Abstract
Diabetic retinopathy is a sight-threatening complication of diabetes mellitus, affecting millions of people worldwide. From a vascular perspective, diabetic retinopathy compromises the structure and function of the blood–retinal barrier, leading to aberrant angiogenesis and vascular leakage, with consequent loss of vision. This review will delve into the vascular abnormalities caused by diabetic retinopathy in the inner blood–retinal barrier, focusing primarily on retinal endothelial cells. It will then discuss how calcium signalling regulates inner blood–retina barrier function and dysfunction, how calcium channels contribute to the development of diabetic retinopathy, and how studying the components of the calcium toolkit may identify new therapeutic targets.
1. Introduction
Diabetes is a chronic metabolic disease characterised by elevated blood glucose concentrations, mainly related to impaired insulin metabolism [1]. While adipose tissue, skeletal muscles, and the liver are primarily affected by the disease due to insulin resistance, further vascular complications can also impact the heart, brain, and eyes [1,2].
Vision can indeed be significantly impaired when hyperglycaemia progresses to diabetic retinopathy (DR), a prevalent complication of diabetes mellitus [3,4]. DR dramatically affects the properties of retinal blood vessels, which supply the retina with oxygen and nutrients while protecting the neuronal environment from peripheral circulation [5]. Specifically, retinal capillaries form the blood–retinal barrier, where endothelial cells (ECs) are connected by tight junctions that restrict the passage of solutes and fluids between the bloodstream and the parenchyma [6,7]. In DR, the properties of ECs are compromised, thereby enhancing vascular permeability and promoting pathological angiogenesis [5,8].
The molecular mechanisms underlying DR have only been partially elucidated. Vascular endothelial growth factor (VEGF) is a key contributor to the vascular alterations observed in the disease. Under pathological conditions, elevated VEGF levels promote endothelial cell migration and proliferation, angiogenesis, extracellular matrix breakdown, and increased vascular permeability [9,10,11,12]. As a result, intravitreal anti-VEGF injections are the primary treatment for retinal disorders. However, up to 40% of patients show little to no response, and even among responders, resistance or adverse effects often arise, impacting treatment adherence [13,14]. Consequently, alternative therapeutic approaches are being actively explored.
As a universal intracellular messenger, calcium (Ca2+) regulates nearly every aspect of cellular life [15]. Both preclinical and clinical studies highlight that Ca2+ signalling becomes dysregulated in a wide range of pathological conditions, including diabetes and eye diseases [16,17,18,19]. EC functions are heavily dependent on Ca2+ channel activities and associated signalling pathways, which are altered in endothelial dysfunction, making them promising therapeutic targets [20,21,22]. Emerging evidence shows that the Ca2+ channel activity of several retinal cell types is dysregulated in hyperglycaemic conditions, contributing to the vascular alterations observed in DR [23]. This review will discuss the structure and function of the inner blood–retinal barrier (iBRB) in health and disease and describe the role of Ca2+ signalling in iBRB function and dysfunction, pointing to Ca2+ channels as emerging therapeutic targets for diabetic retinopathy.
2. Diabetic Retinopathy
DR is the most recurrent microvascular complication of diabetes mellitus [24], thereby representing a primary cause of vision impairment worldwide. The global prevalence of DR among people with diabetes is one-third, and it is estimated that the number of DR patients will rise from 103.12 million in 2020 to 160.50 million by 2045 [25]. From a vascular perspective, DR can be classified into non-proliferative diabetic retinopathy (NPDR) or proliferative diabetic retinopathy (PDR) [26]. NPDR is graded as mild when patients have at least one retinal microaneurysm, but no other findings; moderate when patients present haemorrhages or microaneurysms in one to three retinal quadrants and/or cotton wool spots, hard exudates, or venous beading; and severe when patients develop intraretinal haemorrhages (>20 in each quadrant), venous beading in two or more quadrants, or an intraretinal microvascular abnormality in one or more quadrants. All these stages are characterised by the absence of neovascularisation, which appears when NPDR progresses to PDR [4,26] (Figure 1). PDR is typified by neovascularisation in one or more regions of the eye, such as the iris, the angle of the anterior chamber, the optic disc or elsewhere in the retina, or by vitreous/pre-retinal haemorrhages [27]. Patients with PDR can also develop two types of fibrotic tissues, namely fibrovascular proliferative tissue and avascular proliferative tissue, which contribute to retinal detachment [28,29]. Different terms are then used to classify DR based on the location of microvascular abnormalities, and the presence and extent of vascular leakage. We can therefore observe diabetic maculopathy when the macula is affected and diabetic macular oedema (DMO), when vascular leakage accumulates and exacerbates into oedema. Although the diagnosis and classification of DR rely on vascular changes, as they are easy to visualise, it is important to highlight that the neuroretina functions start declining during the early stages of diabetes before the occurrence of vascular complications. Alterations include distorted colour vision [30], abnormal cone pathway response [31], loss of contrast sensitivity [32], and changes in the microglia [33,34].

Figure 1.
Vascular abnormalities in diabetic retinopathy. Retinae showing the progression of diabetic retinopathy and the vascular abnormalities associated with each phase of the disease. Non-proliferative diabetic retinopathy (NPDR), proliferative diabetic retinopathy (PDR), and intraretinal microvascular abnormality (IRMA). Created in BioRender. Dragoni, S. (2025) https://BioRender.com/kn96utz accessed on 14 April 2025.
3. The Inner Blood–Retina Barrier
The iBRB is a set of capillaries that supply the retinal layers with oxygen and nutrients, and it is formed by ECs that tightly regulate the exchange of fluids, molecules, and cells between the bloodstream and the parenchyma [5,35,36,37]. ECs lie on a basement membrane and interact with astrocytes, pericytes, smooth muscle cells (SMCs), microglia, and neurons to form the neurovascular unit (NVU) [38] (Figure 2). Specialised properties of ECs minimise both paracellular and transcellular permeability. In the blood vessels of most tissues, ECs are held together by adherens junctions formed by vascular endothelial cadherin (VE-cad) and catenins [39,40,41]. However, in the brain and retina, tight junctions further reinforce these connections, establishing a high-resistance barrier that prevents paracellular transport while maintaining apicobasal polarity [6,42,43]. These tight junctions consist of claudins, MARVEL family transmembrane proteins (such as occludin, tricellulin, and MarvelD3), and junctional adhesion molecules (JAMs) [6,40]. Transcellular permeability at the iBRB is also tightly regulated. In retinal ECs, the transporter MFSD2A is overexpressed, limiting caveolin-mediated transcytosis, whereas plasmalemma vesicle-associated protein (PLVAP), which is important for fenestrae formation, is downregulated, limiting molecular transport across ECs [44,45,46]. Several efflux and solute transporters are highly expressed in retinal ECs. Among efflux transporters, the ATP-binding cassette superfamily G member 2 (ABCG2) and MDR1/P-glycoprotein (PGP), which are among the most abundantly expressed transporters in the iBRB, restrict the access of xenobiotics and exogenous molecules, including steroids. Essential nutrients are instead transported by solute transporters, such as GLUT1 and MCT1, down their concentration gradient, or through receptor-mediated vesicular transport, involving the transferrin receptor and low-density lipoprotein receptors [47,48].
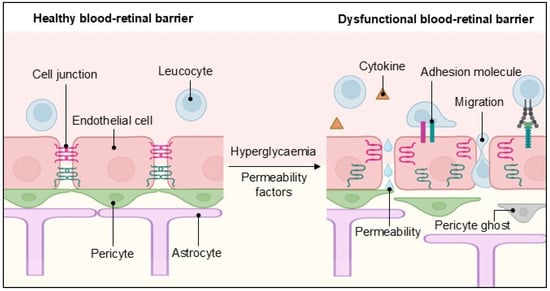
Figure 2.
The inner blood–retinal barrier in health and disease. A schematic representation of the cellular components of the inner blood–retinal barrier and how they change during endothelial dysfunction, leading to vascular permeability, transendothelial migration and cytokine release. Created in BioRender. Dragoni, S. (2025) https://BioRender.com/8l6qtn9 accessed on 17 April 2025.
Additionally, ECs regulate immune responses by expressing lymphocyte adhesion molecules, which mediate leukocyte binding and tissue infiltration. Under normal physiological conditions, ECs exhibit low levels of Intercellular Adhesion Molecule-1 (ICAM-1) and Vascular Cell Adhesion Molecule-1 (VCAM-1), and suppress cytokine-stimulated leukocyte adhesion to endothelium, for example, by secreting TGF-β, thereby minimising leukocyte migration into the retinal environment and preserving the iBRB immune privilege [49]. ECs lie on a basement membrane where pericytes are embedded. Pericytes are phagocytic cells that play a key role in vascular stability by influencing EC growth, vessel remodelling, and angiogenesis [50,51]. Pericytes can uptake fluids, macromolecules, or cell debris by phagocytosis, pinocytosis, and endocytosis [52]. Additionally, pericytes possess contractile properties that help regulate blood flow, a function essential for maintaining the iBRB [53,54,55]. In post-capillary venules, they contribute to immune regulation by expressing chemokines, cytokines, and adhesion molecules that facilitate leukocyte migration [56,57].
Communication among the NVU components is essential for maintaining its integrity. ECs interact with pericytes through gap junctions, membranous interdigitations called “peg-and-socket” connections, and paracrine signalling factors [58], and they share materials and exchange information by transferring microparticles and exosomes between each other. The communication between ECs and pericytes via several signalling pathways is required for angiogenesis. Those include platelet-derived growth factor B/platelet-derived growth factor receptor-β (PDGFB/PDGFR-β) signalling, which induces pericytes to proliferate and migrate toward ECs, thereby guiding pericyte recruitment, transforming growth factor-β/transforming growth factor-β receptor-2 (TGF-β/TGF-βR2) signalling, which is involved in angiogenesis and cell differentiation, as well as Notch pathway and Ang1/Tie2 signalling that regulate vascular maturation, stability, and remodelling [50,51,59]. During retinal development, ECs and pericytes interact with astrocytes to coordinate the expression of tight junction proteins [60].
Astrocytes, as key glial cells, further reinforce the iBRB by secreting inflammatory mediators and trophic factors while also regulating blood flow through neurovascular coupling. Their endfeet tightly envelop ECs, enhancing barrier integrity [61,62]. Astrocytes also contribute to neuronal health by regulating neurotransmitter release, such as glutamate and adenosine. They help maintain ionic and water homeostasis through mechanisms involving Ca2+ signalling, potassium and chloride channels, and aquaporin-4 [63,64]. During retinal vascular development, coordinated signalling between ECs, pericytes, and astrocytes promotes the expression of tight junction proteins necessary for iBRB integrity [60]. Additionally, the basement membrane is a fundamental element in NVU stability, and its thickening disrupts EC interactions with other NVU components, often serving as an early marker of retinal disease [65]. Finally, microglia—macrophage-like cells—are responsible for clearing cellular debris and releasing pro-inflammatory factors [66,67].
4. Endothelial Dysfunction in Diabetic Retinopathy
Disruptions to the NVU structure and function are key factors in the onset and progression of retinal disease. Indeed, during DR, the pericytes of retinal capillaries and the smooth muscle cells of arterioles undergo apoptosis, thereby destabilising the NVU and compromising iBRB integrity [68,69].
From a haemodynamic standpoint, changes in blood flow can be utilised as a diagnostic tool. Despite fluctuations in arterial or intraocular pressure, the retina maintains a constant blood flow through a mechanism known as retinal pressure autoregulation [70]. Impairments in pressure autoregulation may increase retinal capillary pressures, thereby causing shear-induced EC damage [71]. Consistently, abnormalities in retinal blood flow precede the early clinical stages of DR [72,73,74]. These correlate with changes in vessel calibre observed during the progression of DR, where arteriolar vasoconstriction is associated with the early stages of the disease, and is followed by a vasodilation typical of the later stages [75], therefore resulting in reduced and then enhanced blood flow [76], which may relate to the progression to diabetic macular oedema (DMO) and PDR [77,78]. To complete the picture, another critical contributor to the regulation of blood flow is the process of neurovascular coupling, which has been thoroughly discussed elsewhere [79]. In a streptozotocin-induced rat model of type 1 diabetes, abnormal neurovascular coupling contributed to the development of retinopathy through alterations in the nitric oxide (NO) signalling [80].
The breakdown of the iBRB occurs shortly after diabetes induction in animal models [42]. One of its first consequences is certainly vascular leakage, detectable by magnetic resonance imaging, even before clinically recognisable DR lesions appear [81]. The increase in paracellular permeability results from the disruption or decreased expression of cellular junction proteins, which allows fluids and immune cells to cross the barrier, thereby contributing to an inflammatory state (Figure 2).
iBRB breakdown is triggered by the activation of different molecular systems and pathways, including the plasma kallikrein–kinin system (KKS), VEGF, and pro-inflammatory cytokines and chemokines. Bradykinin is a potent vasodilator and a critical component of the plasma KKS [82]. The latter is a proteolytic pathway activated during vascular injury, contributing to inflammation, blood flow, and coagulation [83]. Bradykinin binds to the G protein-coupled receptors B1 and B2 in the plasma membrane of ECs. While B2 is constitutively expressed, B1 is upregulated in injury and inflammatory settings, such as DR [84]. When activated, both receptors lead to the production of NO and prostaglandins, with consequent vessel dilation [85,86]. Bradykinin can also activate Src kinases, leading to the phosphorylation of adherens junction protein VE-cadherin and reversible opening of EC junctions with consequent vascular leakage [87]. Indeed, intravitreal injection of bradykinin or B1 and B2 receptor agonists increased retinal vascular permeability in animal models [84]. Moreover, proteomic studies have identified components of the plasma KKS in the vitreous of patients with advanced DR [88,89].
