

Avenanthramide-C as Alzheimer’s Disease-Modifying Therapy: Early and Sustained Intervention Prevents Disease Progression in Mouse Models

 , , , , and
, , , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mouse Models
2.2. Hippocampal Slice Preparation
2.3. Electrophysiology
2.4. Avn-C Preparation and Administration
2.5. Western Blot
2.6. Immunohistochemistry (IHC)
2.7. Amyloid-β (1-42) Preparation
2.8. BV-2 Cell Culture
2.9. Phagocytosis Assay
2.10. Immunocytochemistry
2.11. Statistical Analysis
3. Results
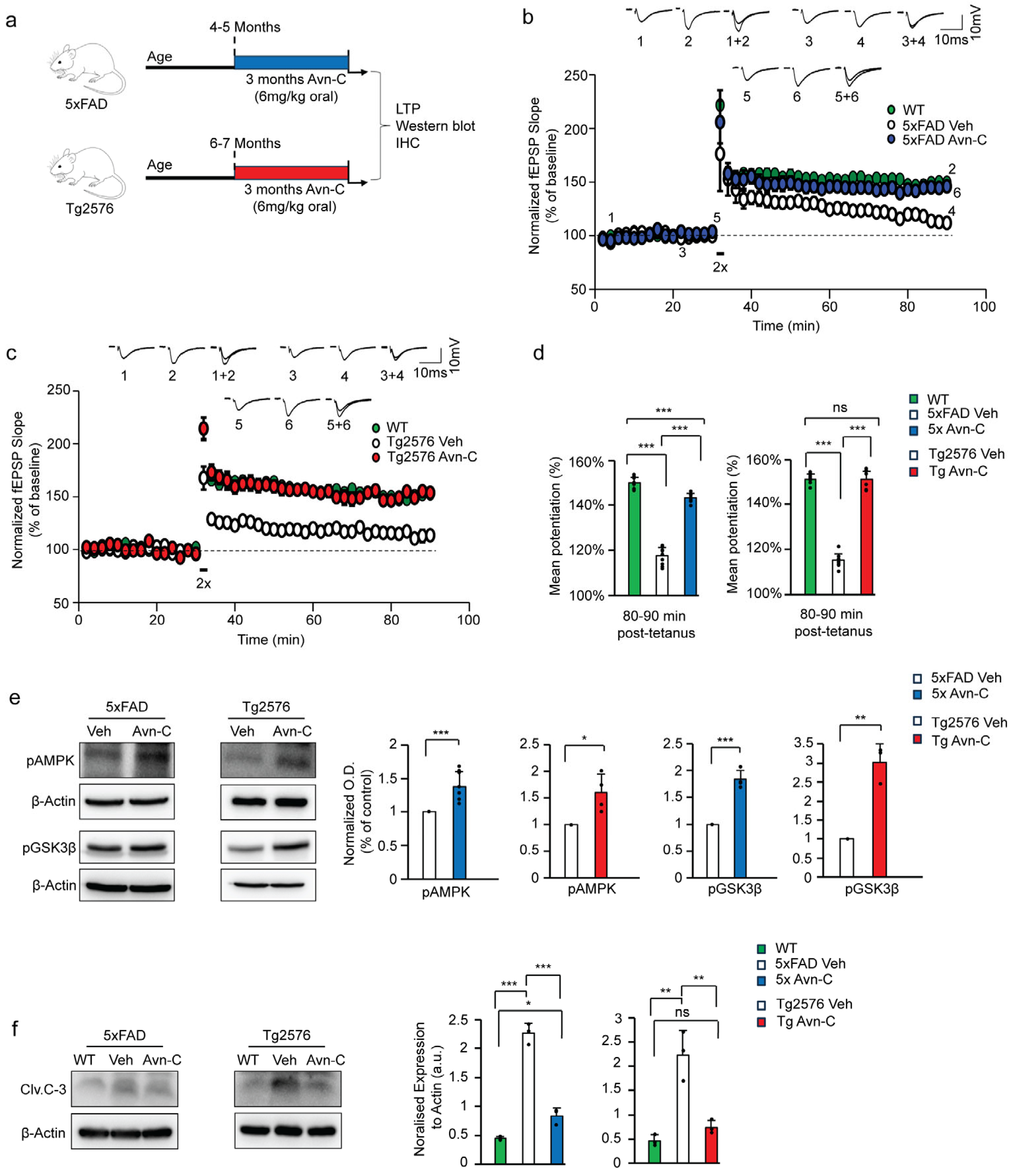
3.1. Long-Term Avn-C Administration from the Early AD Stage Restores and Maintains LTP While Preventing Further Synaptic Impairment
3.2. Sustained Avn-C Administration from Early AD Stage Preserves AMPK Activation, Inhibits GSK3β and Caspase-3, and Prevents Neurodegeneration
3.3. Three-Month Oral Administration of Avn-C from the Early AD Stage Attenuates Amyloidogenic Processing and Aβ1-42 Production
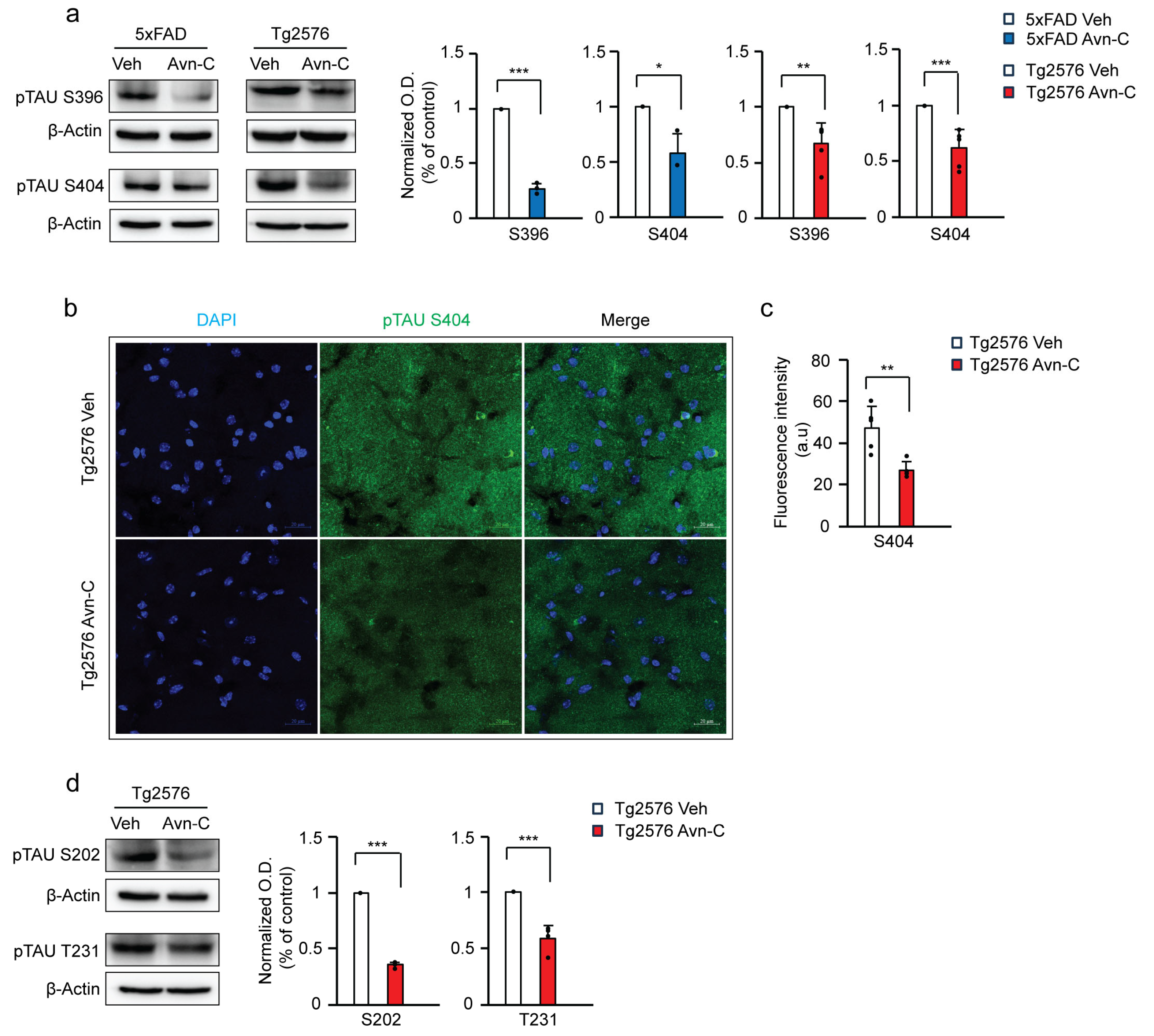
3.4. Long-Term Oral Administration of Avn-C from the Early AD Stage Attenuates GSK3β-Mediated Tau Hyperphosphorylation
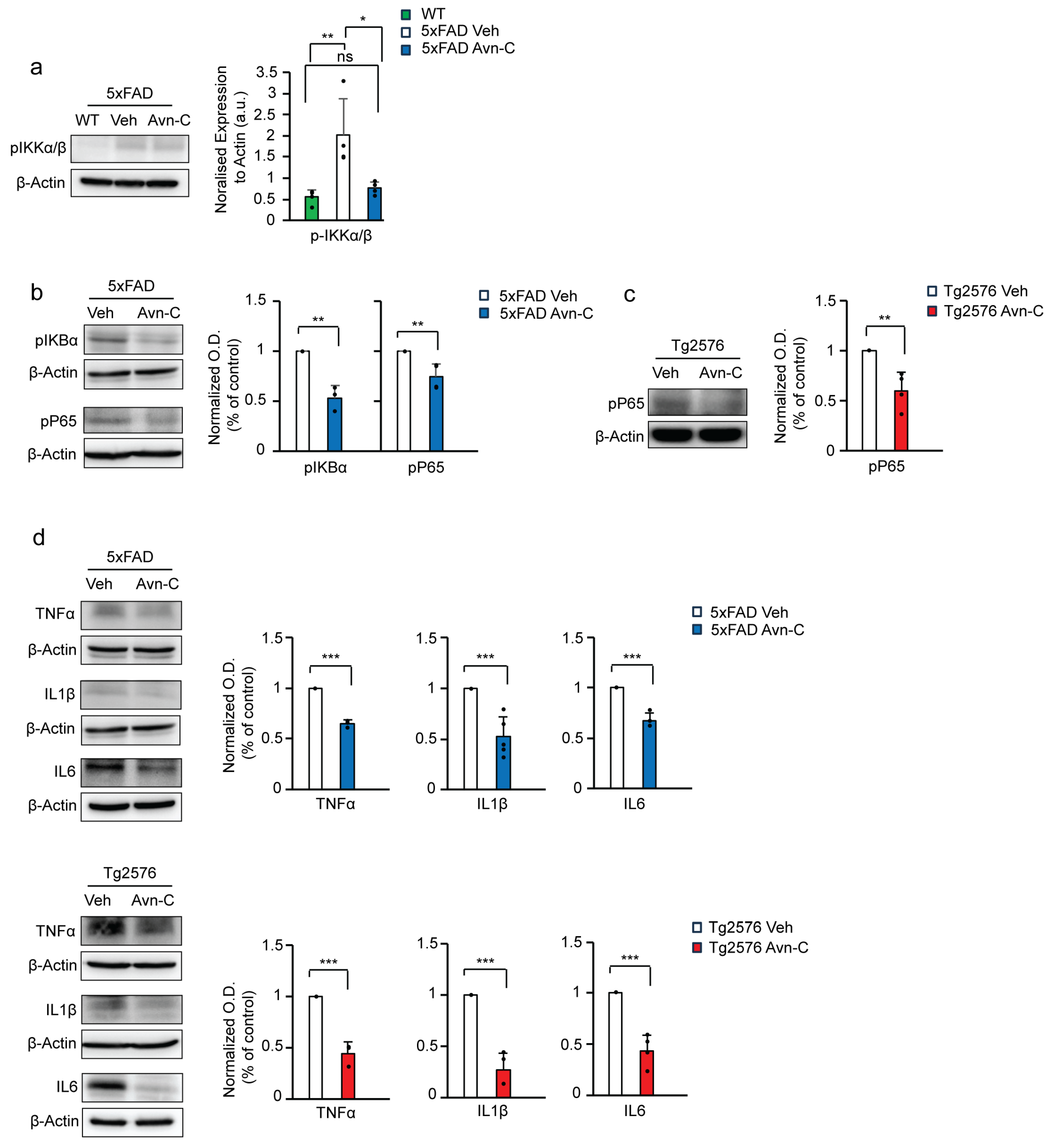
3.5. Avn-C Administration from the Early AD Stage Sustains Anti-Inflammatory Effects and Prevents Neuroinflammation
3.6. Long-Term Avn-C Treatment from the Early AD Stage Modulates Microglial Morphology and Reduces Chronic Activation
3.7. Early and Sustained Avn-C Treatment Reduces Large Amyloid Plaques and Promotes Microglial Barrier Formation Around Smaller Plaques in the Hippocampus
3.8. Avn-C Protects Microglial Cells from Oligo-Aβ1-42-Induced Phagocytic Impairment
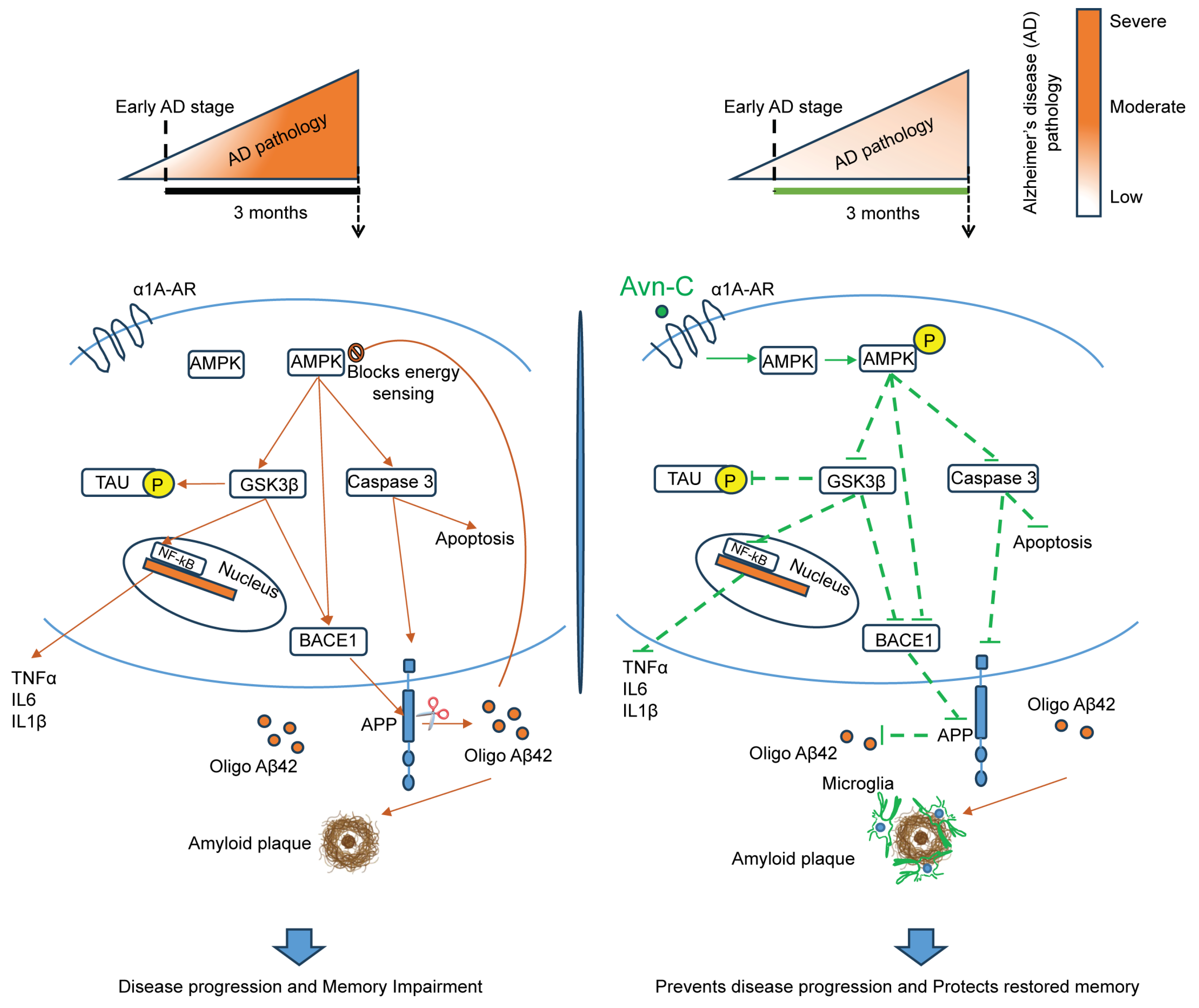
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Avn-C | Avenanthramide-C |
| APP | Amyloid precursor protein |
| Aβ1-42 | Amyloid-beta 1-42 |
| oAβ1-42 | Oligomer Amyloid beta1-42 |
| fEPSP | Field excitatory postsynaptic potential |
| LTP | Long-term potentiation |
| CA | Cornu Ammonis |
| AMPK | Adenosine monophosphate-activated protein kinase |
| GSK3β | Glycogen synthase kinase three β |
| BACE1 | Beta-site amyloid precursor protein cleaving enzyme 1 |
| TNF⍺ | Tumor necrosis factor-alpha |
| IL6 | Interleukin 6 |
| IL1β | Interleukin 1 beta |
| Iba1 | Ionized calcium-binding adaptor molecule 1 |
References
