci. Rep. 2019, 9, 3097. [Google Scholar] [CrossRef]
- You, J.; Wang, M.; Aoki, T.; Tamura, T.; Pickart, C.M. Proteolytic Targeting of Transcriptional Regulator TIP120B by a HECT Domain E3 Ligase. J. Biol. Chem. 2003, 278, 23369–23375. [Google Scholar] [CrossRef]
- Gu, J.; Mao, W.; Ren, W.; Xu, F.; Zhu, Q.; Lu, C.; Lin, Z.; Zhang, Z.; Chu, Y.; Liu, R.; et al. Ubiquitin-protein ligase E3C maintains non-small-cell lung cancer stemness by targeting AHNAK-p53 complex. Cancer Lett. 2019, 443, 125–134. [Google Scholar] [CrossRef]
- Pan, S.-J.; Zhan, S.-K.; Ji, W.-Z.; Pan, Y.-X.; Liu, W.; Li, D.-Y.; Huang, P.; Zhang, X.-X.; Cao, C.-Y.; Zhang, J.; et al. Ubiquitin-protein ligase E3C promotes glioma progression by mediating the ubiquitination and degrading of Annexin A7. Sci. Rep. 2015, 5, 11066. [Google Scholar] [CrossRef]
- Gottlieb, C.D.; Thompson, A.C.S.; Ordureau, A.; Harper, J.W.; Kopito, R.R. Acute unfolding of a single protein immediately stimulates recruitment of ubiquitin protein ligase E3C (UBE3C) to 26S proteasomes. J. Biol. Chem. 2019, 294, 16511–16524. [Google Scholar] [CrossRef] [PubMed]
- Besche, H.C.; Sha, Z.; Kukushkin, N.V.; Peth, A.; Hock, E.-M.; Kim, W.; Gygi, S.; Gutierrez, J.A.; Liao, H.; Dick, L.; et al. Autoubiquitination of the 26S proteasome on Rpn13 regulates breakdown of ubiquitin conjugates. Embo J. 2014, 33, 1159–1176. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hayward, G.S. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 2010, 33, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Nomura, N.; Nagase, T.; Miyajima, N.; Sazuka, T.; Tanaka, A.; Sato, S.; Seki, N.; Kawarabayasi, Y.; Ishikawa, K.I.; Tabata, S. Prediction of the coding sequences of unidentified human genes. II. The coding sequences of 40 new genes (KIAA0041-KIAA0080) deduced by analysis of cDNA clones from human cell line KG-1. DNA Res. 1994, 1, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Bayés, A.; van de Lagemaat, L.N.; Collins, M.O.; Croning, M.D.R.; Whittle, I.R.; Choudhary, J.S.; Grant, S.G.N. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 2011, 14, 19–21. [Google Scholar] [CrossRef]
- Diering, G.H.; Nirujogi, R.S.; Roth, R.H.; Worley, P.F.; Pandey, A.; Huganir, R.L. Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science 2017, 355, 511–515. [Google Scholar] [CrossRef]
- Okada, M.; Ohtake, F.; Nishikawa, H.; Wu, W.; Saeki, Y.; Takana, K.; Ohta, T. Liganded ERα Stimulates the E3 Ubiquitin Ligase Activity of UBE3C to Facilitate Cell Proliferation. Mol. Endocrinol. (Baltim. Md.) 2015, 29, 1646–1657. [Google Scholar] [CrossRef]
- Jiang, J.-H.; Liu, Y.-F.; Ke, A.-W.; Gu, F.-M.; Yu, Y.; Dai, Z.; Gao, Q.; Shi, G.-M.; Liao, B.-Y.; Xie, Y.-H.; et al. Clinical significance of the ubiquitin ligase UBE3C in hepatocellular carcinoma revealed by exome sequencing. Hepatology 2014, 59, 2216–2227. [Google Scholar] [CrossRef]
- Nara, M.; Teshima, K.; Watanabe, A.; Ito, M.; Iwamoto, K.; Kitabayashi, A.; Kume, M.; Hatano, Y.; Takahashi, N.; Iida, S.; et al. Bortezomib reduces the tumorigenicity of multiple myeloma via downregulation of upregulated targets in clonogenic side population cells. PLoS ONE 2013, 8, e56954. [Google Scholar] [CrossRef]
- Tang, L.; Yi, X.-M.; Chen, J.; Chen, F.-J.; Lou, W.; Gao, Y.-L.; Zhou, J.; Su, L.-N.; Xu, X.; Lu, J.-Q.; et al. Ubiquitin ligase UBE3C promotes melanoma progression by increasing epithelial-mesenchymal transition in melanoma cells. Oncotarget 2016, 7, 15738–15746. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.L.; Wen, X.F.; Li, R.B.; Jin, Y.C.; Wang, X.L.; Zhou, L.; Chen, H.X. UBE3C promotes growth and metastasis of renal cell carcinoma via activating Wnt/β-catenin pathway. PLoS ONE 2015, 10, e0115622. [Google Scholar] [CrossRef] [PubMed]
- Pasaje, C.F.A.; Kim, J.H.; Park, B.L.; Park, J.S.; Uh, S.T.; Kim, M.K.; Park, C.S.; Shin, H.D. UBE3C genetic variations as potent markers of nasal polyps in Korean asthma patients. J. Hum. Genet. 2011, 56, 797–800. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, L.T.; Jiang, C.Y. MicroRNA Expression Profiles Identify Biomarker for Differentiating the Embolic Stroke from Thrombotic Stroke. BioMed Res. Int. 2018. [Google Scholar] [CrossRef]