VEGF, which contributes both to angiogenesis and vascular permeability, is also increased in the retina in DR [90]. Its expression is enhanced by hypoxia and causes a decrease in the expression of ZO1, phosphorylation, ubiquitination and subsequent inactivation of occludin via PKCβ, and phosphorylation of VE-cadherin [87], thereby increasing paracellular permeability.
Hyperglycaemic conditions trigger the synthesis of advanced glycation end products (AGEs) and reactive oxygen species (ROS), which activate the transcription factor NF-κB, with consequent expression of pro-inflammatory molecules [91]. Among those, TNF-α leads to the downregulation of claudin-5 and ZO-1, via PKCζ activation [91]. Cytokine IL-1B, which is found in high levels in the diabetic rat retina, induces iBRB breakdown by leukocyte recruitment and histamine release [92], whereas chemokine CCL2, which was found to be higher in the vitreous of DR patients [93], recruits monocytes, which in turn secrete growth factors and pro-inflammatory mediators in a positive feedback manner [94]. Under hyperglycaemic conditions, leukocyte adhesion molecules such as ICAM1 are upregulated, and metalloproteinases, such as MMP-9 and MMP-2, are activated and cause the shedding of the glycocalyx, which covers the luminal surface of ECs and prevents the adhesion of leukocytes and platelets [95,96].
Pericyte loss, considered an early hallmark of DR, contributes to alterations in iBRB structural integrity and precedes other vascular abnormalities, such as the formation of acellular capillaries [97]. Pericyte dropout from vascular capillaries leads to EC dysfunction, with consequent vascular leakage, blood vessel dilation, and formation of microaneurysms [58,98,99]. The molecular mechanisms underlying pericyte loss remain elusive. However, several mechanisms—including the formation and accumulation of AGEs [100], inflammation, ROS production [101,102], and related signalling pathways—have been correlated with pericyte loss in DR. Pericyte death in diabetes may also result from an uncontrolled immune response, as illustrated by an in vitro study demonstrating that retinal autoantibodies can induce pericyte death by activating the complement system [103].
Finally, structural alterations of the basement membrane are observed in the retinal vasculature of both diabetic animals and patients [65], impairing communication between ECs and pericytes and EC autoregulation.
When enhanced permeability persists, fluid accumulation in the retina overpowers reabsorption mechanisms, leading to the development of DMO.
5. Calcium Signalling in the Blood–Retinal Barrier
A growing number of investigations unravelled the role of endothelial Ca2+ signalling at the blood–brain barrier (BBB) [104,105,106,107], which plays a similar role as the iBRB by maintaining the proper microenvironment for neurons to function. At the BBB, an array of intra- and intercellular Ca2+ signals enable microvascular ECs to detect neuronal activity and synaptic transmission, to regulate local changes in cerebral blood flow, to influence BBB permeability, and to undergo angiogenesis [104,107,108,109,110]. Endothelial Ca2+ signals can be triggered by inositol-1,4,5-trisphosphate (InsP3)-dependent Ca2+ release from the endoplasmic reticulum (ER) [111,112] and lysosomal Ca2+ mobilisation through Two-Pore Channels (TPCs) or Transient Receptor Potential (TRP) Mucolipin 1 (TRPML1) channels [113,114], whereas the following reduction in the ER Ca2+ concentration leads to the activation of store-operated channels (SOCs) [114]. SOCE in cerebrovascular ECs has been associated with STIM and Orai proteins [115], which underpin the majority of endothelial SOCs throughout peripheral circulation [116]. TRP channels provide an additional means for Ca2+ entry pathway at the BBB [104,105]. TRP channels are non-selective cation channels which are, with a few exceptions, permeable to both Na+ and Ca2+, thereby regulating the endothelial function by inducing both membrane depolarisation and an increase in intracellular Ca2+ concentration ([Ca2+]i) [104,105,117,118]. Endothelial TRP channels at the BBB can be gated by Gq protein-coupled receptors (GqPCRs) [119,120], neuronal activity [110], reactive oxygen species [121], and dietary agonists [115]. Finally, cerebrovascular ECs are sensitive to laminar shear stress, which causes an increase in [Ca2+]i by activating the mechanosensitive Piezo 1 channels [122]. Conversely, the role of endothelial Ca2+ signalling at the iBRB is yet to be fully understood [38] and will be the subject of this section.
Agonist-Induced Ca2+ Signals at the iBRB
The resting Ca2+ concentration in vascular ECs ranges between 50 and 100 nM [123,124,125] and is maintained by an intricate network of Ca2+ transporting systems that include the following: plasma membrane Ca2+-ATPase (PMCA), Na+-Ca2+ exchanger (NCX), Sarco-Endoplasmic Reticulum Ca2+-ATPase (SERCA), and mitochondrial Ca2+ uniporter (MCU) [20,126]. The Ca2+ clearing machinery also intervenes to fine-tune the spatiotemporal organisation of the intracellular Ca2+ signals [127,128] and to restore the [Ca2+]i to the basal levels upon the removal of an extracellular signal [129,130]. Only SERCA2 was detected at the iBRB [131], while SERCA1 and SERCA3 are unlikely to be expressed [132]. Similarly, it is unknown which PMCA and NCX isoforms are expressed and how they contribute to endothelial Ca2+ dynamics at the iBRB. The molecular mechanisms shaping the Ca2+ response to physiological agonists are also still unclear. Endothelial agonists stimulate either Gq-protein-coupled receptors (GqPCRs; e.g., acetylcholine, ATP, histamine, glutamate) or tyrosine kinase receptors (TKRs; vascular endothelial growth factor receptor 2 or VEGFR2) to recruit, respectively, phospholipase Cβ (PLCβ) and PLCγ. PLC signalling, in turn, leads to the production of InsP3 and diacylglycerol (DAG) from the membrane-bound precursor phosphatidylinositol 4,5-bisphosphate (PIP2) [20,133]. InsP3Rs are expressed [134], and GqPCR activation with the endothelial autacoid, ATP, can stimulate InsP3 production [135] in retinal ECs. A recent investigation revealed that type 1 InsP3R (InsP3R1) supports the ER-to-mitochondria Ca2+ shuttle, which plays a crucial role in the regulation of endothelial bioenergetics [127], in human retinal ECs [136]. Interestingly, InsP3R1 is physically tethered to the voltage-dependent anion channel 1 (VDAC1) by the glucose-regulated protein 75 (GRP-75) and leads to an MCU-dependent increase in mitochondrial Ca2+ concentration [136]. It is, however, still unclear whether InsP3R2 and InsP3R3, which support ER Ca2+ release at the BBB [107], are also expressed at the iBRB. Similarly, the role of TPCs and TRPML1 at this location remains unclear. The purinergic receptor P2 × 4, which was also sporadically detected in acidic vesicles [137], can support retinal neo-angiogenesis [138] but whether this is due to lysosomal Ca2+ mobilisation is yet to be demonstrated.
The retinal angiogenesis assay has long been used as a proxy of the physiological roles played by specific components of the Ca2+ machinery previously identified in cultured vascular ECs. VEGF has long been known to stimulate sprouting angiogenesis through an increase in endothelial [Ca2+]i [133]. In accord, VEGF induces intracellular Ca2+ oscillations in human cultured retinal ECs by activating VEGFR2 [139,140,141]. This Ca2+ signature strongly resembles the Ca2+ signal induced by VEGF in circulating endothelial colony forming cells (ECFCs), which is shaped by ER Ca2+ release through InsP3Rs and lysosomal Ca2+ mobilisation through TPCs and maintained by SOCE [142,143]. It was suggested that the rhythmic ER Ca2+ release through InsP3Rs is triggered by lysosomal Ca2+ release through the Ca2+-induced Ca2+ release mechanism, according to the “trigger hypothesis” formulated by Galione and Churchill [144]. Lysosomal TPCs are gated by the intracellular second messenger, nicotinic acid adenine dinucleotide phosphate (NAADP), as also shown in cerebral microvascular endothelial cells [108].
In our opinion, further work is mandatory to assess whether the blockade of InsP3Rs [118], as well as of TPCs [145], affects VEGF-induced Ca2+ waves at the iBRB. The only indirect evidence that VEGF stimulates InsP3-induced ER Ca2+ release in retinal capillary ECs was provided by Galeano-Otero and coworkers, who showed that the pharmacological blockade of SOCE with GSK-7975A impaired retinal angiogenesis in mouse xenografts [146]. GSK-7975A is a reliable blocker of Orai1, which contributes the Ca2+-permeable pore subunit to most endothelial SOCs [116]. According to this model [147,148,149], ER Ca2+ release through InsP3Rs activates STIM proteins, which detect the fall in free ER Ca2+ and translocate to ER–plasma membrane junctions to bind to and gate Orai1. Therefore, the retinal angiogenic assay suggests that both InsP3Rs and SOCE shape VEGF-induced Ca2+ signals in retinal capillary ECs. Future work on cultured retinal capillary endothelial cells is required to confirm this hypothesis, which could hold promising therapeutic perspectives for DR (see next paragraph).
6. Calcium Signalling in Diabetic Retinopathy
The dysregulation of endothelial Ca2+ signalling has long been associated with both cardiovascular disorders [22,129,150,151,152,153], e.g., hypertension, type 2 diabetes, atherosclerosis, obesity, sepsis, and deep vein thrombosis, and cancer [154,155]. The endothelial Ca2+ machinery at the BBB can also be impaired in brain disorders, including Alzheimer’s disease [156,157,158], ischaemic stroke [159], and traumatic brain injury [160]. It is, therefore, not surprising that the endothelial Ca2+ toolkit is remodelled in DR.
6.1. ER-Dependent Ca2+ Signalling at the iBRB Is Impaired by DR
A recent investigation showed that InsP3R1–GRP–75–VDAC1 signalling was enhanced in in vitro models of DR that were obtained by culturing human retinal capillary ECs in the presence of hyperglycaemia or advanced glycosylation end products. This in turn led to mitochondrial Ca2+ overload followed by exaggerated ROS production, decreased mitochondrial membrane potential, cytochrome c release, caspase-3 activation, and apoptosis [136]. The ER-to-mitochondria Ca2+ shuttle is driven by the basal activity of PLCβ that occurs even in the absence of extracellular stimulation [161]. Intriguingly, the Food and Drug Administration (FDA)-approved drug, LiCl, which inhibits PLC [162], was recently proposed as an adjuvant for anti-VEGF therapy in patients affected by DR [163]. Therefore, the InsP3R1–GRP-75–VDAC1 signalling pathway could also be probed in in vivo models of DR to assess its suitability as an alternative target to treat DR patients showing resistance to anti-VEGF therapies. A pioneering investigation suggested that PLC-dependent InsP3 synthesis was not affected under high glucose conditions [135]. However, a subsequent study reported that hyperglycaemia can enhance the expression of Gαq/PLCβ-mediated signalling through the mitogen-activated protein kinase (MAPK)/phosphatidylinositol 3-kinase (PI3K) pathway [164]. Therefore, future work is mandatory to confirm whether the formation of mitochondria-associated ER membranes (MAMs), which provide the physical substrate for the ER-to-mitochondria Ca2+ shuttle, is enhanced by hyperglycaemia. It should also be noted that the exaggerated oxidative burst of single mitochondria can mediate the retrograde activation of the ROS-sensitive InsP3Rs [165,166], thereby boosting ER Ca2+ release. Therefore, we suggest that agonist-induced Ca2+ mobilisation through InsP3Rs could be increased in DR. This hypothesis could also explain the reported increase in NO production and neurovascular coupling, as InsP3 signalling is a primary determinant of endothelial NO synthase (eNOS) at the BBB [111].
In addition to InsP3-induced ER Ca2+ release, hyperglycaemia could also enhance endothelial SOCE [167], thereby potentially impacting on the angiogenic response to VEGF. Early studies showed that VEGF-induced endothelial Ca2+ signals are dysregulated in cancer [168] and primary myelofibrosis [169]. An exaggerated Ca2+ response to VEGF could contribute to explaining the detrimental effects of VEGF on the iBRB structure in DR. Additionally, unveiling the Ca2+ signalling machinery driving VEGF-induced Ca2+ oscillations could suggest alternative targets to circumvent the resistance to anti-VEGF therapies. Several FDA-approved drugs were also shown to inhibit SOCE [170,171], and the pharmacological blockade of SOCE was proposed as an anti-angiogenic strategy in cancer patients resistant to VEGF inhibitors [172,173]. In this scenario, assessing the molecular make-up of endothelial SOCs at the iBRB is also mandatory, as Orai1 may not be the sole Ca2+-permeable channel gated by STIM proteins in response to the ER Ca2+ depletion [116]. In accord, VEGF-induced endothelial SOCE can also be sustained by TRP Canonical 1 (TRPC1) [116] and TRPC4 [174]. Moreover, VEGF-dependent Ca2+ entry can be further sustained by DAG-regulated TRP channels, including TRPC3 [118] and TRPC6 [175].