- Carlesimo, G.A.; Oscar-Berman, M. Memory deficits in Alzheimer’s patients: A comprehensive review. Neuropsychol. Rev. 1992, 3, 119–169. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.B.; Albert, M.S.; Butters, N.; Payne, M. Differential patterns of memory loss among patients with Alzheimer’s disease, Huntington’s disease, and alcoholic Korsakoff’s syndrome. Arch. Neurol. 1986, 43, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimer’s Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci. Ther. 2011, 17, 514–524. [Google Scholar] [CrossRef]
- Kulkarni, B.; Cruz-Martins, N.; Kumar, D. Microglia in Alzheimer’s Disease: An Unprecedented Opportunity as Prospective Drug Target. Mol. Neurobiol. 2022, 59, 2678–2693. [Google Scholar] [CrossRef]
- Zhou, L.-J.; Peng, J.; Xu, Y.-N.; Zeng, W.-J.; Zhang, J.; Wei, X.; Mai, C.-L.; Lin, Z.-J.; Liu, Y.; Murugan, M.; et al. Microglia Are Indispensable for Synaptic Plasticity in the Spinal Dorsal Horn and Chronic Pain. Cell Rep. 2019, 27, 3844–3859.e3846. [Google Scholar] [CrossRef]
- Chapman, P.F.; White, G.L.; Jones, M.W.; Cooper-Blacketer, D.; Marshall, V.J.; Irizarry, M.; Younkin, L.; Good, M.A.; Bliss, T.V.P.; Hyman, B.T.; et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat. Neurosci. 1999, 2, 271–276. [Google Scholar] [CrossRef]
- Cummings, J.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer’s disease drug development pipeline: 2023. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2023, 9, e12385. [Google Scholar] [CrossRef]
- Hansen, R.A.; Gartlehner, G.; Webb, A.P.; Morgan, L.C.; Moore, C.G.; Jonas, D.E. Efficacy and safety of donepezil, galantamine, and rivastigmine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. Clin. Interv. Aging 2008, 3, 211–225. [Google Scholar]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.K.; Day, G.S. Anti-amyloid therapies for Alzheimer disease: Finally, good news for patients. Mol. Neurodegener. 2023, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Atri, A.; Shaughnessy, L.W.; Locascio, J.J.; Growdon, J.H. Long-term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2008, 22, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Marinangeli, C.; Didier, S.; Ahmed, T.; Caillerez, R.; Domise, M.; Laloux, C.; Bégard, S.; Carrier, S.; Colin, M.; Marchetti, P.; et