


Cellular Senescence, Mitochondrial Dysfunction, and Their Link to Cardiovascular Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
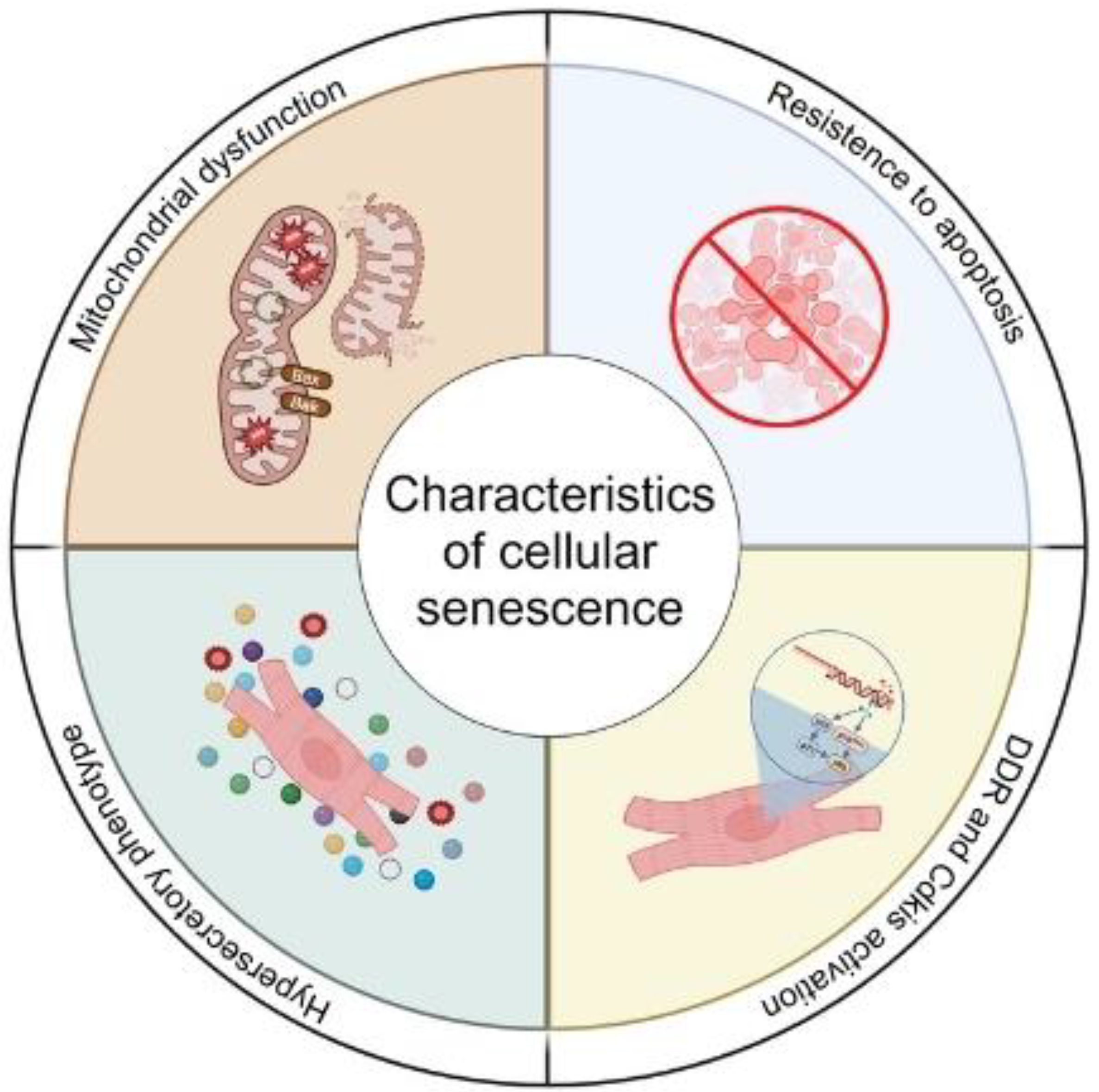
1. Cellular Senescence
2. Mitochondrial Abnormalities in Cardiovascular Diseases
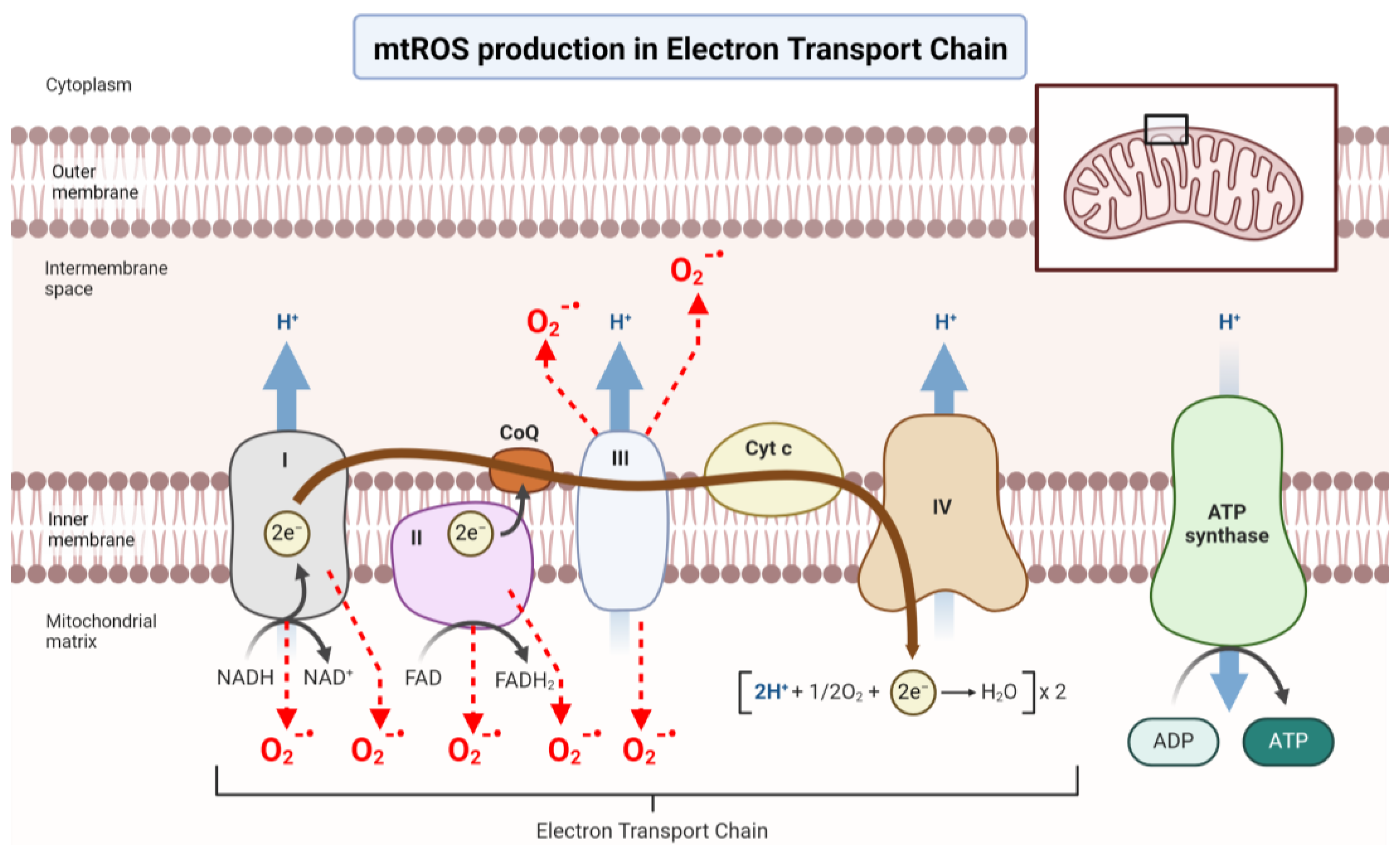
2.1. Energy Starvation and Oxidative Stress
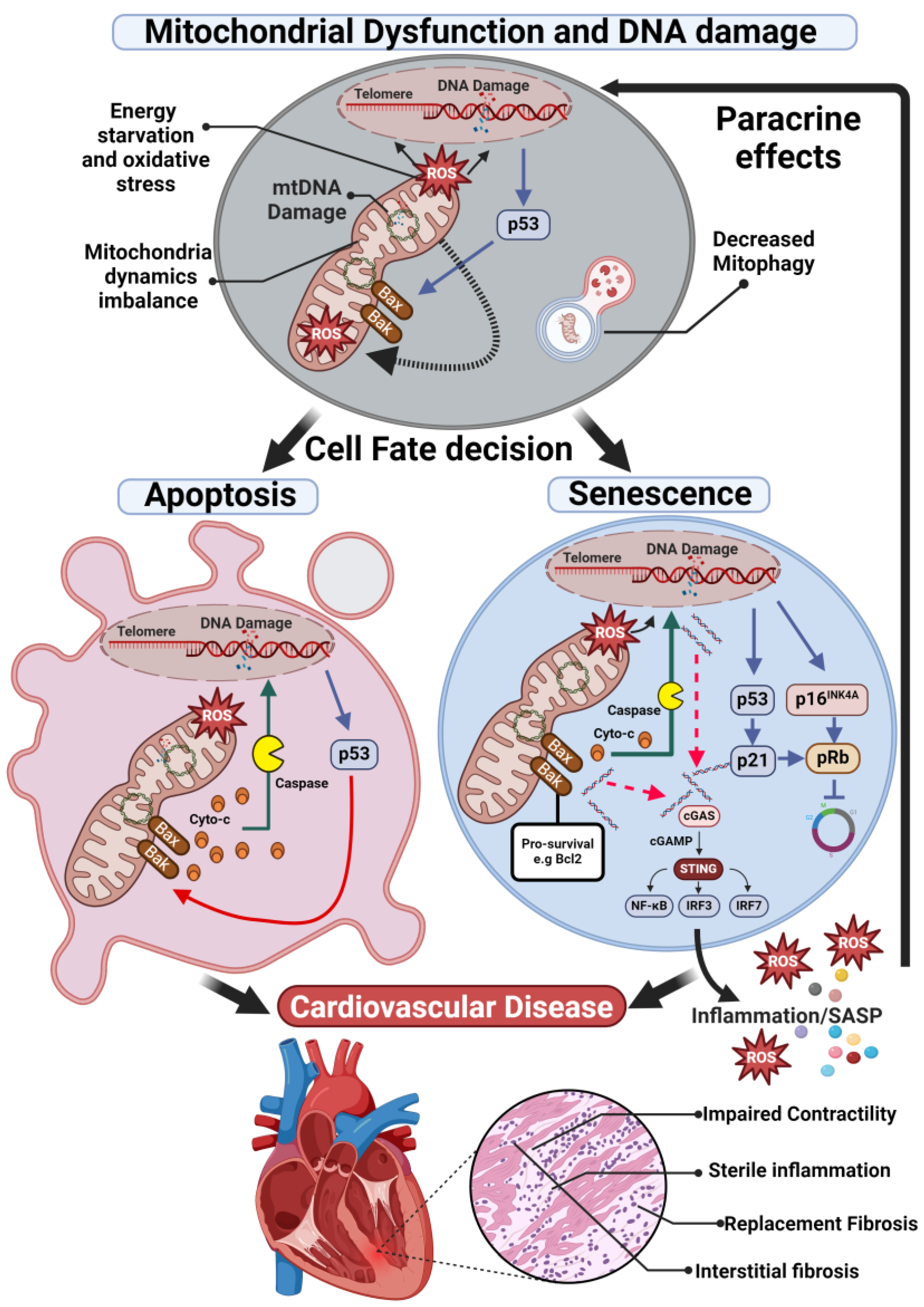
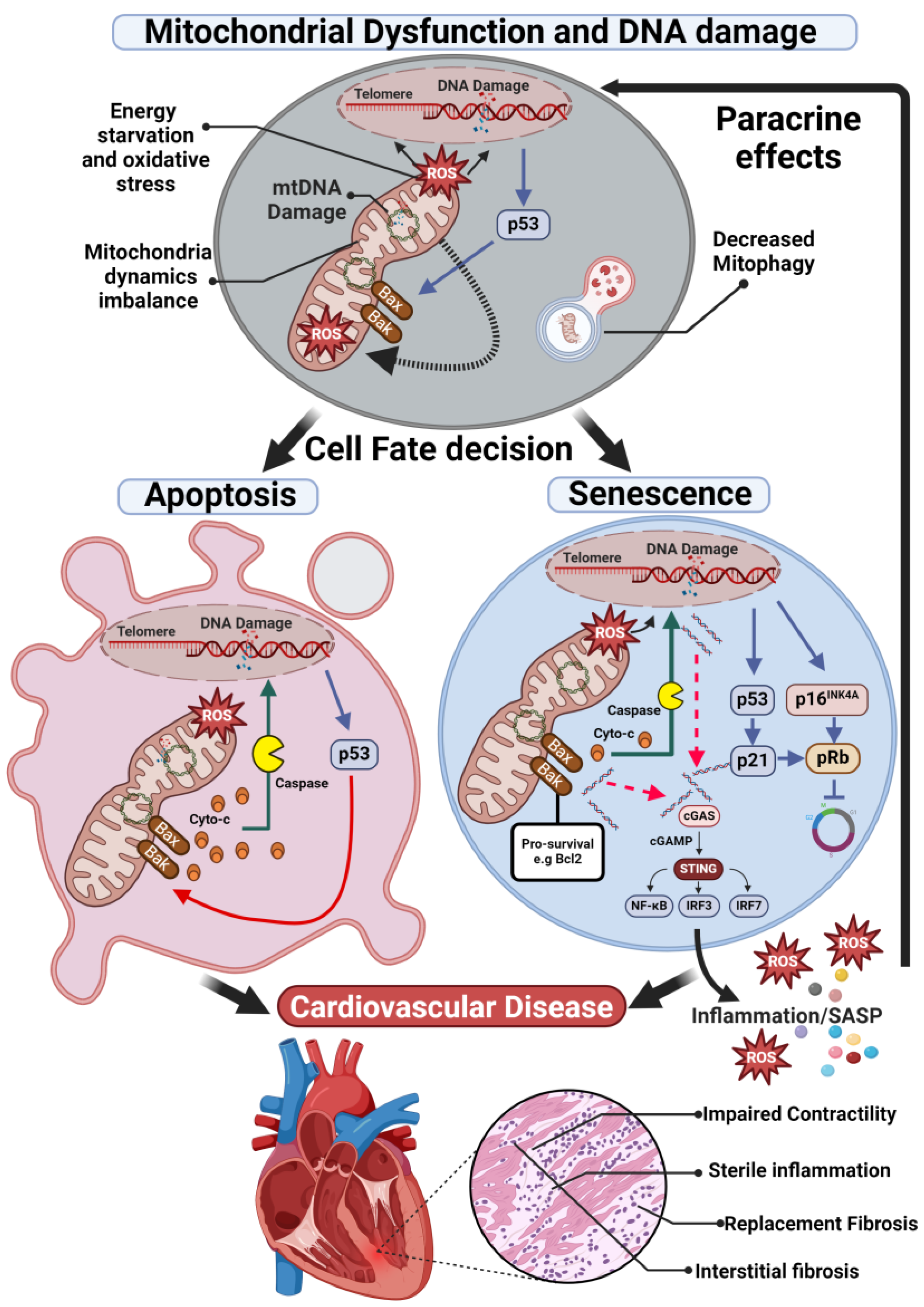
2.2. Mitochondria Dynamics Imbalance
2.3. Cell Apoptosis and Mitophagy
3. Mitochondria Dysfunction, Senescence, and Inflammation in CVD
4. Mitochondrial and Senescent Cells Targeted Therapies for CVD
4.1. Therapeutically Targeting Mitochondrial Dysfunction
4.2. Senolytics, Senomorphics and Future Approaches
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Redgrave, R.; Dookun, E.; Booth, L.; Folaranm, O.; Tual-Chalot, S.; Gill, J.; Owens, A.; Spyridopoulos, I.; Passos, J.; Richardson, G. Senescent cardiomyocytes contribute to cardiac dysfunction following myocardial infarction. NPJ Aging 2023, 9, 15. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef]
- Richardson, G.D. Simultaneous Assessment of Cardiomyocyte DNA Synthesis and Ploidy: A Method to Assist Quantification of Cardiomyocyte Regeneration and Turnover. J. Vis. Exp. 2016. [CrossRef]
- de Lange, T.; Shiue, L.; Myers, R.M.; Cox, D.R.; Naylor, S.L.; Killery, A.M.; Varmus, H.E. Structure and variability of human chromosome ends. Mol. Cell. Biol. 1990, 10, 518–527. [Google Scholar] [CrossRef]
- Szostak, J.W.; Blackburn, E.H. Cloning yeast telomeres on linear plasmid vectors. Cell 1982, 29, 245–255. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Anderson, R.; Richardson, G.D.; Passos, J.F. Mechanisms driving the ageing heart. Exp. Gerontol. 2018, 109, 5–15. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Dookun, E.; Walaszczyk, A.; Redgrave, R.; Palmowski, P.; Tual-Chalot, S.; Suwana, A.; Chapman, J.; Jirkovsky, E.; Donastorg Sosa, L.; Gill, E.; et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell 2020, 19, e13249. [Google Scholar] [CrossRef] [PubMed]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef]
- Salerno, N.; Marino, F.; Scalise, M.; Salerno, L.; Molinaro, C.; Filardo, A.; Chiefalo, A.; Panuccio, G.; De Angelis, A.; Urbanek, K.; et al. Pharmacological clearance of senescent cells improves cardiac remodeling and function after myocardial infarction in female aged mice. Mech. Ageing Dev. 2022, 208, 111740. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef]