6.2. Endothelial TRP Channels at the iBRB
TRP channels are the best-characterised Ca2+ entry pathway in retinal capillary ECs [38]. The TRP superfamily includes non-selective cation channels that are variably permeable to monovalent (Na+ and K+) and divalent (Ca2+, Mg2+, Zn2+, Fe2+) cations and are sub-divided into six sub-families based upon their sequence homology: canonical (TRPC1-7), melastatin (TRPM1-8), vanilloid (TRPV1-6), ankyrin (TRPA1), polycystin (TRPP), and TRPML1-3. The TRPP sub-family consists of eight members, but only TRPP2, TRPP3, and TRPP5 can be regarded as true ion channels [176,177,178]. TRP channels modulate the endothelial function by promoting both membrane depolarisation and Ca2+ entry, which can in turn induce InsP3Rs-mediated ER Ca2+ release through Ca2+-induced Ca2+ release [169,179]. Endothelial TRP channels serve as polymodal sensors that integrate multiple physical and chemical signals generated from both the surrounding microenvironment and the circulating blood flow, including second messengers, e.g., DAG and arachidonic acid (AA), reactive oxygen species, e.g., hydrogen peroxide (H2O2) and 4-Hydroxynonenal (4-HNE), intracellular ions, e.g., an increase [Ca2+]i and a decrease in cytosolic Mg2+. Additional endothelial TRP channels are sensitive to dietary agonists, e.g., capsaicin, menthol, and allyl isothiocyanate (AITC); gasotransmitters, e.g., NO and hydrogen sulfide (H2S); changes in temperature; and mechanical deformations of the plasma membrane, e.g., laminar shear stress, membrane stretch, and osmotic swelling [166,177,178,180,181,182,183]. TRP channels are also widely expressed at the BBB [105], where they regulate CBF and endothelial permeability. For instance, in mouse microcirculation, capillary endothelial TRPA1 channels are activated by neuronal activity through 4-HNE production, thereby initiating a slow inter-endothelial Ca2+ wave that increases CBF by stimulating endothelium-dependent hyperpolarisation (EDH) [184]. Similarly, TRPA1 may be activated by 4-HNE [185] or H2S [120] in human cerebrovascular ECs to induce NO release, which is the primary vasorelaxing mechanism in human brain microcirculation [186]. In addition, the endothelial TRPC3 and TRPC4 channels, which are both gated by the second messenger DAG [118], regulate BBB permeability and lead to neuronal disorders when their expression is altered [187,188]. TRPV4 can also regulate the BBB integrity under physiological [189], but not inflammatory [190], conditions. Finally, the mechanosensitive TRPP2 isoform, which serves as a Ca2+-conducting pathway in combination with TRPC1, could drive stretch-induced BBB damage upon traumatic brain injury [191]. These pieces of evidence led to the hypothesis of a critical role for TRP channels at the iBRB [38].
6.3. The Physiopathological Role of Endothelial TRP Channels at the iBRB
While the role of TRP channels in phototransduction is well understood, their expression and function within the retinal vasculature remain poorly characterised [192]. Despite this limited understanding, existing evidence indicates that TRP channels are critically involved in both normal and disease-related processes in the retinal vasculature (Figure 3). RT-PCR analyses have confirmed the presence of all TRP channel subtypes in the whole mouse retina [193]. Specifically, TRPC1, TRPC3, TRPC4, and TRPC6 have been identified in human retinal ECs, while TRPV1 and TRPV4 are expressed not only in human retinal ECs but also in primary bovine retinal ECs and intact retinal vessels in mice and rats. Additionally, retinal VSMCs in rats express TRPC1, TRPM7, TRPV1, TRPV2, TRPV4, and TRPP1 (Dragoni et al., 2025) (Table 1) [38].

Figure 3.
The role of TRP channels in iBRB functions. In retinal ECs, TRPC1/4/5/6, TRPM7 and TRPV1/4 are involved in angiogenesis, whereas Ca2+ entry through TRPV4 induces vascular permeability. In VSMCs, TRPV2 regulates blood flow. Created in BioRender. Dragoni, S. (2025) https://BioRender.com/s9eg1dn, accessed on 18 April 2025.

Table 1.
Endothelial Transient Receptor Potential channels at the inner blood–retinal barrier. AA: arachidonic acid; DAG: diacylglycerol; ERK: extracellular signal-regulated kinase; STIM1: stromal interaction molecule 1; and VSMCs: vascular smooth muscle cells.
TRPV2—which stimulates angiogenesis and modulates permeability at the BBB [206], where it is the most abundant TRP isoform [207]—is critical in fine-tuning autoregulation at the iBRB (Table 1). As mentioned above, diabetes-related impairments in the mechanism of pressure autoregulation lead to elevated capillary pressure, with consequent EC damage [71]. On the vascular smooth muscle cells of retinal arterioles, TRPV2 channels trigger the myogenic response, thereby contributing to blood flow autoregulation [194]. In diabetic rats, downregulation of TRPV2 and its inability to be activated by stretch impair the myogenic reactivity of retinal arterioles [196]. A decreased expression of TRPV2 was also observed in vascular SMCs from diabetic donors. Interestingly, non-diabetic rats heterozygous for TRPV2, besides lacking the myogenic reaction, also develop the vascular abnormalities typical of DR, such as vascular leakage, formation of acellular capillaries, neovascularisation and upregulation of inflammatory factors [196], suggesting the TRPV2 loss can act as a trigger in the onset of DR.
TRPV4 is another member of the TRPV sub-family that controls both vascular leakage and angiogenesis. TRPV4 can be physiologically activated by laminar shear stress [208] or downstream of the endothelial GqPCRs, which stimulate PLCβ to cleave PIP2 and gate TRPV4 [119]. Vascular leakage is a critical feature of DR, and it is caused by loss or malfunction of NVU components with consequent iBRB breakdown [209]. It has been established that Ca2+ entry plays a pivotal role in the onset of leakage [210]. Several studies have investigated the role of TRPV4 in vascular permeability in health and disease, generating interesting but contradictory results. In diabetic rats, activation of TRPV4 participates in the onset of oedema and TRPV4-selective antagonists resolved iBRB breakdown in diabetic rats (Table 1) [198,199]. From a molecular perspective, TRPV4 activation in human retinal ECs resulted in the degradation of the tight junction protein occludin, the disruption of cortical F-actin and the reduction of VE-Cadherin and β-catenin colocalisation, which were associated with a decrease in the monolayer impedance and consequent increased permeability [211]. These results are consistent with the increase in vascular permeability observed in vivo in WT mice after activation of TRPV4 [198]. However, a different study showed a decrease in TRPV4 expression in both bovine retinal ECs exposed to high glucose and retinal vessels of STZ-induced diabetic rats [200]. It should be noted that contradictory results have always characterised the role of TRPV4 in vascular leakage throughout peripheral and pulmonary circulations. One reason might be related to the different cell and animal models used [211] which showed that TRPV4 can protect [212,213] or disrupt [214,215] the integrity of the endothelial monolayer in different vascular beds or based on the level of channel activation in the different components of the NVU [198,216,217]. TRPV4-mediated Ca2+ entry was found to activate eNOS in both mouse [111,119,218] and human [219] cerebrovascular endothelial cells. Future work could assess whether TRPV4 activation at the iBRB also leads to NO production, which could further promote hyperpermeability.
TRPV4 is also critical to retinal angiogenesis in cooperation with TRPV1. Aberrant angiogenesis within the retina leads to the progression of NPDR to PDR [5]. The formation of new blood vessels requires the proliferation and migration of ECs, which then assemble into proper vascular tubes. TRPV1 is less expressed than TRPV4 at the BBB [207], but in the peripheral circulation, TRPV1-mediated Ca2+ entry supports the angiogenic sprouting [183]. TRPV1 and TRPV4 are highly expressed in bovine retinal microvascular ECs, with TRPV1 broadly distributed in the cytoplasm and scattered in the plasma membrane (Table 1) [139], and TRPV4 mainly localised in the plasma membrane (Table 1) [211]. Interestingly, a study found that TRPV1 and TRPV4 form heteromeric channels which regulate the tubulogenic phase of the angiogenesis process in vitro and the neovascularisation in vivo in a model of oxygen-induced retinopathy (OIR) [139]. TRPV1 and TRPV4 are sensitive to multiple physiological agonists (Table 1), but the physiological gating signal for the heteromeric TRPV1/TRPV4 channel in bovine retinal ECs is unknown. Interestingly, they are both activated by PIP2 depletion (Table 1), thereby potentially placing this Ca2+ entry pathway under the control of PLC signalling. A recent investigation demonstrated that TRPV4 is mechanically activated during the pathological neovascularization of the retina [220]. TRPV1 does not serve as a mechanosensory but it could do when assembled with TRPV4.
Retinal angiogenesis can also be supported by TRPC channels, as shown in the remainder of the peripheral circulation [118,221,222]. Indeed, under hyperglycaemic conditions, the expression of TRPC1 and TRPC6 increased, and inhibition of these channels led to a reduction in VEGF expression as well as in proliferation, migration and tube formation of human retinal ECs [202]. Consistently, trpc1/4/5/6 quadruple knockdown mice subjected to 30 weeks of hyperglycaemia were protected from the vascular abnormalities of DR, such as pericyte loss and retinal thinning [23]. TRPC1 and TRPC6 were not reported to assemble into a heteromeric channel and, therefore, are likely to serve as independent Ca2+ entry pathways (Table 1).
Additionally, the OIR model also suggested the critical role of TRPC4 in VEGF-induced angiogenesis [203]. Injection of siRNA against mTRPC4 at P12 in OIR mice decreased retinal neovascularisation and the number of neovascular tufts and avascular areas. At a molecular level, downregulation of TRPC4 via siRNA impaired VEGF-induced migration and tubulogenesis in human retinal ECs and inhibited the activation of the VEGF effectors ERK, p38, and AKT [203]. The mechanism by which VEGF can activate TRPC4 is unclear, but it could involve the STIM1-dependent recruitment of a supermolecular complex including Orai1, TRPC1 and TRPC4, as shown in the pulmonary microcirculation [116,174]. TRPM7, which is sensitive to the intracellular Mg2+ concentration [205], has also been involved in retinal angiogenesis, but it is unclear whether it plays any role in retinopathy (Figure 3).
6.4. Is There a Link Between Neuroinflammation and Impairment of Ca2+ Signalling at the iBRB in DR?
Aberrant Ca2+ signalling in astrocytes has long been known as a crucial player of neuroinflammation by initiating inflammatory immune responses following stress, brain injury, or disease-related triggers [223]. Emerging evidence suggested that the hypoxic conditions associated with DR could also increase the spiking in retinal astrocytes [224]. Dysregulated Ca2+ signalling could then lead to the massive release of pro-angiogenic factors, pro-inflammatory, and pro-fibrotic mediators that promote neurodegeneration and hypervascularization, thereby favouring DR progression [225]. Additionally, the increased production of ROS that occurs in the retinal microenvironment could promote neurodegeneration by activating ROS-sensitive Ca2+ signalling pathways and inducing cytosolic Ca2+ overload [226,227]. Intriguingly, excessive Ca2+ signalling could in turn further increase redox signalling and exacerbate ROS-induced oxidative stress [227]. Conversely, it is still unclear whether neuroinflammation may interfere with the endothelial Ca2+ machinery at the iBRB. Clearly, the massive release of VEGF from gliotic Müller cells will stimulate aberrant angiogenesis through an increase in [Ca2+]i, as discussed above. Neuroinflammation is also sustained by multiple neurovascular mediators that are released by Müller cells under hyperglycaemic conditions, including interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α) [225]. IL-6 and TNF-α were shown to increase the resting [Ca2+]i in endothelial cells from other vascular beds [228,229,230]. Future work will have to assess whether these pro-inflammatory cytokines can also disrupt the intracellular Ca2+ dynamics and the Ca2+-dependent functions, i.e., barrier integrity and NO production, at the iBRB. Interestingly, a recent study showed that complement component C3 disrupts the BBB integrity through the C3a/C3aR signalling pathways, which leads to a robust increase in [Ca2+]i [231]. The complement pathway plays a critical role in the neuroinflammatory process associated with DR [232], but it is still unknown whether it also affects the intracellular Ca2+ homeostasis at the iBRB.
7. Conclusions
The clinical causes and molecular pathways driving diabetic retinopathy have been studied for over five decades. Nevertheless, effective and safe therapies are still far away. Intravitreal injections of anti-VEGF and corticosteroids have offered some hope for managing diabetic macular oedema. However, around 50% of patients show little to no benefit from these therapies [13,14,233]. Moreover, laser photocoagulation, which is the current standard approach for treating proliferative diabetic retinopathy, can lead to retinal damage and vision loss. Therefore, new therapeutic options must be investigated.