al. AMP-Activated Protein Kinase Is Essential for the Maintenance of Energy Levels during Synaptic Activation. iScience 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Sayas, C.L.; Ávila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, S.; Li, M.; Ke, D.; Wang, Q.; Yang, Y.; Liu, G.P.; Wang, X.C.; Liu, E.; Wang, J.Z. Human tau accumulation promotes glycogen synthase kinase-3β acetylation and thus upregulates the kinase: A vicious cycle in Alzheimer neurodegeneration. EBioMedicine 2022, 78, 103970. [Google Scholar] [CrossRef]
- Greco, S.J.; Sarkar, S.; Johnston, J.M.; Tezapsidis, N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem. Biophys. Res. Commun. 2009, 380, 98–104. [Google Scholar] [CrossRef]
- Mair, W.; Morantte, I.; Rodrigues, A.P.; Manning, G.; Montminy, M.; Shaw, R.J.; Dillin, A. Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature 2011, 470, 404–408. [Google Scholar] [CrossRef]
- Du, L.L.; Chai, D.M.; Zhao, L.N.; Li, X.H.; Zhang, F.C.; Zhang, H.B.; Liu, L.B.; Wu, K.; Liu, R.; Wang, J.Z.; et al. AMPK activation ameliorates Alzheimer’s disease-like pathology and spatial memory impairment in a streptozotocin-induced Alzheimer’s disease model in rats. J. Alzheimer’s Dis. 2015, 43, 775–784. [Google Scholar] [CrossRef]
- Cho, S.Y.; Kim, E.W.; Park, S.J.; Phillips, B.U.; Jeong, J.; Kim, H.; Heath, C.J.; Kim, D.; Jang, Y.; López-Cruz, L.; et al. Reconsidering repurposing: Long-term metformin treatment impairs cognition in Alzheimer’s model mice. Transl. Psychiatry 2024, 14, 34. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Pratico, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, V.S.; Samidurai, M.; Park, H.J.; Wang, M.; Park, R.Y.; Yu, S.Y.; Kang, H.K.; Hong, S.; Choi, W.S.; Lee, Y.Y.; et al. Avenanthramide-C Restores Impaired Plasticity and Cognition in Alzheimer’s Disease Model Mice. Mol. Neurobiol. 2020, 57, 315–330. [Google Scholar] [CrossRef]
- Lee, Y.-Y.; Wang, M.; Son, Y.; Yang, E.-J.; Kang, M.-S.; Kim, H.-J.; Kim, H.-S.; Jo, J. Oat Extract Avenanthramide-C Reverses Hippocampal Long-Term Potentiation Decline in Tg2576 Mice. Molecules 2021, 26, 6105. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.O.; Milbury, P.E.; Collins, F.W.; Blumberg, J.B. Avenanthramides Are Bioavailable and Have Antioxidant Activity in Humans after Acute Consumption of an Enriched Mixture from Oats12. J. Nutr. 2007, 137, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Mango, D.; Saidi, A.; Cisale, G.Y.; Feligioni, M.; Corbo, M.; Nisticò, R. Targeting Synaptic Plasticity in Experimental Models of Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 778. [Google Scholar] [CrossRef]
- Westi, E.W.; Molhemi, S.; Hansen, C.T.; Skoven, C.S.; Knopper, R.W.; Ahmad, D.A.; Rindshøj, M.B.; Ameen, A.O.; Hansen, B.; Kohlmeier, K.A.; et al. Comprehensive Analysis of the 5xFAD Mouse Model of Alzheimer’s Disease Using dMRI, Immunohistochemistry, and Neuronal and Glial Functional Metabolic Mapping. Biomolecules 2024, 14, 1294. [Google Scholar] [CrossRef]
- Huh, S.; Baek, S.-J.; Lee, K.-H.; Whitcomb, D.J.; Jo, J.; Choi, S.-M.; Kim, D.H.; Park, M.-S.; Lee, K.H.; Kim, B.C. The reemergence of long-term potentiation in aged Alzheimer’s disease mouse model. Sci. Rep. 2016, 6, 29152. [Google Scholar] [CrossRef]
- Woods, A.; Vertommen, D.; Neumann, D.; Türk, R.; Bayliss, J.; Schlattner, U.; Wallimann, T.; Carling, D.; Rider, M.H. Identification of Phosphorylation Sites in AMP-activated Protein Kinase (AMPK) for Upstream AMPK Kinases and Study of Their Roles by Site-directed Mutagenesis. J. Biol. Chem. 2003, 278, 28434–28442. [Google Scholar] [CrossRef]
- Lin-Marq, N.; Borel, C.; Antonarakis, S.E. Peutz-Jeghers LKB1 mutants fail to activate GSK-3beta, preventing it from inhibiting Wnt signaling. Mol. Genet. Genom. 2005, 273, 184–196. [Google Scholar] [CrossRef]
- Peineau, S.; Bradley, C.; Taghibiglou, C.; Doherty, A.; Bortolotto, Z.A.; Wang, Y.T.; Collingridge, G.L. The role of GSK-3 in synaptic plasticity. Br. J. Pharmacol. 2008, 153 (Suppl. S1), S428–S437. [Google Scholar] [CrossRef]
- Louneva, N.; Cohen, J.W.; Han, L.Y.; Talbot, K.; Wilson, R.S.; Bennett, D.A.; Trojanowski, J.Q.; Arnold, S.E. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. Am. J. Pathol. 2008, 173, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, X.; Xia, W.; Zhang, Y.; Wang, C. Targeting Amyloidogenic Processing of APP in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- da Silva Correia, A.; Schmitz, M.; Fischer, A.