- Balint, B.; Yin, H.; Nong, Z.; Arpino, J.M.; O’Neil, C.; Rogers, S.R.; Randhawa, V.K.; Fox, S.A.; Chevalier, J.; Lee, J.J.; et al. Seno-destructive smooth muscle cells in the ascending aorta of patients with bicuspid aortic valve disease. EBioMedicine 2019, 43, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Witham, M.D.; Granic, A.; Miwa, S.; Passos, J.F.; Richardson, G.D.; Sayer, A.A. New Horizons in cellular senescence for clinicians. Age Ageing 2023, 52, afad127. [Google Scholar] [CrossRef]
- Booth, L.K.; Redgrave, R.E.; Tual-Chalot, S.; Spyridopoulos, I.; Phillips, H.M.; Richardson, G.D. Heart Disease and Ageing: The Roles of Senescence, Mitochondria, and Telomerase in Cardiovascular Disease. Subcell. Biochem. 2023, 103, 45–78. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.K.; Redgrave, R.E.; Folaranmi, O.; Gill, J.H.; Richardson, G.D. Anthracycline-induced cardiotoxicity and senescence. Front. Aging 2022, 3, 1058435. [Google Scholar] [CrossRef]
- Dookun, E.; Passos, J.F.; Arthur, H.M.; Richardson, G.D. Therapeutic Potential of Senolytics in Cardiovascular Disease. Cardiovasc. Drugs Ther. 2022, 36, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Owens, W.A.; Walaszczyk, A.; Spyridopoulos, I.; Dookun, E.; Richardson, G.D. Senescence and senolytics in cardiovascular disease: Promise and potential pitfalls. Mech. Ageing Dev. 2021, 198, 111540. [Google Scholar] [CrossRef]
- Hoffmann, J.; Richardson, G.; Haendeler, J.; Altschmied, J.; Andrés, V.; Spyridopoulos, I. Telomerase as a Therapeutic Target in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1047–1061. [Google Scholar] [CrossRef]
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 2020, 8, 624216. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Q.; Feng, X.; Liu, Y.; Zhou, Y. Mitochondrial Dysfunction in Cardiovascular Diseases: Potential Targets for Treatment. Front. Cell Dev. Biol. 2022, 10, 841523. [Google Scholar] [CrossRef]
- Stamerra, C.A.; Di Giosia, P.; Giorgini, P.; Ferri, C.; Sukhorukov, V.N.; Sahebkar, A. Mitochondrial Dysfunction and Cardiovascular Disease: Pathophysiology and Emerging Therapies. Oxid. Med. Cell. Longev. 2022, 2022, 9530007. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Ganote, C.E. Mitochondrial structure and function in acute myocardial ischemic injury. Circ. Res. 1976, 38, I80–I91. [Google Scholar] [PubMed]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.F.; Schönekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 1994, 267, H742–H750. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Liu, Y.Y.; Liu, G.H.; Lu, H.B.; Mao, C.Y. l-Carnitine and heart disease. Life Sci. 2018, 194, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef]