Dysregulation of Ca2+ channels characterises a plethora of diseases, and Ca2+ channel blockers are the standard treatment for certain cardiovascular conditions, such as hypertension and arrhythmia. Targeting the intracellular Ca2+ toolkit is currently under intense investigation in the search for alternative treatment of life-threatening disorders, such as cancer and severe combined immunodeficiency. Ca2+ entry is a pivotal trigger for mechanisms that lead to inner blood–retinal barrier disruption, such as vascular leakage and pathological angiogenesis. Limiting Ca2+ channel activity has been shown to be protective against diabetic retinopathy or hyperglycaemia-induced damage. Therefore, further investigation into the role of the components of the Ca2+ toolkit may prove to be a promising strategy for identifying new therapeutic targets for diabetic retinopathy and eye diseases. A first mandatory step is the characterisation of the angiogenic Ca2+ signalling pathways recruited by VEGF, e.g., InsP3Rs, TPCs, SOCE, and/or TRPC channels. The growing availability of FDA-approved drugs that can be repurposed to block Orai1, which is the primary pore-forming subunit of the endothelial SOCs, could then lead to further investigations assessing their efficacy in in vivo models of DR. The following FDA-drugs were shown to inhibit SOCE: flecainide, propranolol, lithium chloride, leflunomide, and teriflunomide [170,171]. Furthermore, CalciMedica developed a selective Orai1 inhibitor, termed AuxoraTM, that showed a safety profile and tolerability in a Phase 2b clinical trial for the treatment of acute pancreatitis (AP) with accompanying systemic inflammatory response syndrome (SIRS) [234,235]. Additionally, TPCs are also sensitive to several FDA-approved drugs, which were suggested as an alternative approach to prevent SARS-CoV-2 infection during the COVID-19 pandemic [236,237]. The following FDA drugs also target lysosomal TPCs: salmeterol, PF-543, racecadotril [238], pimozide, fluphenazine [239], clomiphene, raloxifene [240], verapamil, diltiazem, and nimodipine [240]. Then, it will be worth of assessing whether endothelial TRP channels could also serve as an effective molecular target to treat DR. Many endogenous mediators and synthetic compounds were found to serve as agonists or inhibitors of TRP-mediated Ca2+ signals. TRPV2 loss could be, at least partially, rescued by TRPV2 agonists, such as cannabidiol and the anti-gout medication probenecid [206,241]. Excessive Ca2+ entry through TRPV4 channels could be hampered by the selective antagonist GSK2798745 [242]. Phase 2 clinical trials demonstrated that GSK2798745 showed a benign safety profile in healthy volunteers and in patients suffering from cardiogenic lung oedema (https://clinicaltrials.gov/study/NCT02497937?term=trpv4&rank=3, accessed on 5 March 2025). Intriguingly, GSK2798745 also blocked TRPV4-mediated Ca2+ entry in human retinal microvascular endothelial cells [243]. Therefore, further pre-clinical and clinical investigations could assess whether the manipulation of SOCE or TRP signalling provides an alternative therapeutic strategy for DR. As anticipated, this approach will benefit of the straightforward elucidation of the pro-angiogenic Ca2+ signalling machinery recruited by VEGF at the iBRB.
Author Contributions
Conceptualization, S.D. and F.M.; writing—original draft preparation, S.D. and F.M.; writing—review and editing, S.D. and F.M.; visualization, S.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Antar, S.A.; Ashour, N.A.; Sharaky, M.; Khattab, M.; Ashour, N.A.; Zaid, R.T.; Roh, E.J.; Elkamhawy, A.; Al-Karmalawy, A.A. Diabetes mellitus: Classification, mediators, and complications; A gate to identify potential targets for the development of new effective treatments. Biomed. Pharmacother. 2023, 168, 115734. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, W.; Liu, J.; Xie, M.; Liu, Q.; Li, S. Vascular complications of diabetes: A narrative review. Medicine 2023, 102, e35285. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Thool, A.R. A Narrative Review of Retinopathy in Diabetic Patients. Cureus 2024, 16, e52308. [Google Scholar] [CrossRef]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.P.; Simo, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef]
- O’Leary, F.; Campbell, M. The blood-retina barrier in health and disease. FEBS J. 2023, 290, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Hudson, N.; Campbell, M. Tight Junctions of the Neurovascular Unit. Front. Mol. Neurosci. 2021, 14, 752781. [Google Scholar] [CrossRef]
- Liebner, S.; Kniesel, U.; Kalbacher, H.; Wolburg, H. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur. J. Cell Biol. 2000, 79, 707–717. [Google Scholar] [CrossRef]
- Cunha-Vaz, J.; Faria de Abreu, J.R.; Campos, A.J. Early breakdown of the blood-retinal barrier in diabetes. Br. J. Ophthalmol. 1975, 59, 649–656. [Google Scholar] [CrossRef]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Ghalehbandi, S.; Yuzugulen, J.; Pranjol, M.Z.I.; Pourgholami, M.H. The role of VEGF in cancer-induced angiogenesis and research progress of drugs targeting VEGF. Eur. J. Pharmacol. 2023, 949, 175586. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Barber, A.J.; Hollinger, L.A.; Wolpert, E.B.; Gardner, T.W. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J. Biol. Chem. 1999, 274, 23463–23467. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Barber, A.J.; Khin, S.; Lieth, E.; Tarbell, J.M.; Gardner, T.W. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: Vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group. Diabetes 1998, 47, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Wallsh, J.O.; Gallemore, R.P. Anti-VEGF-Resistant Retinal Diseases: A Review of the Latest Treatment Options. Cells 2021, 10, 1049. [Google Scholar] [CrossRef]
- Shahzad, H.; Mahmood, S.; McGee, S.; Hubbard, J.; Haque, S.; Paudyal, V.; Denniston, A.K.; Hill, L.J.; Jalal, Z. Non-adherence and non-persistence to intravitreal anti-vascular endothelial growth factor (anti-VEGF) therapy: A systematic review and meta-analysis. Syst. Rev. 2023, 12, 92. [Google Scholar] [CrossRef]
- Bootman, M.D.; Bultynck, G. Fundamentals of Cellular Calcium Signaling: A Primer. Cold Spring Harb. Perspect. Biol. 2020, 12, a038802. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium signalling remodelling and disease. Biochem. Soc. Trans. 2012, 40, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef]
- Koivisto, A.P.; Belvisi, M.G.; Gaudet, R.; Szallasi, A. Advances in TRP channel drug discovery: From target validation to clinical studies. Nat. Rev. Drug Discov. 2022, 21, 41–59. [Google Scholar] [CrossRef]
- Silvestri, R.; Nicoli, V.; Gangadharannambiar, P.; Crea, F.; Bootman, M.D. Calcium signalling pathways in prostate cancer initiation and progression. Nat. Rev. Urol. 2023, 20, 524–543. [Google Scholar] [CrossRef]
- Moccia, F.; Brunetti, V.; Soda, T.; Berra-Romani, R.; Scarpellino, G. Cracking the Endothelial Calcium (Ca2+) Code: A Matter of Timing and Spacing. Int. J. Mol. Sci. 2023, 24, 16765. [Google Scholar] [CrossRef]
- McCarron, J.G.; Wilson, C.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Lee, M.D. Heterogeneity and emergent behaviour in the vascular endothelium. Curr. Opin. Pharmacol. 2019, 45, 23–32. [Google Scholar] [CrossRef]
- Suzuki, Y.; Giles, W.R.; Zamponi, G.W.; Kondo, R.; Imaizumi, Y.; Yamamura, H. Ca2+ signaling in vascular smooth muscle and endothelial cells in blood vessel remodeling: A review. Inflamm. Regen. 2024, 44, 50. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, R.; Schlotterer, A.; Schumacher, D.; Matka, C.; Mathar, I.; Dietrich, N.; Medert, R.; Kriebs, U.; Lin, J.; Nawroth, P.; et al. TRPC proteins contribute to development of diabetic retinopathy and regulate glyoxalase 1 activity and methylglyoxal accumulation. Mol. Metab. 2018, 9, 156–167. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef]
- Teo, Z.L.; Tham, Y.C.; Yu, M.; Chee, M.L.; Rim, T.H.; Cheung, N.; Bikbov, M.M.; Wang, Y.X.; Tang, Y.; Lu, Y.; et al. Global Prevalence of Diabetic Retinopathy and Projection of Burden through 2045: Systematic Review and Meta-analysis. Ophthalmology 2021, 128, 1580–1591. [Google Scholar] [CrossRef]
- Morya, A.K.; Ramesh, P.V.; Nishant, P.; Kaur, K.; Gurnani, B.; Heda, A.; Salodia, S. Diabetic retinopathy: A review on its pathophysiology and novel treatment modalities. World J. Methodol. 2024, 14, 95881. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Zaveri, J.; Becker, N. Proliferative diabetic retinopathy (PDR). Dis. Mon. 2021, 67, 101140. [Google Scholar] [CrossRef] [PubMed]
- McMeel, J.W. Diabetic retinopathy: Fibrotic proliferation and retinal detachment. Trans. Am. Ophthalmol. Soc. 1971, 69, 440–493. [Google Scholar]
- Roy, S.; Amin, S.; Roy, S. Retinal fibrosis in diabetic retinopathy. Exp. Eye Res. 2016, 142, 71–75. [Google Scholar] [CrossRef]
- Feitosa-Santana, C.; Oiwa, N.N.; Paramei, G.V.; Bimler, D.; Costa, M.F.; Lago, M.; Nishi, M.; Ventura, D.F. Color space distortions in patients with type 2 diabetes mellitus. Vis. Neurosci. 2006, 23, 663–668. [Google Scholar] [CrossRef]
- Alvarez, Y.; Chen, K.; Reynolds, A.L.; Waghorne, N.; O’Connor, J.J.; Kennedy, B.N. Predominant cone photoreceptor dysfunction in a hyperglycaemic model of non-proliferative diabetic retinopathy. Dis. Model. Mech. 2010, 3, 236–245. [Google Scholar] [CrossRef] [PubMed]
- O’Neill-Biba, M.; Sivaprasad, S.; Rodriguez-Carmona, M.; Wolf, J.E.; Barbur, J.L. Loss of chromatic sensitivity in AMD and diabetes: A comparative study. Ophthalmic Physiol. Opt. 2010, 30, 705–716. [Google Scholar] [CrossRef]
- Chang, R.C.; Shi, L.; Huang, C.C.; Kim, A.J.; Ko, M.L.; Zhou, B.; Ko, G.Y. High-Fat Diet-Induced Retinal Dysfunction. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2367–2380. [Google Scholar] [CrossRef]
- Zeng, X.X.; Ng, Y.K.; Ling, E.A. Neuronal and microglial response in the retina of streptozotocin-induced diabetic rats. Vis. Neurosci. 2000, 17, 463–471. [Google Scholar] [CrossRef]
- Cunha-Vaz, J.; Bernardes, R.; Lobo, C. Blood-retinal barrier. Eur. J. Ophthalmol. 2011, 21 (Suppl. S6), S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Runkle, E.A.; Antonetti, D.A. The blood-retinal barrier: Structure and functional significance. Methods Mol. Biol. 2011, 686, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Coranguez, M.; Ramos, C.; Antonetti, D.A. The inner blood-retinal barrier: Cellular basis and development. Vision Res. 2017, 139, 123–137. [Google Scholar] [CrossRef]
- Dragoni, S.; Moccia, F.; Bootman, M.D. The Roles of Transient Receptor Potential (TRP) Channels Underlying Aberrant Calcium Signaling in Blood-Retinal Barrier Dysfunction. Cold Spring Harb. Perspect. Biol. 2025, 17, a041763. [Google Scholar] [CrossRef]
- Bazzoni, G.; Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef]
- Bazzoni, G.; Martinez-Estrada, O.M.; Orsenigo, F.; Cordenonsi, M.; Citi, S.; Dejana, E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J. Biol. Chem. 2000, 275, 20520–20526. [Google Scholar] [CrossRef]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef]
- Erickson, K.K.; Sundstrom, J.M.; Antonetti, D.A. Vascular permeability in ocular disease and the role of tight junctions. Angiogenesis 2007, 10, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Turowski, P. Polarised VEGFA Signalling at Vascular Blood-Neural Barriers. Int. J. Mol. Sci. 2018, 19, 1378. [Google Scholar] [CrossRef] [PubMed]
- Wisniewska-Kruk, J.; van der Wijk, A.E.; van Veen, H.A.; Gorgels, T.G.; Vogels, I.M.; Versteeg, D.; Van Noorden, C.J.; Schlingemann, R.O.; Klaassen, I. Plasmalemma Vesicle-Associated Protein Has a Key Role in Blood-Retinal Barrier Loss. Am. J. Pathol. 2016, 186, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Enyong, E.N.; Gurley, J.M.; De Ieso, M.L.; Stamer, W.D.; Elliott, M.H. Caveolar and non-Caveolar Caveolin-1 in ocular homeostasis and disease. Prog. Retin. Eye Res. 2022, 91, 101094. [Google Scholar] [CrossRef]