-L.; da Silva Correia, S.; Simonetti, F.L.; Saher, G.; Goya-Maldonado, R.; Arora, A.S.; Fischer, A.; Outeiro, T.F.; et al. Cellular prion protein acts as mediator of amyloid beta uptake by caveolin-1 causing cellular dysfunctions in vitro and in vivo. Alzheimer’s Dement. 2024, 20, 6776–6792. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zimmermann, H.R.; Ma, T. Therapeutic Potential of AMP-Activated Protein Kinase in Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 68, 33–38. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef]
- Cai, H.; Wang, Y.; McCarthy, D.; Wen, H.; Borchelt, D.R.; Price, D.L.; Wong, P.C. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 2001, 4, 233–234. [Google Scholar] [CrossRef]
- Liu, Q.F.; Kanmani, S.; Lee, J.; Kim, G.-W.; Jeon, S.; Koo, B.-S. Neoline Improves Memory Impairment and Reduces Amyloid-β Level and Tau Phosphorylation Through AMPK Activation in the Mouse Alzheimer’s Disease Model. J. Alzheimer’s Dis. 2021, 81, 507–516. [Google Scholar] [CrossRef]
- Choi, H.-J.; Park, J.-H.; Jeong, Y.J.; Hwang, J.-W.; Lee, S.; Lee, H.; Seol, E.; Kim, I.-w.; Cha, B.-Y.; Seo, J.; et al. Donepezil ameliorates Aβ pathology but not tau pathology in 5xFAD mice. Mol. Brain 2022, 15, 63. [Google Scholar] [CrossRef]
- Godemann, R.; Biernat, J.; Mandelkow, E.; Mandelkow, E.M. Phosphorylation of tau protein by recombinant GSK-3beta: Pronounced phosphorylation at select Ser/Thr-Pro motifs but no phosphorylation at Ser262 in the repeat domain. FEBS Lett. 1999, 454, 157–164. [Google Scholar] [CrossRef]
- DaRocha-Souto, B.; Coma, M.; Pérez-Nievas, B.G.; Scotton, T.C.; Siao, M.; Sánchez-Ferrer, P.; Hashimoto, T.; Fan, Z.; Hudry, E.; Barroeta, I.; et al. Activation of glycogen synthase kinase-3 beta mediates β-amyloid induced neuritic damage in Alzheimer’s disease. Neurobiol. Dis. 2012, 45, 425–437. [Google Scholar] [CrossRef]
- Israël, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [PubMed]
- Finco, T.S.; Beg, A.A.; Baldwin, A.S., Jr. Inducible phosphorylation of I kappa B alpha is not sufficient for its dissociation from NF-kappa B and is inhibited by protease inhibitors. Proc. Natl. Acad. Sci. USA 1994, 91, 11884–11888. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Parent, L.; Maniatis, T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell 1996, 84, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S., Jr. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef]