- Casademont, J.; Miró, O. Electron transport chain defects in heart failure. Heart Fail. Rev. 2002, 7, 131–139. [Google Scholar] [CrossRef]
- Arbustini, E.; Diegoli, M.; Fasani, R.; Grasso, M.; Morbini, P.; Banchieri, N.; Bellini, O.; Dal Bello, B.; Pilotto, A.; Magrini, G.; et al. Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am. J. Pathol. 1998, 153, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Sheeran, F.L.; Pepe, S. Mitochondrial Bioenergetics and Dysfunction in Failing Heart. Adv. Exp. Med. Biol. 2017, 982, 65–80. [Google Scholar] [CrossRef]
- Dai, D.F.; Chen, T.; Wanagat, J.; Laflamme, M.; Marcinek, D.J.; Emond, M.J.; Ngo, C.P.; Prolla, T.A.; Rabinovitch, P.S. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010, 9, 536–544. [Google Scholar] [CrossRef]
- Kang, S.; Louboutin, J.P.; Datta, P.; Landel, C.P.; Martinez, D.; Zervos, A.S.; Strayer, D.S.; Fernandes-Alnemri, T.; Alnemri, E.S. Loss of HtrA2/Omi activity in non-neuronal tissues of adult mice causes premature aging. Cell Death Differ. 2013, 20, 259–269. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg. Species 2016, 1, 9–21. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox Signal 2006, 8, 1775–1789. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993, 9, 246–249. [Google Scholar] [CrossRef]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial Dysfunction Accounts for the Stochastic Heterogeneity in Telomere-Dependent Senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Jurk, D.; Marques, F.D.M.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Malaquin, N.; Carrier-Leclerc, A.; Dessureault, M.; Rodier, F. DDR-mediated crosstalk between DNA-damaged cells and their microenvironment. Front. Genet. 2015, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Martini, H.; Passos, J.F. Cellular senescence: All roads lead to mitochondria. FEBS J. 2023, 290, 1186–1202. [Google Scholar] [CrossRef] [PubMed]
- Hertz, D.G. Trauma and nostalgia: New aspects on the coping of aging Holocaust survivors. Isr. J. Psychiatry Relat. Sci. 1990, 27, 189–198. [Google Scholar] [PubMed]
- Youdim, M.B.; Bakhle, Y.S. Monoamine oxidase: Isoforms and inhibitors in Parkinson’s disease and depressive illness. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S287–S296. [Google Scholar] [CrossRef]
- Villeneuve, C.; Guilbeau-Frugier, C.; Sicard, P.; Lairez, O.; Ordener, C.; Duparc, T.; De Paulis, D.; Couderc, B.; Spreux-Varoquaux, O.; Tortosa, F.; et al. p53-PGC-1α pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: Role in chronic left ventricular dysfunction in mice. Antioxid. Redox Signal 2013, 18, 5–18. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Birch, J.; Fielder, E.; Rahmatika, D.; Taylor, J.; Chapman, J.; Lagnado, A.; Carroll, B.M.; Miwa, S.; Richardson, G.; et al. Rapamycin improves healthspan but not inflammaging in nfκb1−/− mice. Aging Cell 2019, 18, e12882. [Google Scholar] [CrossRef]