- Zhang, C.L.; Wang, H.L.; Li, P.C.; Hong, C.D.; Chen, A.Q.; Qiu, Y.M.; Zeng, A.P.; Zhou, Y.F.; Hu, B.; Li, Y.N. Mfsd2a overexpression alleviates vascular dysfunction in diabetic retinopathy. Pharmacol. Res. 2021, 171, 105755. [Google Scholar] [CrossRef]
- Yefimova, M.G.; Jeanny, J.C.; Guillonneau, X.; Keller, N.; Nguyen-Legros, J.; Sergeant, C.; Guillou, F.; Courtois, Y. Iron, ferritin, transferrin, and transferrin receptor in the adult rat retina. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2343–2351. [Google Scholar]
- Burdo, J.R.; Antonetti, D.A.; Wolpert, E.B.; Connor, J.R. Mechanisms and regulation of transferrin and iron transport in a model blood-brain barrier system. Neuroscience 2003, 121, 883–890. [Google Scholar] [CrossRef]
- Liversidge, J.; Sewell, H.F.; Forrester, J.V. Interactions between lymphocytes and cells of the blood-retina barrier: Mechanisms of T lymphocyte adhesion to human retinal capillary endothelial cells and retinal pigment epithelial cells in vitro. Immunology 1990, 71, 390–396. [Google Scholar]
- Gerhardt, H.; Betsholtz, C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003, 314, 15–23. [Google Scholar] [CrossRef]
- Arboleda-Velasquez, J.F.; Primo, V.; Graham, M.; James, A.; Manent, J.; D’Amore, P.A. Notch signaling functions in retinal pericyte survival. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5191–5199. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.E. Brain macrophages: On the role of pericytes and perivascular cells. Brain Res. Brain Res. Rev. 1999, 31, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Trost, A.; Lange, S.; Schroedl, F.; Bruckner, D.; Motloch, K.A.; Bogner, B.; Kaser-Eichberger, A.; Strohmaier, C.; Runge, C.; Aigner, L.; et al. Brain and Retinal Pericytes: Origin, Function and Role. Front. Cell Neurosci. 2016, 10, 20. [Google Scholar] [CrossRef]
- Trost, A.; Bruckner, D.; Rivera, F.J.; Reitsamer, H.A. Pericytes in the Retina. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2019; Volume 1122, pp. 1–26. [Google Scholar] [CrossRef]
- Alarcon-Martinez, L.; Yemisci, M.; Dalkara, T. Pericyte morphology and function. Histol. Histopathol. 2021, 36, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Proebstl, D.; Voisin, M.B.; Woodfin, A.; Whiteford, J.; D’Acquisto, F.; Jones, G.E.; Rowe, D.; Nourshargh, S. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J. Exp. Med. 2012, 209, 1219–1234. [Google Scholar] [CrossRef]
- Stark, K.; Eckart, A.; Haidari, S.; Tirniceriu, A.; Lorenz, M.; von Bruhl, M.L.; Gartner, F.; Khandoga, A.G.; Legate, K.R.; Pless, R.; et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs. Nat. Immunol. 2013, 14, 41–51. [Google Scholar] [CrossRef]
- Huang, H. Pericyte-Endothelial Interactions in the Retinal Microvasculature. Int. J. Mol. Sci. 2020, 21, 7413. [Google Scholar] [CrossRef]
- Walshe, T.E.; Connell, P.; Cryan, L.; Ferguson, G.; Gardiner, T.; Morrow, D.; Redmond, E.M.; O’Brien, C.; Cahill, P.A. Microvascular retinal endothelial and pericyte cell apoptosis in vitro: Role of hedgehog and Notch signaling. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4472–4483. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, J.H.; Yu, Y.S.; Kim, D.H.; Kim, K.W. Recruitment of pericytes and astrocytes is closely related to the formation of tight junction in developing retinal vessels. J. Neurosci. Res. 2009, 87, 653–659. [Google Scholar] [CrossRef]
- Newman, E.A. Glial cell regulation of neuronal activity and blood flow in the retina by release of gliotransmitters. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140195. [Google Scholar] [CrossRef]
- Kugler, E.C.; Greenwood, J.; MacDonald, R.B. The “Neuro-Glial-Vascular” Unit: The Role of Glia in Neurovascular Unit Formation and Dysfunction. Front. Cell Dev. Biol. 2021, 9, 732820. [Google Scholar] [CrossRef] [PubMed]
- Florence, C.M.; Baillie, L.D.; Mulligan, S.J. Dynamic volume changes in astrocytes are an intrinsic phenomenon mediated by bicarbonate ion flux. PLoS ONE 2012, 7, e51124. [Google Scholar] [CrossRef] [PubMed]
- Lafrenaye, A.D.; Simard, J.M. Bursting at the Seams: Molecular Mechanisms Mediating Astrocyte Swelling. Int. J. Mol. Sci. 2019, 20, 330. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Kim, D. Retinal capillary basement membrane thickening: Role in the pathogenesis of diabetic retinopathy. Prog. Retin. Eye Res. 2021, 82, 100903. [Google Scholar] [CrossRef] [PubMed]
- Kinuthia, U.M.; Wolf, A.; Langmann, T. Microglia and Inflammatory Responses in Diabetic Retinopathy. Front. Immunol. 2020, 11, 564077. [Google Scholar] [CrossRef]
- Fan, W.; Huang, W.; Chen, J.; Li, N.; Mao, L.; Hou, S. Retinal microglia: Functions and diseases. Immunology 2022, 166, 268–286. [Google Scholar] [CrossRef]
- Fu, D.; Wu, M.; Zhang, J.; Du, M.; Yang, S.; Hammad, S.M.; Wilson, K.; Chen, J.; Lyons, T.J. Mechanisms of modified LDL-induced pericyte loss and retinal injury in diabetic retinopathy. Diabetologia 2012, 55, 3128–3140. [Google Scholar] [CrossRef]
- Martinet, W.; De Bie, M.; Schrijvers, D.M.; De Meyer, G.R.; Herman, A.G.; Kockx, M.M. 7-ketocholesterol induces protein ubiquitination, myelin figure formation, and light chain 3 processing in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2296–2301. [Google Scholar] [CrossRef]
- Johnson, P.C. Autoregulation of blood flow. Circ. Res. 1986, 59, 483–495. [Google Scholar] [CrossRef]
- Curtis, T.M.; Gardiner, T.A.; Stitt, A.W. Microvascular lesions of diabetic retinopathy: Clues towards understanding pathogenesis? Eye 2009, 23, 1496–1508. [Google Scholar] [CrossRef]
- Kohner, E.M.; Patel, V.; Rassam, S.M. Role of blood flow and impaired autoregulation in the pathogenesis of diabetic retinopathy. Diabetes 1995, 44, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Pournaras, C.J.; Rungger-Brandle, E.; Riva, C.E.; Hardarson, S.H.; Stefansson, E. Regulation of retinal blood flow in health and disease. Prog. Retin. Eye Res. 2008, 27, 284–330. [Google Scholar] [CrossRef] [PubMed]
- Hanaguri, J.; Yokota, H.; Watanabe, M.; Yamagami, S.; Kushiyama, A.; Kuo, L.; Nagaoka, T. Retinal blood flow dysregulation precedes neural retinal dysfunction in type 2 diabetic mice. Sci. Rep. 2021, 11, 18401. [Google Scholar] [CrossRef] [PubMed]
- Schmetterer, L.; Wolzt, M. Ocular blood flow and associated functional deviations in diabetic retinopathy. Diabetologia 1999, 42, 387–405. [Google Scholar] [CrossRef]
- Ciulla, T.A.; Harris, A.; Latkany, P.; Piper, H.C.; Arend, O.; Garzozi, H.; Martin, B. Ocular perfusion abnormalities in diabetes. Acta Ophthalmol. Scand. 2002, 80, 468–477. [Google Scholar] [CrossRef]
- Grunwald, J.E.; Riva, C.E.; Baine, J.; Brucker, A.J. Total retinal volumetric blood flow rate in diabetic patients with poor glycemic control. Investig. Ophthalmol. Vis. Sci. 1992, 33, 356–363. [Google Scholar]
- Klein, R.; Myers, C.E.; Lee, K.E.; Gangnon, R.; Klein, B.E. Changes in retinal vessel diameter and incidence and progression of diabetic retinopathy. Arch. Ophthalmol. 2012, 130, 749–755. [Google Scholar] [CrossRef]
- Kur, J.; Newman, E.A.; Chan-Ling, T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog. Retin. Eye Res. 2012, 31, 377–406. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Newman, E.A. Inhibition of inducible nitric oxide synthase reverses the loss of functional hyperemia in diabetic retinopathy. Glia 2010, 58, 1996–2004. [Google Scholar] [CrossRef]
- Trick, G.L.; Liggett, J.; Levy, J.; Adamsons, I.; Edwards, P.; Desai, U.; Tofts, P.S.; Berkowitz, B.A. Dynamic contrast enhanced MRI in patients with diabetic macular edema: Initial results. Exp. Eye Res. 2005, 81, 97–102. [Google Scholar] [CrossRef]
- Joseph, K.; Kaplan, A.P. Formation of bradykinin: A major contributor to the innate inflammatory response. Adv. Immunol. 2005, 86, 159–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Feener, E.P. Plasma kallikrein-kinin system and diabetic retinopathy. Biol. Chem. 2013, 394, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Abdouh, M.; Talbot, S.; Couture, R.; Hassessian, H.M. Retinal plasma extravasation in streptozotocin-diabetic rats mediated by kinin B1 and B2 receptors. Br. J. Pharmacol. 2008, 154, 136–143. [Google Scholar] [CrossRef]
- Hardy, P.; Abran, D.; Hou, X.; Lahaie, I.; Peri, K.G.; Asselin, P.; Varma, D.R.; Chemtob, S. A major role for prostacyclin in nitric oxide-induced ocular vasorelaxation in the piglet. Circ. Res. 1998, 83, 721–729. [Google Scholar] [CrossRef]
- Jeppesen, P.; Aalkjaer, C.; Bek, T. Bradykinin relaxation in small porcine retinal arterioles. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1891–1896. [Google Scholar]
- Orsenigo, F.; Giampietro, C.; Ferrari, A.; Corada, M.; Galaup, A.; Sigismund, S.; Ristagno, G.; Maddaluno, L.; Koh, G.Y.; Franco, D.; et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat. Commun. 2012, 3, 1208. [Google Scholar] [CrossRef]
- Kim, T.; Kim, S.J.; Kim, K.; Kang, U.B.; Lee, C.; Park, K.S.; Yu, H.G.; Kim, Y. Profiling of vitreous proteomes from proliferative diabetic retinopathy and nondiabetic patients. Proteomics 2007, 7, 4203–4215. [Google Scholar] [CrossRef]
- Gao, B.B.; Chen, X.; Timothy, N.; Aiello, L.P.; Feener, E.P. Characterization of the vitreous proteome in diabetes without diabetic retinopathy and diabetes with proliferative diabetic retinopathy. J. Proteome Res. 2008, 7, 2516–2525. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P.; Avery, R.L.; Arrigg, P.G.; Keyt, B.A.; Jampel, H.D.; Shah, S.T.; Pasquale, L.R.; Thieme, H.; Iwamoto, M.A.; Park, J.E.; et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994, 331, 1480–1487. [Google Scholar] [CrossRef]
- Aveleira, C.A.; Lin, C.M.; Abcouwer, S.F.; Ambrosio, A.F.; Antonetti, D.A. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 2010, 59, 2872–2882. [Google Scholar] [CrossRef]
- Bamforth, S.D.; Lightman, S.L.; Greenwood, J. Interleukin-1 beta-induced disruption of the retinal vascular barrier of the central nervous system is mediated through leukocyte recruitment and histamine. Am. J. Pathol. 1997, 150, 329–340. [Google Scholar] [PubMed]
- Banerjee, S.; Savant, V.; Scott, R.A.; Curnow, S.J.; Wallace, G.R.; Murray, P.I. Multiplex bead analysis of vitreous humor of patients with vitreoretinal disorders. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2203–2207. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rangasamy, S.; McGuire, P.G.; Franco Nitta, C.; Monickaraj, F.; Oruganti, S.R.; Das, A. Chemokine mediated monocyte trafficking into the retina: Role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS ONE 2014, 9, e108508. [Google Scholar] [CrossRef]
- Nieuwdorp, M.; van Haeften, T.W.; Gouverneur, M.C.; Mooij, H.L.; van Lieshout, M.H.; Levi, M.; Meijers, J.C.; Holleman, F.; Hoekstra, J.B.; Vink, H.; et al. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes 2006, 55, 480–486. [Google Scholar] [CrossRef]
- Lipowsky, H.H.; Gao, L.; Lescanic, A. Shedding of the endothelial glycocalyx in arterioles, capillaries, and venules and its effect on capillary hemodynamics during inflammation. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2235–H2245. [Google Scholar] [CrossRef]
- Hammes, H.P.; Lin, J.; Renner, O.; Shani, M.; Lundqvist, A.; Betsholtz, C.; Brownlee, M.; Deutsch, U. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes 2002, 51, 3107–3112. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, T.A.; Archer, D.B.; Curtis, T.M.; Stitt, A.W. Arteriolar involvement in the microvascular lesions of diabetic retinopathy: Implications for pathogenesis. Microcirculation 2007, 14, 25–38. [Google Scholar] [CrossRef]