- Mathes, E.; O’Dea, E.L.; Hoffmann, A.; Ghosh, G. NF-kappaB dictates the degradation pathway of IkappaBalpha. EMBO J. 2008, 27, 1357–1367. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef]
- Constanzo, J.; Midavaine, É.; Fouquet, J.; Lepage, M.; Descoteaux, M.; Kirby, K.; Tremblay, L.; Masson-Côté, L.; Geha, S.; Longpré, J.-M.; et al. Brain irradiation leads to persistent neuroinflammation and long-term neurocognitive dysfunction in a region-specific manner. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 102, 109954. [Google Scholar] [CrossRef]
- Reddaway, J.; Richardson, P.E.; Bevan, R.J.; Stoneman, J.; Palombo, M. Microglial morphometric analysis: So many options, so little consistency. Front. Neuroinform. 2023, 17, 1211188. [Google Scholar] [CrossRef]
- Davis, B.M.; Salinas-Navarro, M.; Cordeiro, M.F.; Moons, L.; De Groef, L. Characterizing microglia activation: A spatial statistics approach to maximize information extraction. Sci. Rep. 2017, 7, 1576. [Google Scholar] [CrossRef]
- Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef]
- Shi, F.J.; Xie, H.; Zhang, C.Y.; Qin, H.F.; Zeng, X.W.; Lou, H.; Zhang, L.; Xu, G.T.; Zhang, J.F.; Xu, G.X. Is Iba-1 protein expression a sensitive marker for microglia activation in experimental diabetic retinopathy? Int. J. Ophthalmol. 2021, 14, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Tanaka, K.; Suzuki, S.; Dembo, T.; Fukuuchi, Y. Enhanced Expression of Iba1, Ionized Calcium-Binding Adapter Molecule 1, After Transient Focal Cerebral Ischemia In Rat Brain. Stroke 2001, 32, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef]
- Qiao, O.; Ji, H.; Zhang, Y.; Zhang, X.; Zhang, X.; Liu, N.; Huang, L.; Liu, C.; Gao, W. New insights in drug development for Alzheimer’s disease based on microglia function. Biomed. Pharmacother. 2021, 140, 111703. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 92, 252–264. [Google Scholar] [CrossRef]
- Pan, X.-d.; Zhu, Y.-g.; Lin, N.; Zhang, J.; Ye, Q.-y.; Huang, H.-p.; Chen, X.-c. Microglial phagocytosis induced by fibrillar β-amyloid is attenuated by oligomeric β-amyloid: Implications for Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 45. [Google Scholar] [CrossRef]