- Chiao, Y.A.; Zhang, H.; Sweetwyne, M.; Whitson, J.; Ting, Y.S.; Basisty, N.; Pino, L.K.; Quarles, E.; Nguyen, N.H.; Campbell, M.D.; et al. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. Elife 2020, 9, e55513. [Google Scholar] [CrossRef]
- Windecker, S. Chapter 30—Acute Coronary Syndromes. In Clinical Critical Care Medicine; Albert, R.K., Slutsky, A.S., Ranieri, V.M., Takala, J., Torres, A., Eds.; Mosby: Philadelphia, PA, USA, 2006; pp. 301–318. [Google Scholar]
- Gintant, G.; Sager, P.T.; Stockbridge, N. Evolution of strategies to improve preclinical cardiac safety testing. Nat. Rev. Drug Discov. 2016, 15, 457–471. [Google Scholar] [CrossRef]
- Laverty, H.; Benson, C.; Cartwright, E.; Cross, M.; Garland, C.; Hammond, T.; Holloway, C.; McMahon, N.; Milligan, J.; Park, B.; et al. How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br. J. Pharmacol. 2011, 163, 675–693. [Google Scholar] [CrossRef]
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Post-marketing withdrawal of analgesic medications because of adverse drug reactions: A systematic review. Expert. Opin. Drug Saf. 2018, 17, 63–72. [Google Scholar] [CrossRef]
- Ewer, M.S.; Ewer, S.M. Cardiotoxicity of anticancer treatments. Nat. Rev. Cardiol. 2015, 12, 547–558. [Google Scholar] [CrossRef]
- Chow, E.J.; Leger, K.J.; Bhatt, N.S.; Mulrooney, D.A.; Ross, C.J.; Aggarwal, S.; Bansal, N.; Ehrhardt, M.J.; Armenian, S.H.; Scott, J.M.; et al. Paediatric cardio-oncology: Epidemiology, screening, prevention, and treatment. Cardiovasc. Res. 2019, 115, 922–934. [Google Scholar] [CrossRef]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef]
- Doroshow, J.H.; Davies, K.J. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J. Biol. Chem. 1986, 261, 3068–3074. [Google Scholar] [CrossRef]
- Parker, M.A.; King, V.; Howard, K.P. Nuclear magnetic resonance study of doxorubicin binding to cardiolipin containing magnetically oriented phospholipid bilayers. Biochim. Biophys. Acta (BBA) Biomembr. 2001, 1514, 206–216. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Abe, K.; Ikeda, M.; Ide, T.; Tadokoro, T.; Miyamoto, H.D.; Furusawa, S.; Tsutsui, Y.; Miyake, R.; Ishimaru, K.; Watanabe, M.; et al. Doxorubicin causes ferroptosis and cardiotoxicity by intercalating into mitochondrial DNA and disrupting Alas1-dependent heme synthesis. Sci. Signal 2022, 15, eabn8017. [Google Scholar] [CrossRef]