- Zhang, Q.; Yan, X.; Han, H.; Wang, Y.; Sun, J. Pericyte in retinal vascular diseases: A multifunctional regulator and potential therapeutic target. FASEB J. 2024, 38, e23679. [Google Scholar] [CrossRef]
- Stitt, A.W.; Li, Y.M.; Gardiner, T.A.; Bucala, R.; Archer, D.B.; Vlassara, H. Advanced glycation end products (AGEs) co-localize with AGE receptors in the retinal vasculature of diabetic and of AGE-infused rats. Am. J. Pathol. 1997, 150, 523–531. [Google Scholar]
- Maeda, S.; Matsui, T.; Ojima, A.; Takeuchi, M.; Yamagishi, S. Sulforaphane inhibits advanced glycation end product-induced pericyte damage by reducing expression of receptor for advanced glycation end products. Nutr. Res. 2014, 34, 807–813. [Google Scholar] [CrossRef]
- Garcia-Quintans, N.; Sanchez-Ramos, C.; Prieto, I.; Tierrez, A.; Arza, E.; Alfranca, A.; Redondo, J.M.; Monsalve, M. Oxidative stress induces loss of pericyte coverage and vascular instability in PGC-1alpha-deficient mice. Angiogenesis 2016, 19, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Smith, D.; Li, Q.; Sheibani, N.; Huang, S.; Kern, T.; Nagaraj, R.H.; Lin, F. Antibody-mediated retinal pericyte injury: Implications for diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5520–5526. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Sanford, M.; Shi, H.; Tarantini, S. The role of endothelial TRP channels in age-related vascular cognitive impairment and dementia. Front. Aging Neurosci. 2023, 15, 1149820. [Google Scholar] [CrossRef]
- Kuppusamy, M.; Ottolini, M.; Sonkusare, S.K. Role of TRP ion channels in cerebral circulation and neurovascular communication. Neurosci. Lett. 2021, 765, 136258. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Wang, N.; Decrock, E.; Bol, M.; Gadicherla, A.K.; Culot, M.; Cecchelli, R.; Bultynck, G.; Leybaert, L. Endothelial calcium dynamics, connexin channels and blood-brain barrier function. Prog. Neurobiol. 2013, 108, 1–20. [Google Scholar] [CrossRef]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Soda, T.; Moccia, F. Endothelial signaling at the core of neurovascular coupling: The emerging role of endothelial inward-rectifier K+ (Kir2.1) channels and N-methyl-d-aspartate receptors in the regulation of cerebral blood flow. Int. J. Biochem. Cell Biol. 2021, 135, 105983. [Google Scholar] [CrossRef]
- Scarpellino, G.; Brunetti, V.; Berra-Romani, R.; De Sarro, G.; Guerra, G.; Soda, T.; Moccia, F. The Unexpected Role of the Endothelial Nitric Oxide Synthase at the Neurovascular Unit: Beyond the Regulation of Cerebral Blood Flow. Int. J. Mol. Sci. 2024, 25, 9071. [Google Scholar] [CrossRef]
- Alvarado, M.G.; Thakore, P.; Earley, S. Transient Receptor Potential Channel Ankyrin 1: A Unique Regulator of Vascular Function. Cells 2021, 10, 1167. [Google Scholar] [CrossRef]
- Longden, T.A.; Mughal, A.; Hennig, G.W.; Harraz, O.F.; Shui, B.; Lee, F.K.; Lee, J.C.; Reining, S.; Kotlikoff, M.I.; Konig, G.M.; et al. Local IP3 receptor-mediated Ca2+ signals compound to direct blood flow in brain capillaries. Sci. Adv. 2021, 7, eabh0101. [Google Scholar] [CrossRef]
- Mughal, A.; Hennig, G.W.; Heppner, T.; Tsoukias, N.M.; Hill-Eubanks, D.; Nelson, M.T. Electrocalcium coupling in brain capillaries: Rapidly traveling electrical signals ignite local calcium signals. Proc. Natl. Acad. Sci. USA 2024, 121, e2415047121. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, V.; Berra-Romani, R.; Conca, F.; Soda, T.; Biella, G.R.; Gerbino, A.; Moccia, F.; Scarpellino, G. Lysosomal TRPML1 triggers global Ca2+ signals and nitric oxide release in human cerebrovascular endothelial cells. Front. Physiol. 2024, 15, 1426783. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Pellavio, G.; Botta, L.; Orgiu, M.; Forcaia, G.; Sancini, G.; Laforenza, U.; Moccia, F. Group 1 metabotropic glutamate receptors trigger glutamate-induced intracellular Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. Cell. Mol. Life Sci. 2020, 77, 2235–2253. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Angelone, T. Targeting endothelial ion signalling to rescue cerebral blood flow in cerebral disorders. Vasc. Pharmacol. 2022, 145, 106997. [Google Scholar] [CrossRef]
- Moccia, F.; Brunetti, V.; Perna, A.; Guerra, G.; Soda, T.; Berra-Romani, R. The Molecular Heterogeneity of Store-Operated Ca2+ Entry in Vascular Endothelial Cells: The Different roles of Orai1 and TRPC1/TRPC4 Channels in the Transition from Ca2+-Selective to Non-Selective Cation Currents. Int. J. Mol. Sci. 2023, 24, 3259. [Google Scholar] [CrossRef]
- Dragoni, S.; Guerra, G.; Fiorio Pla, A.; Bertoni, G.; Rappa, A.; Poletto, V.; Bottino, C.; Aronica, A.; Lodola, F.; Cinelli, M.P.; et al. A functional transient receptor potential vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J. Cell. Physiol. 2015, 230, 95–104. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Guerra, G.; Borghesi, A.; Stronati, M.; Rosti, V.; Tanzi, F.; et al. Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor-induced intracellular Ca2+ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 2013, 22, 2561–2580. [Google Scholar] [CrossRef]
- Harraz, O.F.; Longden, T.A.; Hill-Eubanks, D.; Nelson, M.T. PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. eLife 2018, 7, e38689. [Google Scholar] [CrossRef]
- Soda, T.; Brunetti, V.; De Sarro, G.; Biella, G.; Moccia, F.; Berra-Romani, R.; Scarpellino, G. Transient Receptor Potential Ankyrin 1 (TRPA1) Mediates Hydrogen Sulfide-induced Ca2+ Entry and Nitric Oxide Production in Human Cerebrovascular Endothelium. Curr. Neuropharmacol. 2025. [Google Scholar] [CrossRef]
- Park, L.; Wang, G.; Moore, J.; Girouard, H.; Zhou, P.; Anrather, J.; Iadecola, C. The key role of transient receptor potential melastatin-2 channels in amyloid-beta-induced neurovascular dysfunction. Nat. Commun. 2014, 5, 5318. [Google Scholar] [CrossRef]
- Lim, X.R.; Abd-Alhaseeb, M.M.; Ippolito, M.; Koide, M.; Senatore, A.J.; Plante, C.; Hariharan, A.; Weir, N.; Longden, T.A.; Laprade, K.A.; et al. Endothelial Piezo1 channel mediates mechano-feedback control of brain blood flow. Nat. Commun. 2024, 15, 8686. [Google Scholar] [CrossRef] [PubMed]
- Laskey, R.E.; Adams, D.J.; Cannell, M.; van Breemen, C. Calcium entry-dependent oscillations of cytoplasmic calcium concentration in cultured endothelial cell monolayers. Proc. Natl. Acad. Sci. USA 1992, 89, 1690–1694. [Google Scholar] [CrossRef]
- Fleming, I.; Fisslthaler, B.; Busse, R. Calcium signaling in endothelial cells involves activation of tyrosine kinases and leads to activation of mitogen-activated protein kinases. Circ. Res. 1995, 76, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, M.; Oshima, T.; Matsuura, H.; Inoue, T.; Kambe, M.; Kajiyama, G. Differential effects of extracellular Mg2+ on thrombin-induced and capacitative Ca2+ entry in human coronary arterial endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3356–3361. [Google Scholar] [CrossRef]
- Bachkoenig, O.A.; Gottschalk, B.; Malli, R.; Graier, W.F. An unexpected effect of risperidone reveals a nonlinear relationship between cytosolic Ca2+ and mitochondrial Ca2+ uptake. Curr. Top. Membr. 2022, 90, 13–35. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Lee, M.D.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Girkin, J.M.; Saunter, C.D.; McCarron, J.G. Mitochondrial ATP production provides long-range control of endothelial inositol trisphosphate-evoked calcium signaling. J. Biol. Chem. 2019, 294, 737–758. [Google Scholar] [CrossRef]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca2+ oscillations and nitric oxide release by inducing NAADP- and InsP3 -dependent Ca2+ release in mouse brain endothelial cells. J. Cell. Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Guzman-Silva, A.; Vargaz-Guadarrama, A.; Flores-Alonso, J.C.; Alonso-Romero, J.; Trevino, S.; Sanchez-Gomez, J.; Coyotl-Santiago, N.; Garcia-Carrasco, M.; Moccia, F. Type 2 Diabetes Alters Intracellular Ca2+ Handling in Native Endothelium of Excised Rat Aorta. Int. J. Mol. Sci. 2019, 21, 250. [Google Scholar] [CrossRef]
- Moccia, F.; Berra-Romani, R.; Baruffi, S.; Spaggiari, S.; Signorelli, S.; Castelli, L.; Magistretti, J.; Taglietti, V.; Tanzi, F. Ca2+ uptake by the endoplasmic reticulum Ca2+-ATPase in rat microvascular endothelial cells. Biochem. J. 2002, 364, 235–244. [Google Scholar] [CrossRef]
- Liu, G.; Wu, F.; Wu, H.; Wang, Y.; Jiang, X.; Hu, P.; Tong, X. Inactivation of cysteine 674 in the sarcoplasmic/endoplasmic reticulum calcium ATPase 2 causes retinopathy in the mouse. Exp. Eye Res. 2021, 207, 108559. [Google Scholar] [CrossRef]
- Krizaj, D. Serca isoform expression in the mammalian retina. Exp. Eye Res. 2005, 81, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca2+ Signaling, Angiogenesis and Vasculogenesis: Just What It Takes to Make a Blood Vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, S.; Fujii, A.; Nakade, S.; Mikoshiba, K. Immunohistochemical localization of inositol 1,4,5-trisphosphate receptors in non-neural tissues, with special reference to epithelia, the reproductive system, and muscular tissues. Cell Tissue Res. 1996, 285, 235–251. [Google Scholar] [CrossRef]
- Li, W.; Wang, W.; Liu, X. Comparative study of high-glucose effect on phosphatidylcholine hydrolysis of cultured retinal capillary pericytes and endothelial cells. Biochim. Biophys. Acta 1994, 1222, 339–347. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.Y.; Shao, J.; Zhu, L.; Xie, T.H.; Cai, J.; Wang, W.; Cai, M.X.; Wang, Z.L.; Yao, Y.; et al. GRP75 Modulates Endoplasmic Reticulum-Mitochondria Coupling and Accelerates Ca2+-Dependent Endothelial Cell Apoptosis in Diabetic Retinopathy. Biomolecules 2022, 12, 1778. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Endolysosomal Ca2+ signaling in cardiovascular health and disease. Int. Rev. Cell Mol. Biol. 2021, 363, 203–269. [Google Scholar] [CrossRef]
- Palinski, W.; Monti, M.; Camerlingo, R.; Iacobucci, I.; Bocella, S.; Pinto, F.; Iannuzzi, C.; Mansueto, G.; Pignatiello, S.; Fazioli, F.; et al. Lysosome purinergic receptor P2X4 regulates neoangiogenesis induced by microvesicles from sarcoma patients. Cell Death Dis. 2021, 12, 797. [Google Scholar] [CrossRef]
- O’Leary, C.; McGahon, M.K.; Ashraf, S.; McNaughten, J.; Friedel, T.; Cincola, P.; Barabas, P.; Fernandez, J.A.; Stitt, A.W.; McGeown, J.G.; et al. Involvement of TRPV1 and TRPV4 Channels in Retinal Angiogenesis. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3297–3309. [Google Scholar] [CrossRef] [PubMed]
- Sivaraj, K.K.; Li, R.; Albarran-Juarez, J.; Wang, S.; Tischner, D.; Grimm, M.; Swiercz, J.M.; Offermanns, S.; Wettschureck, N. Endothelial Galphaq/11 is required for VEGF-induced vascular permeability and angiogenesis. Cardiovasc. Res. 2015, 108, 171–180. [Google Scholar] [CrossRef][Green Version]
- Wang, C.; Dai, X.; Wu, S.; Xu, W.; Song, P.; Huang, K.; Zou, M.H. FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes are involved in angiogenesis and neoangiogenesis. Nat. Commun. 2021, 12, 2616. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Di Nezza, F.; Pellavio, G.; Faris, P.S.; Negri, S.; De Luca, A.; Laforenza, U.; Ambrosone, L.; Rosti, V.; et al. Nicotinic acid adenine dinucleotide phosphate activates two-pore channel TPC1 to mediate lysosomal Ca2+ release in endothelial colony-forming cells. J. Cell. Physiol. 2021, 236, 688–705. [Google Scholar] [CrossRef]
- Churchill, G.C.; Galione, A. NAADP induces Ca2+ oscillations via a two-pool mechanism by priming IP3- and cADPR-sensitive Ca2+ stores. EMBO J. 2001, 20, 2666–2671. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef]