- Duff, K.; Beglinger, L.J.; Van Der Heiden, S.; Moser, D.J.; Arndt, S.; Schultz, S.K.; Paulsen, J.S. Short-term practice effects in amnestic mild cognitive impairment: Implications for diagnosis and treatment. Int. Psychogeriatr. 2008, 20, 986–999. [Google Scholar] [CrossRef]
- Jelic, V.; Kivipelto, M.; Winblad, B. Clinical trials in mild cognitive impairment: Lessons for the future. J. Neurol. Neurosurg. Psychiatry 2006, 77, 429–438. [Google Scholar] [CrossRef]
- Rao, Y.L.; Ganaraja, B.; Murlimanju, B.V.; Joy, T.; Krishnamurthy, A.; Agrawal, A. Hippocampus and its involvement in Alzheimer’s disease: A review. 3 Biotech 2022, 12, 55. [Google Scholar] [CrossRef]
- Seixas da Silva, G.S.; Melo, H.M.; Lourenco, M.V.; Lyra, E.S.N.M.; de Carvalho, M.B.; Alves-Leon, S.V.; de Souza, J.M.; Klein, W.L.; da-Silva, W.S.; Ferreira, S.T.; et al. Amyloid-β oligomers transiently inhibit AMP-activated kinase and cause metabolic defects in hippocampal neurons. J. Biol. Chem. 2017, 292, 7395–7406. [Google Scholar] [CrossRef]
- Luo, Y.; Smith, J.V.; Paramasivam, V.; Burdick, A.; Curry, K.J.; Buford, J.P.; Khan, I.; Netzer, W.J.; Xu, H.; Butko, P. Inhibition of amyloid-β aggregation and caspase-3 activation by the Ginkgo biloba extract EGb761. Proc. Natl. Acad. Sci. USA 2002, 99, 12197–12202. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; Noguchi, K.; Sato, K.; Hoshino, T.; Imahori, K. Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 1993, 90, 7789–7793. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Hernández, F. GSK-3 inhibitors for Alzheimer’s disease. Expert Rev. Neurother. 2007, 7, 1527–1533. [Google Scholar] [CrossRef]
- Li, W.; Sperry, J.B.; Crowe, A.; Trojanowski, J.Q.; Smith, A.B., 3rd; Lee, V.M. Inhibition of tau fibrillization by oleocanthal via reaction with the amino groups of tau. J. Neurochem. 2009, 110, 1339–1351. [Google Scholar] [CrossRef]
- Velander, P.; Wu, L.; Henderson, F.; Zhang, S.; Bevan, D.R.; Xu, B. Natural product-based amyloid inhibitors. Biochem. Pharmacol. 2017, 139, 40–55. [Google Scholar] [CrossRef]