- Lebrecht, D.; Kokkori, A.; Ketelsen, U.P.; Setzer, B.; Walker, U.A. Tissue-specific mtDNA lesions and radical-associated mitochondrial dysfunction in human hearts exposed to doxorubicin. J. Pathol. 2005, 207, 436–444. [Google Scholar] [CrossRef]
- Mitry, M.A.; Laurent, D.; Keith, B.L.; Sira, E.; Eisenberg, C.A.; Eisenberg, L.M.; Joshi, S.; Gupte, S.; Edwards, J.G. Accelerated cardiomyocyte senescence contributes to late-onset doxorubicin-induced cardiotoxicity. Am. J. Physiol. Cell Physiol. 2020, 318, C380–C391. [Google Scholar] [CrossRef]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Will, Y.; Dykens, J.A.; Nadanaciva, S.; Hirakawa, B.; Jamieson, J.; Marroquin, L.D.; Hynes, J.; Patyna, S.; Jessen, B.A. Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib, and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol. Sci. 2008, 106, 153–161. [Google Scholar] [CrossRef]
- Bouitbir, J.; Alshaikhali, A.; Panajatovic, M.V.; Abegg, V.F.; Paech, F.; Krähenbühl, S. Mitochondrial oxidative stress plays a critical role in the cardiotoxicity of sunitinib: Running title: Sunitinib and oxidative stress in hearts. Toxicology 2019, 426, 152281. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; De Francesco, E.M.; Pasqua, T.; Granieri, M.C.; De Bartolo, A.; Gallo Cantafio, M.E.; Muoio, M.G.; Gentile, M.; Neri, A.; Angelone, T.; et al. Mitochondrial Determinants of Anti-Cancer Drug-Induced Cardiotoxicity. Biomedicines 2022, 10, 520. [Google Scholar] [CrossRef]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2021, 178, 2060–2076. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Liu, Y.; Yin, H. Mitochondrial Dynamics: Biogenesis, Fission, Fusion, and Mitophagy in the Regulation of Stem Cell Behaviors. Stem Cells Int. 2019, 2019, 9757201. [Google Scholar] [CrossRef]
- Fenton, A.R.; Jongens, T.A.; Holzbaur, E.L.F. Mitochondrial dynamics: Shaping and remodeling an organelle network. Curr. Opin. Cell Biol. 2021, 68, 28–36. [Google Scholar] [CrossRef]
- Luan, Y.; Ren, K.D.; Luan, Y.; Chen, X.; Yang, Y. Mitochondrial Dynamics: Pathogenesis and Therapeutic Targets of Vascular Diseases. Front. Cardiovasc. Med. 2021, 8, 770574. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. Mitofusin 2: A mitochondria-shaping protein with signaling roles beyond fusion. Antioxid. Redox Signal 2008, 10, 621–633. [Google Scholar] [CrossRef]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Dorn, G.W., 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Kikuchi, R.; Ngoh, G.A.; Coughlan, K.A.; Dominguez, I.; Stanley, W.C.; Walsh, K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ. Res. 2012, 111, 1012–1026. [Google Scholar] [CrossRef]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [CrossRef]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef]