- Galeano-Otero, I.; Del Toro, R.; Khatib, A.M.; Rosado, J.A.; Ordonez-Fernandez, A.; Smani, T. SARAF and Orai1 Contribute to Endothelial Cell Activation and Angiogenesis. Front. Cell Dev. Biol. 2021, 9, 639952. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef]
- Emrich, S.M.; Yoast, R.E.; Trebak, M. Physiological Functions of CRAC Channels. Annu. Rev. Physiol. 2022, 84, 355–379. [Google Scholar] [CrossRef]
- Tanwar, J.; Trebak, M.; Motiani, R.K. Cardiovascular and Hemostatic Disorders: Role of STIM and Orai Proteins in Vascular Disorders. Adv. Exp. Med. Biol. 2017, 993, 425–452. [Google Scholar] [CrossRef]
- Yuan, Q.; Yang, J.; Santulli, G.; Reiken, S.R.; Wronska, A.; Kim, M.M.; Osborne, B.W.; Lacampagne, A.; Yin, Y.; Marks, A.R. Maintenance of normal blood pressure is dependent on IP3R1-mediated regulation of eNOS. Proc. Natl. Acad. Sci. USA 2016, 113, 8532–8537. [Google Scholar] [CrossRef]
- Lin, Q.; Zhao, L.; Jing, R.; Trexler, C.; Wang, H.; Li, Y.; Tang, H.; Huang, F.; Zhang, F.; Fang, X.; et al. Inositol 1,4,5-Trisphosphate Receptors in Endothelial Cells Play an Essential Role in Vasodilation and Blood Pressure Regulation. J. Am. Heart Assoc. 2019, 8, e011704. [Google Scholar] [CrossRef]
- Wilson, C.; Zhang, X.; Lee, M.D.; MacDonald, M.; Heathcote, H.R.; Alorfi, N.M.N.; Buckley, C.; Dolan, S.; McCarron, J.G. Disrupted endothelial cell heterogeneity and network organization impair vascular function in prediabetic obesity. Metabolism 2020, 111, 154340. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chu, Y.; Lan, Y.; Wang, S.; Zhang, Y.; Liu, Y.; Wang, X.; Yu, F.; Ma, X. Loss of Endothelial TRPC1 Induces Aortic Hypercontractility and Hypertension. Circ. Res. 2025, 136, 508–523. [Google Scholar] [CrossRef]
- Scarpellino, G.; Genova, T.; Quarta, E.; Distasi, C.; Dionisi, M.; Fiorio Pla, A.; Munaron, L. P2X Purinergic Receptors Are Multisensory Detectors for Micro-Environmental Stimuli That Control Migration of Tumoral Endothelium. Cancers 2022, 14, 2743. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef]
- Peters, E.C.; Gee, M.T.; Pawlowski, L.N.; Kath, A.M.; Polk, F.D.; Vance, C.J.; Sacoman, J.L.; Pires, P.W. Amyloid-beta disrupts unitary calcium entry through endothelial NMDA receptors in mouse cerebral arteries. J. Cereb. Blood Flow. Metab. 2022, 42, 145–161. [Google Scholar] [CrossRef]
- Lim, X.R.; Willemse, L.; Harraz, O.F. Amyloid beta Abeta(1-40) activates Piezo1 channels in brain capillary endothelial cells. Biophys. J. 2024. [Google Scholar] [CrossRef] [PubMed]
- Mughal, A.; Sackheim, A.M.; Koide, M.; Bonson, G.; Ebner, G.; Hennig, G.; Lockette, W.; Nelson, M.T.; Freeman, K. Pathogenic soluble tau peptide disrupts endothelial calcium signaling and vasodilation in the brain microvasculature. J. Cereb. Blood Flow. Metab. 2024, 44, 680–688. [Google Scholar] [CrossRef]
- Zong, P.; Feng, J.; Li, C.X.; Jellison, E.R.; Yue, Z.; Miller, B.; Yue, L. Activation of endothelial TRPM2 exacerbates blood-brain barrier degradation in ischemic stroke. Cardiovasc. Res. 2024, 120, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, R.; Qayum, S.; Pliss, A.; Kuzmin, A.N.; Muthaiah, V.P.K.; Kaliyappan, K.; Prasad, P.N.; Mahajan, S.D. Mitochondrial Dysfunction and Apoptosis in Brain Microvascular Endothelial Cells Following Blast Traumatic Brain Injury. Cell. Mol. Neurobiol. 2023, 43, 3639–3651. [Google Scholar] [CrossRef]
- Ivanova, A.; Atakpa-Adaji, P.; Rao, S.; Marti-Solano, M.; Taylor, C.W. Dual regulation of IP3 receptors by IP3 and PIP2 controls the transition from local to global Ca2+ signals. Mol. Cell 2024, 84, 3997–4015. [Google Scholar] [CrossRef]
- Sade, Y.; Toker, L.; Kara, N.Z.; Einat, H.; Rapoport, S.; Moechars, D.; Berry, G.T.; Bersudsky, Y.; Agam, G. IP3 accumulation and/or inositol depletion: Two downstream lithium’s effects that may mediate its behavioral and cellular changes. Transl. Psychiatry 2016, 6, e968. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Baccouche, B.; Olayinka, O.; Serikbaeva, A.; Kazlauskas, A. The Role of the Wnt Pathway in VEGF/Anti-VEGF-Dependent Control of the Endothelial Cell Barrier. Investig. Ophthalmol. Vis. Sci. 2021, 62, 17. [Google Scholar] [CrossRef] [PubMed]
- Descorbeth, M.; Anand-Srivastava, M.B. Role of growth factor receptor transactivation in high glucose-induced increased levels of Gq/11alpha and signaling in vascular smooth muscle cells. J. Mol. Cell. Cardiol. 2010, 49, 221–233. [Google Scholar] [CrossRef]
- Booth, D.M.; Varnai, P.; Joseph, S.K.; Hajnoczky, G. Oxidative bursts of single mitochondria mediate retrograde signaling toward the ER. Mol. Cell 2021, 81, 3866–3876. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? Int. J. Mol. Sci. 2021, 22, 9821. [Google Scholar] [CrossRef]
- Daskoulidou, N.; Zeng, B.; Berglund, L.M.; Jiang, H.; Chen, G.L.; Kotova, O.; Bhandari, S.; Ayoola, J.; Griffin, S.; Atkin, S.L.; et al. High glucose enhances store-operated calcium entry by upregulating ORAI/STIM via calcineurin-NFAT signalling. J. Mol. Med. 2015, 93, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Laforenza, U.; Cattaneo, F.; Ruffinatti, F.A.; Poletto, V.; Massa, M.; Tancredi, R.; Zuccolo, E.; Khdar, D.A.; Riccardi, A.; et al. VEGF-induced intracellular Ca2+ oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 2017, 8, 95223–95246. [Google Scholar] [CrossRef]
- Dragoni, S.; Reforgiato, M.; Zuccolo, E.; Poletto, V.; Lodola, F.; Ruffinatti, F.A.; Bonetti, E.; Guerra, G.; Barosi, G.; Rosti, V.; et al. Dysregulation of VEGF-induced proangiogenic Ca2+ oscillations in primary myelofibrosis-derived endothelial colony-forming cells. Exp. Hematol. 2015, 43, 1019–1030.e3. [Google Scholar] [CrossRef]
- Rahman, S.; Rahman, T. Unveiling some FDA-approved drugs as inhibitors of the store-operated Ca2+ entry pathway. Sci. Rep. 2017, 7, 12881. [Google Scholar] [CrossRef]
- Moccia, F.; Brunetti, V.; Soda, T.; Faris, P.; Scarpellino, G.; Berra-Romani, R. Store-Operated Ca2+ Entry as a Putative Target of Flecainide for the Treatment of Arrhythmogenic Cardiomyopathy. J. Clin. Med. 2023, 12, 5295. [Google Scholar] [CrossRef]
- Moccia, F.; Poletto, V. May the remodeling of the Ca2+ toolkit in endothelial progenitor cells derived from cancer patients suggest alternative targets for anti-angiogenic treatment? Biochim. Biophys. Acta 2015, 1853, 1958–1973. [Google Scholar] [CrossRef] [PubMed]
- Poletto, V.; Rosti, V.; Biggiogera, M.; Guerra, G.; Moccia, F.; Porta, C. The role of endothelial colony forming cells in kidney cancer’s pathogenesis, and in resistance to anti-VEGFR agents and mTOR inhibitors: A speculative review. Crit. Rev. Oncol. Hematol. 2018, 132, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, D.L.; Wu, S.; Chen, H.; Alexeyev, M.; St Croix, C.M.; Pitt, B.R.; Uhlig, S.; Stevens, T. Orai1 determines calcium selectivity of an endogenous TRPC heterotetramer channel. Circ.Res. 2012, 110, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Hamdollah Zadeh, M.A.; Glass, C.A.; Magnussen, A.; Hancox, J.C.; Bates, D.O. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation 2008, 15, 605–614. [Google Scholar] [CrossRef]
- Tsagareli, M.G.; Nozadze, I. An overview on transient receptor potential channels superfamily. Behav. Pharmacol. 2020, 31, 413–434. [Google Scholar] [CrossRef]
- Thakore, P.; Earley, S. Transient Receptor Potential Channels and Endothelial Cell Calcium Signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial Transient Receptor Potential Channels and Vascular Remodeling: Extracellular Ca2+ Entry for Angiogenesis, Arteriogenesis and Vasculogenesis. Front. Physiol. 2019, 10, 1618. [Google Scholar] [CrossRef]
- Heathcote, H.R.; Lee, M.D.; Zhang, X.; Saunter, C.D.; Wilson, C.; McCarron, J.G. Endothelial TRPV4 channels modulate vascular tone by Ca2+ -induced Ca2+ release at inositol 1,4,5-trisphosphate receptors. Br. J. Pharmacol. 2019, 176, 3297–3317. [Google Scholar] [CrossRef]
- Scarpellino, G.; Munaron, L.; Cantelmo, A.R.; Fiorio Pla, A. Calcium-Permeable Channels in Tumor Vascularization: Peculiar Sensors of Microenvironmental Chemical and Physical Cues. Rev. Physiol. Biochem. Pharmacol. 2022, 182, 111–137. [Google Scholar] [CrossRef]
- Solano, A.S.; Lavanderos, B.; Metwally, E.; Earley, S. Transient Receptor Potential Channels in Vascular Mechanotransduction. Am. J. Hypertens. 2025, 38, 151–160. [Google Scholar] [CrossRef]
- Jackson, W.F. Endothelial Ion Channels and Cell-Cell Communication in the Microcirculation. Front. Physiol. 2022, 13, 805149. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Rosti, V.; Antognazza, M.R.; Lodola, F.; Moccia, F. Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis. Cells 2020, 9, 1341. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.; Alvarado, M.G.; Ali, S.; Mughal, A.; Pires, P.W.; Yamasaki, E.; Pritchard, H.A.; Isakson, B.E.; Tran, C.H.T.; Earley, S. Brain endothelial cell TRPA1 channels initiate neurovascular coupling. eLife 2021, 10, e63040. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Brunetti, V.; Pellavio, G.; Soda, T.; Laforenza, U.; Scarpellino, G.; Moccia, F. Allyl Isothiocianate Induces Ca2+ Signals and Nitric Oxide Release by Inducing Reactive Oxygen Species Production in the Human Cerebrovascular Endothelial Cell Line hCMEC/D3. Cells 2023, 12, 1732. [Google Scholar] [CrossRef] [PubMed]
- Hoiland, R.L.; Caldwell, H.G.; Howe, C.A.; Nowak-Fluck, D.; Stacey, B.S.; Bailey, D.M.; Paton, J.F.R.; Green, D.J.; Sekhon, M.S.; Macleod, D.B.; et al. Nitric oxide is fundamental to neurovascular coupling in humans. J. Physiol. 2020, 598, 4927–4939. [Google Scholar] [CrossRef]
- Ryu, H.J.; Kim, J.E.; Kim, Y.J.; Kim, J.Y.; Kim, W.I.; Choi, S.Y.; Kim, M.J.; Kang, T.C. Endothelial transient receptor potential conical channel (TRPC)-3 activation induces vasogenic edema formation in the rat piriform cortex following status epilepticus. Cell. Mol. Neurobiol. 2013, 33, 575–585. [Google Scholar] [CrossRef]
- Li, W.; Ehrich, M. Transient alterations of the blood-brain barrier tight junction and receptor potential channel gene expression by chlorpyrifos. J. Appl. Toxicol. 2013, 33, 1187–1191. [Google Scholar] [CrossRef]
- Narita, K.; Sasamoto, S.; Koizumi, S.; Okazaki, S.; Nakamura, H.; Inoue, T.; Takeda, S. TRPV4 regulates the integrity of the blood-cerebrospinal fluid barrier and modulates transepithelial protein transport. FASEB J. 2015, 29, 2247–2259. [Google Scholar] [CrossRef]
- Rosenkranz, S.C.; Shaposhnykov, A.; Schnapauff, O.; Epping, L.; Vieira, V.; Heidermann, K.; Schattling, B.; Tsvilovskyy, V.; Liedtke, W.; Meuth, S.G.; et al. TRPV4-Mediated Regulation of the Blood Brain Barrier Is Abolished During Inflammation. Front. Cell Dev. Biol. 2020, 8, 849. [Google Scholar] [CrossRef]
- Berrout, J.; Jin, M.; O’Neil, R.G. Critical role of TRPP2 and TRPC1 channels in stretch-induced injury of blood-brain barrier endothelial cells. Brain Res. 2012, 1436, 1–12. [Google Scholar] [CrossRef]
- Thebault, S. Minireview: Insights into the role of TRP channels in the retinal circulation and function. Neurosci. Lett. 2021, 765, 136285. [Google Scholar] [CrossRef] [PubMed]
- Gilliam, J.C.; Wensel, T.G. TRP channel gene expression in the mouse retina. Vision. Res. 2011, 51, 2440–2452. [Google Scholar] [CrossRef]
- McGahon, M.K.; Fernandez, J.A.; Dash, D.P.; McKee, J.; Simpson, D.A.; Zholos, A.V.; McGeown, J.G.; Curtis, T.M. TRPV2 Channels Contribute to Stretch-Activated Cation Currents and Myogenic Constriction in Retinal Arterioles. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5637–5647. [Google Scholar] [CrossRef]
- Kojima, I.; Nagasawa, M. Trpv2. Handb. Exp. Pharmacol. 2014, 222, 247–272. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, M.; Esquiva, G.; McGahon, M.K.; Hombrebueno, J.M.R.; Augustine, J.; Canning, P.; Edgar, K.S.; Barabas, P.; Friedel, T.; Cincola, P.; et al. Loss of TRPV2-mediated blood flow autoregulation recapitulates diabetic retinopathy in rats. JCI Insight 2022, 7, e155128. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid Ligands Targeting TRP Channels. Front. Mol. Neurosci. 2018, 11, 487. [Google Scholar] [CrossRef]