- Park, G.; Nhan, H.S.; Tyan, S.H.; Kawakatsu, Y.; Zhang, C.; Navarro, M.; Koo, E.H. Caspase Activation and Caspase-Mediated Cleavage of APP Is Associated with Amyloid β-Protein-Induced Synapse Loss in Alzheimer’s Disease. Cell Rep. 2020, 31, 107839. [Google Scholar] [CrossRef]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body Fluid Cytokine Levels in Mild Cognitive Impairment and Alzheimer’s Disease: A Comparative Overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, J.; Rowan, M.J.; Anwyl, R. β-amyloid inhibition of long-term potentiation is mediated via tumor necrosis factor. Eur. J. Neurosci. 2005, 22, 2827–2832. [Google Scholar] [CrossRef]
- Guo, W.; Wise, M.L.; Collins, F.W.; Meydani, M. Avenanthramides, polyphenols from oats, inhibit IL-1β-induced NF-κB activation in endothelial cells. Free Radic. Biol. Med. 2008, 44, 415–429. [Google Scholar] [CrossRef]
- Wang, C.; Zong, S.; Cui, X.; Wang, X.; Wu, S.; Wang, L.; Liu, Y.; Lu, Z. The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Front. Immunol. 2023, 14, 1117172. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 2015, 6, 6176. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-L.; Chen, W.-T.; Lin, Y.-Y.; Lu, P.-H.; Hsieh, S.-L.; Cheng, I.H.-J. Amelioration of amyloid-β-induced deficits by DcR3 in an Alzheimer’s disease model. Mol. Neurodegener. 2017, 12, 30. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nathan, A.B.P.; Aziz, A.; Choi, S.; Lee, S.; Jeon, S.; Kim, H.-S.; Cho, J.; Jo, J. Avenanthramide-C as Alzheimer’s Disease-Modifying Therapy: Early and Sustained Intervention Prevents Disease Progression in Mouse Models. Cells 2025, 14, 826. https://doi.org/10.3390/cells14110826
Nathan ABP, Aziz A, Choi S, Lee S, Jeon S, Kim H-S, Cho J, Jo J. Avenanthramide-C as Alzheimer’s Disease-Modifying Therapy: Early and Sustained Intervention Prevents Disease Progression in Mouse Models. Cells. 2025; 14(11):826. https://doi.org/10.3390/cells14110826
Chicago/Turabian StyleNathan, Alen Benhur Pravin, Areeba Aziz, Semyeong Choi, Seunghee Lee, Seyoung Jeon, Hyung-Seok Kim, Jonghyun Cho, and Jihoon Jo. 2025. "Avenanthramide-C as Alzheimer’s Disease-Modifying Therapy: Early and Sustained Intervention Prevents Disease Progression in Mouse Models" Cells 14, no. 11: 826. https://doi.org/10.3390/cells14110826
APA StyleNathan, A. B. P., Aziz, A., Choi, S., Lee, S., Jeon, S., Kim, H.-S., Cho, J., & Jo, J. (2025). Avenanthramide-C as Alzheimer’s Disease-Modifying Therapy: Early and Sustained Intervention Prevents Disease Progression in Mouse Models. Cells, 14(11), 826. https://doi.org/10.3390/cells14110826