- Morris, S.; Molina-Riquelme, I.; Barrientos, G.; Bravo, F.; Aedo, G.; Gómez, W.; Lagos, D.; Verdejo, H.; Peischard, S.; Seebohm, G.; et al. Inner mitochondrial membrane structure and fusion dynamics are altered in senescent human iPSC-derived and primary rat cardiomyocytes. Biochim. Biophys. Acta (BBA) Bioenerg. 2023, 1864, 148949. [Google Scholar] [CrossRef]
- Lee, H.C.; Yin, P.H.; Chi, C.W.; Wei, Y.H. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. J. Biomed. Sci. 2002, 9, 517–526. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, S.Y.; Lim, W.C.; Kim, S.; Park, Y.Y.; Sun, X.; Youle, R.J.; Cho, H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 2007, 282, 22977–22983. [Google Scholar] [CrossRef]
- Yu, R.; Lendahl, U.; Nistér, M.; Zhao, J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front. Endocrinol. 2020, 11, 374. [Google Scholar] [CrossRef]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef]
- Sihag, S.; Cresci, S.; Li, A.Y.; Sucharov, C.C.; Lehman, J.J. PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J. Mol. Cell. Cardiol. 2009, 46, 201–212. [Google Scholar] [CrossRef]
- Wang, J.; Li, S.; Wang, J.; Wu, F.; Chen, Y.; Zhang, H.; Guo, Y.; Lin, Y.; Li, L.; Yu, X.; et al. Spermidine alleviates cardiac aging by improving mitochondrial biogenesis and function. Aging 2020, 12, 650–671. [Google Scholar] [CrossRef]
- Xiong, S.; Salazar, G.; Patrushev, N.; Ma, M.; Forouzandeh, F.; Hilenski, L.; Alexander, R.W. Peroxisome proliferator-activated receptor γ coactivator-1α is a central negative regulator of vascular senescence. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 988–998. [Google Scholar] [CrossRef]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef]
- Brady, N.R.; Hamacher-Brady, A.; Gottlieb, R.A. Proapoptotic BCL-2 family members and mitochondrial dysfunction during ischemia/reperfusion injury, a study employing cardiac HL-1 cells and GFP biosensors. Biochim. Biophys. Acta 2006, 1757, 667–678. [Google Scholar] [CrossRef]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 2010, 123, 917–926. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Newmeyer, D.D.; Farschon, D.M.; Reed, J.C. Cell-free apoptosis in Xenopus egg extracts: Inhibition by Bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell 1994, 79, 353–364. [Google Scholar] [CrossRef]