- Yang, B.-Z.; Han, S.; Kranzler, H.R.; Farrer, L.A.; Gelernter, J. A genomewide linkage scan of cocaine dependence and major depressive episode in two populations. Neuropsychopharmacology 2011, 36, 2422–2430. [Google Scholar] [CrossRef]
- Garriock, H.A.; Kraft, J.B.; Shyn, S.I.; Peters, E.J.; Yokoyama, J.S.; Jenkins, G.D.; Reinalda, M.S.; Slager, S.L.; McGrath, P.J.; Hamilton, S.P. A Genomewide Association Study of Citalopram Response in Major Depressive Disorder. Biol. Psychiatry 2010, 67, 133–138. [Google Scholar] [CrossRef]
- Filatova, E.V.; Shadrina, M.I.; Alieva, A.K.; Kolacheva, A.A.; Slominsky, P.A.; Ugrumov, M.V. Expression analysis of genes of ubiquitin-proteasome protein degradation system in MPTP-induced mice models of early stages of Parkinson’s disease. Dokl. Biochem. Biophys. 2014, 456, 116–118. [Google Scholar] [CrossRef]
- Papoulidis, I.; Papageorgiou, E.; Siomou, E.; Oikonomidou, E.; Thomaidis, L.; Vetro, A.; Zuffardi, O.; Liehr, T.; Manolakos, E.; Vassilis, P. A patient with partial trisomy 21 and 7q deletion expresses mild Down syndrome phenotype. Gene 2014, 536, 441–443. [Google Scholar] [CrossRef]
- Kuo, C.-L.; Goldberg, A.L. Ubiquitinated proteins promote the association of proteasomes with the deubiquitinating enzyme Usp14 and the ubiquitin ligase Ube3c. Proc. Natl. Acad. Sci. USA 2017, 114, E3404–E3413. [Google Scholar] [CrossRef]
- Chu, B.W.; Kovary, K.M.; Guillaume, J.; Chen, L.; Teruel, M.N.; Wandless, T.J. The E3 ubiquitin ligase UBE3C enhances proteasome processivity by ubiquitinating partially proteolyzed substrates. J. Biol. Chem. 2013, 288, 34575–34587. [Google Scholar] [CrossRef]
- You, J.; Pickart, C.M. A HECT Domain E3 Enzyme Assembles Novel Polyubiquitin Chains. J. Biol. Chem. 2001, 276, 19871–19878. [Google Scholar] [CrossRef] [PubMed]
- Fei, C.; Li, Z.; Li, C.; Chen, Y.; Chen, Z.; He, X.; Mao, L.; Wang, X.; Zeng, R.; Li, L. Smurf1-Mediated Lys29-Linked Nonproteolytic Polyubiquitination of Axin Negatively Regulates Wnt/β-Catenin Signaling. Mol. Cell. Biol. 2013, 33, 4095–4105. [Google Scholar] [CrossRef] [PubMed]
- Karim, M.; Biquand, E.; Declercq, M.; Jacob, Y.; van der Werf, S.; Demeret, C. Nonproteolytic K29-Linked Ubiquitination of the PB2 Replication Protein of Influenza A Viruses by Proviral Cullin 4-Based E3 Ligases. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, T.; Li, X.; Yang, M.; Tang, S.; Zhu, X.; Gu, Y.; Su, X.; Xia, M.; Li, W.; et al. Lys29-linkage of ASK1 by Skp1−Cullin 1−Fbxo21 ubiquitin ligase complex is required for antiviral innate response. eLife 2016, 5. [Google Scholar] [CrossRef]
- Van de Weijer, M.L.; Krshnan, L.; Liberatori, S.; Guerrero, E.N.; Robson-Tull, J.; Hahn, L.; Lebbink, R.J.; Wiertz, E.J.H.J.; Fischer, R.; Ebner, D.; et al. Quality Control of ER Membrane Proteins by the RNF185/Membralin Ubiquitin Ligase Complex. Mol. Cell 2020, 79, 768–781.e7. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambrozkiewicz, M.C.; Cuthill, K.J.; Harnett, D.; Kawabe, H.; Tarabykin, V. Molecular Evolution, Neurodevelopmental Roles and Clinical Significance of HECT-Type UBE3 E3 Ubiquitin Ligases. Cells 2020, 9, 2455. https://doi.org/10.3390/cells9112455
Ambrozkiewicz MC, Cuthill KJ, Harnett D, Kawabe H, Tarabykin V. Molecular Evolution, Neurodevelopmental Roles and Clinical Significance of HECT-Type UBE3 E3 Ubiquitin Ligases. Cells. 2020; 9(11):2455. https://doi.org/10.3390/cells9112455
Chicago/Turabian StyleAmbrozkiewicz, Mateusz C., Katherine J. Cuthill, Dermot Harnett, Hiroshi Kawabe, and Victor Tarabykin. 2020. "Molecular Evolution, Neurodevelopmental Roles and Clinical Significance of HECT-Type UBE3 E3 Ubiquitin Ligases" Cells 9, no. 11: 2455. https://doi.org/10.3390/cells9112455
APA StyleAmbrozkiewicz, M. C., Cuthill, K. J., Harnett, D., Kawabe, H., & Tarabykin, V. (2020). Molecular Evolution, Neurodevelopmental Roles and Clinical Significance of HECT-Type UBE3 E3 Ubiquitin Ligases. Cells, 9(11), 2455. https://doi.org/10.3390/cells9112455