- Arredondo Zamarripa, D.; Noguez Imm, R.; Bautista Cortes, A.M.; Vazquez Ruiz, O.; Bernardini, M.; Fiorio Pla, A.; Gkika, D.; Prevarskaya, N.; Lopez-Casillas, F.; Liedtke, W.; et al. Dual contribution of TRPV4 antagonism in the regulatory effect of vasoinhibins on blood-retinal barrier permeability: Diabetic milieu makes a difference. Sci. Rep. 2018, 8, 9652. [Google Scholar] [CrossRef]
- Orduna Rios, M.; Noguez Imm, R.; Hernandez Godinez, N.M.; Bautista Cortes, A.M.; Lopez Escalante, D.D.; Liedtke, W.; Martinez Torres, A.; Concha, L.; Thebault, S. TRPV4 inhibition prevents increased water diffusion and blood-retina barrier breakdown in the retina of streptozotocin-induced diabetic mice. PLoS ONE 2019, 14, e0212158. [Google Scholar] [CrossRef]
- Monaghan, K.; McNaughten, J.; McGahon, M.K.; Kelly, C.; Kyle, D.; Yong, P.H.; McGeown, J.G.; Curtis, T.M. Hyperglycemia and Diabetes Downregulate the Functional Expression of TRPV4 Channels in Retinal Microvascular Endothelium. PLoS ONE 2015, 10, e0128359. [Google Scholar] [CrossRef]
- White, J.P.; Cibelli, M.; Urban, L.; Nilius, B.; McGeown, J.G.; Nagy, I. TRPV4: Molecular Conductor of a Diverse Orchestra. Physiol. Rev. 2016, 96, 911–973. [Google Scholar] [CrossRef]
- Lang, H.B.; Xie, R.X.; Huang, M.L.; Fang, L.Y.; Tang, Y.B.; Zhang, F. The Effect and Mechanism of TRPC1, 3, and 6 on the Proliferation, Migration, and Lumen Formation of Retinal Vascular Endothelial Cells Induced by High Glucose. Ophthalmic Res. 2020, 63, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Song, H.B.; Jun, H.O.; Kim, J.H.; Fruttiger, M.; Kim, J.H. Suppression of transient receptor potential canonical channel 4 inhibits vascular endothelial growth factor-induced retinal neovascularization. Cell Calcium 2015, 57, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cheng, X.; Tian, J.; Xiao, Y.; Tian, T.; Xu, F.; Hong, X.; Zhu, M.X. TRPC channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol. Ther. 2020, 209, 107497. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, X.; Liao, L.; Chen, J.; Wang, Y.; Yao, M.; Zhu, L.; Li, J.; Wang, X.; Chen, A.F.; et al. The TRPM7 channel reprograms cellular glycolysis to drive tumorigenesis and angiogenesis. Cell Death Dis. 2023, 14, 183. [Google Scholar] [CrossRef]
- Luo, H.; Rossi, E.; Saubamea, B.; Chasseigneaux, S.; Cochois, V.; Choublier, N.; Smirnova, M.; Glacial, F.; Perriere, N.; Bourdoulous, S.; et al. Cannabidiol Increases Proliferation, Migration, Tubulogenesis, and Integrity of Human Brain Endothelial Cells through TRPV2 Activation. Mol. Pharm. 2019, 16, 1312–1326. [Google Scholar] [CrossRef]
- Luo, H.; Saubamea, B.; Chasseigneaux, S.; Cochois, V.; Smirnova, M.; Glacial, F.; Perriere, N.; Chaves, C.; Cisternino, S.; Decleves, X. Molecular and Functional Study of Transient Receptor Potential Vanilloid 1-4 at the Rat and Human Blood-Brain Barrier Reveals Interspecies Differences. Front. Cell Dev. Biol. 2020, 8, 578514. [Google Scholar] [CrossRef]
- Geng, L.; Zhang, C.; He, C.; Zhang, K.; Kan, H.; Mao, A.; Ma, X. Physiological levels of fluid shear stress modulate vascular function through TRPV4 sparklets. Acta Biochim. Biophys. Sin. 2022, 54, 1268–1277. [Google Scholar] [CrossRef]
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef]
- Tiruppathi, C.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 2006, 13, 693–708. [Google Scholar] [CrossRef]
- Phuong, T.T.T.; Redmon, S.N.; Yarishkin, O.; Winter, J.M.; Li, D.Y.; Krizaj, D. Calcium influx through TRPV4 channels modulates the adherens contacts between retinal microvascular endothelial cells. J. Physiol. 2017, 595, 6869–6885. [Google Scholar] [CrossRef]
- Ke, S.K.; Chen, L.; Duan, H.B.; Tu, Y.R. Opposing actions of TRPV4 channel activation in the lung vasculature. Respir. Physiol. Neurobiol. 2015, 219, 43–50. [Google Scholar] [CrossRef]
- Akazawa, Y.; Yuki, T.; Yoshida, H.; Sugiyama, Y.; Inoue, S. Activation of TRPV4 strengthens the tight-junction barrier in human epidermal keratinocytes. Skin Pharmacol. Physiol. 2013, 26, 15–21. [Google Scholar] [CrossRef]
- Willette, R.N.; Bao, W.; Nerurkar, S.; Yue, T.L.; Doe, C.P.; Stankus, G.; Turner, G.H.; Ju, H.; Thomas, H.; Fishman, C.E.; et al. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J. Pharmacol. Exp. Ther. 2008, 326, 443–452. [Google Scholar] [CrossRef]
- Villalta, P.C.; Rocic, P.; Townsley, M.I. Role of MMP2 and MMP9 in TRPV4-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L652–L659. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yang, Z.; Jacoby, R.A.; Wu, S.M.; Pang, J.J. The expression and function of TRPV4 channels in primate retinal ganglion cells and bipolar cells. Cell Death Dis. 2019, 10, 364. [Google Scholar] [CrossRef] [PubMed]
- Redmon, S.N.; Yarishkin, O.; Lakk, M.; Jo, A.; Mustafic, E.; Tvrdik, P.; Krizaj, D. TRPV4 channels mediate the mechanoresponse in retinal microglia. Glia 2021, 69, 1563–1582. [Google Scholar] [CrossRef] [PubMed]
- Harraz, O.F.; Longden, T.A.; Dabertrand, F.; Hill-Eubanks, D.; Nelson, M.T. Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion. Proc. Natl. Acad. Sci. USA 2018, 115, E3569–E3577. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic Acid Evokes an Increase in Intracellular Ca2+ Concentration and Nitric Oxide Production in Endothelial Cells from Human Brain Microcirculation. Cells 2019, 8, 689. [Google Scholar] [CrossRef]
- Cappelli, H.C.; Guarino, B.D.; Kanugula, A.K.; Adapala, R.K.; Perera, V.; Smith, M.A.; Paruchuri, S.; Thodeti, C.K. Transient receptor potential vanilloid 4 channel deletion regulates pathological but not developmental retinal angiogenesis. J. Cell. Physiol. 2021, 236, 3770–3779. [Google Scholar] [CrossRef]
- Moccia, F.; Dragoni, S.; Cinelli, M.; Montagnani, S.; Amato, B.; Rosti, V.; Guerra, G.; Tanzi, F. How to utilize Ca2+ signals to rejuvenate the repairative phenotype of senescent endothelial progenitor cells in elderly patients affected by cardiovascular diseases: A useful therapeutic support of surgical approach? BMC Surg. 2013, 13 (Suppl. S2), S46. [Google Scholar] [CrossRef]
- Smani, T.; Gomez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.M.; Rosado, J.A. TRP Channels in Angiogenesis and Other Endothelial Functions. Front. Physiol. 2018, 9, 1731. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, M.M.; Prakriya, M. Calcium signaling at the interface between astrocytes and brain inflammation. Curr. Opin. Neurobiol. 2025, 90, 102940. [Google Scholar] [CrossRef] [PubMed]
- Shahulhameed, S.; Swain, S.; Jana, S.; Chhablani, J.; Ali, M.J.; Pappuru, R.R.; Tyagi, M.; Vishwakarma, S.; Sailaja, N.; Chakrabarti, S.; et al. A Robust Model System for Retinal Hypoxia: Live Imaging of Calcium Dynamics and Gene Expression Studies in Primary Human Mixed Retinal Culture. Front. Neurosci. 2019, 13, 1445. [Google Scholar] [CrossRef] [PubMed]
- Llorian-Salvador, M.; Cabeza-Fernandez, S.; Gomez-Sanchez, J.A.; de la Fuente, A.G. Glial cell alterations in diabetes-induced neurodegeneration. Cell. Mol. Life Sci. 2024, 81, 47. [Google Scholar] [CrossRef]
- Rohowetz, L.J.; Kraus, J.G.; Koulen, P. Reactive Oxygen Species-Mediated Damage of Retinal Neurons: Drug Development Targets for Therapies of Chronic Neurodegeneration of the Retina. Int. J. Mol. Sci. 2018, 19, 3362. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Interplay of mitochondrial calcium signalling and reactive oxygen species production in the brain. Biochem. Soc. Trans. 2024, 52, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, B.J.; Singer, H.A.; Adam, A.P.; Ginnan, R.G. CaMKIIdelta is upregulated by pro-inflammatory cytokine IL-6 in a JAK/STAT3-dependent manner to promote angiogenesis. FASEB J. 2021, 35, e21437. [Google Scholar] [CrossRef]
- Li, X.; Kerindongo, R.P.; Preckel, B.; Kalina, J.O.; Hollmann, M.W.; Zuurbier, C.J.; Weber, N.C. Canagliflozin inhibits inflammasome activation in diabetic endothelial cells-Revealing a novel calcium-dependent anti-inflammatory effect of canagliflozin on human diabetic endothelial cells. Biomed. Pharmacother. 2023, 159, 114228. [Google Scholar] [CrossRef]
- Li, X.; Wang, M.; Wolfsgruber, M.; Klatt, O.C.; Hollmann, M.W.; Preckel, B.; Zuurbier, C.J.; Weber, N.C. Empagliflozin prevents TNF-alpha induced endothelial dysfunction under flow -the potential involvement of calcium and sodium-hydrogen exchanger. Eur. J. Pharmacol. 2025, 986, 177147. [Google Scholar] [CrossRef]
- Propson, N.E.; Roy, E.R.; Litvinchuk, A.; Kohl, J.; Zheng, H. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J. Clin. Investig. 2021, 131, e140966. [Google Scholar] [CrossRef]
- Shahulhameed, S.; Vishwakarma, S.; Chhablani, J.; Tyagi, M.; Pappuru, R.R.; Jakati, S.; Chakrabarti, S.; Kaur, I. A Systematic Investigation on Complement Pathway Activation in Diabetic Retinopathy. Front. Immunol. 2020, 11, 154. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.A.; Lois, N.; Royle, P.; Clar, C.; Shyangdan, D.; Waugh, N. Current treatments in diabetic macular oedema: Systematic review and meta-analysis. BMJ Open 2013, 3, e002269. [Google Scholar] [CrossRef]
- Berlansky, S.; Sallinger, M.; Grabmayr, H.; Humer, C.; Bernhard, A.; Fahrner, M.; Frischauf, I. Calcium Signals during SARS-CoV-2 Infection: Assessing the Potential of Emerging Therapies. Cells 2022, 11, 253. [Google Scholar] [CrossRef]
- Miller, J.; Bruen, C.; Schnaus, M.; Zhang, J.; Ali, S.; Lind, A.; Stoecker, Z.; Stauderman, K.; Hebbar, S. Auxora versus standard of care for the treatment of severe or critical COVID-19 pneumonia: Results from a randomized controlled trial. Crit. Care 2020, 24, 502. [Google Scholar] [CrossRef] [PubMed]
- Scorza, S.; Brunetti, V.; Scarpellino, G.; Certini, M.; Gerbino, A.; Moccia, F. Targeting the Ca2+ signaling toolkit as an alternative strategy to mitigate SARS-CoV-2-induced cardiovascular adverse events. Vasc. Pharmacol. 2025, 158, 107458. [Google Scholar] [CrossRef]
- Gunaratne, G.S.; Marchant, J.S. The ins and outs of virus trafficking through acidic Ca2+ stores. Cell Calcium 2022, 102, 102528. [Google Scholar] [CrossRef] [PubMed]
- Gunaratne, G.S.; Johns, M.E.; Hintz, H.M.; Walseth, T.F.; Marchant, J.S. A screening campaign in sea urchin egg homogenate as a platform for discovering modulators of NAADP-dependent Ca2+ signaling in human cells. Cell Calcium 2018, 75, 42–52. [Google Scholar] [CrossRef]
- Penny, C.J.; Vassileva, K.; Jha, A.; Yuan, Y.; Chee, X.; Yates, E.; Mazzon, M.; Kilpatrick, B.S.; Muallem, S.; Marsh, M.; et al. Mining of Ebola virus entry inhibitors identifies approved drugs as two-pore channel pore blockers. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1151–1161. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef]
- O’Connor, B.; Robbins, N.; Koch, S.E.; Rubinstein, J. TRPV2 channel-based therapies in the cardiovascular field. Molecular underpinnings of clinically relevant therapies. Prog. Biophys. Mol. Biol. 2021, 159, 118–125. [Google Scholar] [CrossRef]
- Pero, J.E.; McAtee, J.J.; Behm, D.J.; Briand, J.; Graczyk-Millbrandt, G.; Erhard, K.; Roberts, A.D.; Rivero, R.A.; Holt, D.A.; Lawhorn, B.G. Identification, Synthesis, and Characterization of a Major Circulating Human Metabolite of TRPV4 Antagonist GSK2798745. ACS Med. Chem. Lett. 2021, 12, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Nishinaka, A.; Tanaka, M.; Ohara, K.; Sugaru, E.; Shishido, Y.; Sugiura, A.; Moriguchi, Y.; Toui, A.; Nakamura, S.; Shimada, K.; et al. TRPV4 channels promote vascular permeability in retinal vascular disease. Exp. Eye Res. 2023, 228, 109405. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).