- Takemura, G.; Kanoh, M.; Minatoguchi, S.; Fujiwara, H. Cardiomyocyte apoptosis in the failing heart--a critical review from definition and classification of cell death. Int. J. Cardiol. 2013, 167, 2373–2386. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- González, A.; Fortuño, M.A.; Querejeta, R.; Ravassa, S.; López, B.; López, N.; Díez, J. Cardiomyocyte apoptosis in hypertensive cardiomyopathy. Cardiovasc. Res. 2003, 59, 549–562. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd. Mitochondrial pruning by Nix and BNip3: An essential function for cardiac-expressed death factors. J. Cardiovasc. Transl. Res. 2010, 3, 374–383. [Google Scholar] [CrossRef]
- Diwan, A.; Wansapura, J.; Syed, F.M.; Matkovich, S.J.; Lorenz, J.N.; Dorn, G.W., 2nd. Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation 2008, 117, 396–404. [Google Scholar] [CrossRef]
- Rottenberg, H.; Hoek, J.B. The Mitochondrial Permeability Transition: Nexus of Aging, Disease and Longevity. Cells 2021, 10, 79. [Google Scholar] [CrossRef]
- Morciano, G.; Bonora, M.; Campo, G.; Aquila, G.; Rizzo, P.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Mechanistic Role of mPTP in Ischemia-Reperfusion Injury. Adv. Exp. Med. Biol. 2017, 982, 169–189. [Google Scholar] [CrossRef]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Song, M.; Gong, G.; Burelle, Y.; Gustafsson, Å.B.; Kitsis, R.N.; Matkovich, S.J.; Dorn, G.W., 2nd. Interdependence of Parkin-Mediated Mitophagy and Mitochondrial Fission in Adult Mouse Hearts. Circ. Res. 2015, 117, 346–351. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Song, M.; Chen, Y.; Gong, G.; Murphy, E.; Rabinovitch, P.S.; Dorn, G.W., 2nd. Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ. Res. 2014, 115, 348–353. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef]
- Saito, T.; Asai, K.; Sato, S.; Hayashi, M.; Adachi, A.; Sasaki, Y.; Takano, H.; Mizuno, K.; Shimizu, W. Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy 2016, 12, 579–587. [Google Scholar] [CrossRef]
- Campos, J.C.; Bozi, L.H.; Bechara, L.R.; Lima, V.M.; Ferreira, J.C. Mitochondrial Quality Control in Cardiac Diseases. Front. Physiol. 2016, 7, 479. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Roth, L.; Schrijvers, D.M.; De Meyer, G.R.Y.; Martinet, W. Defective Autophagy in Atherosclerosis: To Die or to Senesce? Oxid. Med. Cell. Longev. 2018, 2018, 7687083. [Google Scholar] [CrossRef]
- Sergin, I.; Bhattacharya, S.; Emanuel, R.; Esen, E.; Stokes, C.J.; Evans, T.D.; Arif, B.; Curci, J.A.; Razani, B. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci. Signal 2016, 9, ra2. [Google Scholar] [CrossRef]
- Swaminathan, B.; Goikuria, H.; Vega, R.; Rodríguez-Antigüedad, A.; López Medina, A.; Freijo Mdel, M.; Vandenbroeck, K.; Alloza, I. Autophagic marker MAP1LC3B expression levels are associated with carotid atherosclerosis symptomatology. PLoS ONE 2014, 9, e115176. [Google Scholar] [CrossRef]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxid. Med. Cell. Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef]
- Sen, A.; Kallabis, S.; Gaedke, F.; Jüngst, C.; Boix, J.; Nüchel, J.; Maliphol, K.; Hofmann, J.; Schauss, A.C.; Krüger, M.; et al. Mitochondrial membrane proteins and VPS35 orchestrate selective removal of mtDNA. Nat. Commun. 2022, 13, 6704. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Otsu, K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem. J. 2018, 475, 839–852. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- King, K.R.; Aguirre, A.D.; Ye, Y.X.; Sun, Y.; Roh, J.D.; Ng, R.P., Jr.; Kohler, R.H.; Arlauckas, S.P.; Iwamoto, Y.; Savol, A.; et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017, 23, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Luo, W.; Zhang, L.; Wu, W.; Yuan, L.; Xu, H.; Song, J.; Fujiwara, K.; Abe, J.I.; LeMaire, S.A.; et al. STING-IRF3 Triggers Endothelial Inflammation in Response to Free Fatty Acid-Induced Mitochondrial Damage in Diet-Induced Obesity. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 920–929. [Google Scholar] [CrossRef]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Münzel, T. Endothelial Dysfunction, Oxidative Stress, and Risk of Cardiovascular Events in Patients With Coronary Artery Disease. Circulation 2001, 104, 2673–2678. [Google Scholar] [CrossRef]
- McCarthy, C.G.; Wenceslau, C.F.; Goulopoulou, S.; Ogbi, S.; Baban, B.; Sullivan, J.C.; Matsumoto, T.; Webb, R.C. Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc. Res. 2015, 107, 119–130. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, X.; Shi, H.; Yu, Y.; Yu, Y.; Li, M.; Chen, R. NLRP3 inflammasome, an immune-inflammatory target in pathogenesis and treatment of cardiovascular diseases. Clin. Transl. Med. 2020, 10, 91–106. [Google Scholar] [CrossRef]
- Keshavarz-Bahaghighat, H.; Darwesh, A.M.; Sosnowski, D.K.; Seubert, J.M. Mitochondrial Dysfunction and Inflammaging in Heart Failure: Novel Roles of CYP-Derived Epoxylipids. Cells 2020, 9, 1565. [Google Scholar] [CrossRef]
- Liberale, L.; Montecucco, F.; Tardif, J.C.; Libby, P.; Camici, G.G. Inflamm-ageing: The role of inflammation in age-dependent cardiovascular disease. Eur. Heart J. 2020, 41, 2974–2982. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Geng, J.; Zhao, J.; Ni, Q.; Zhao, C.; Zheng, Y.; Chen, X.; Wang, L. Trimethylamine N-Oxide Exacerbates Cardiac Fibrosis via Activating the NLRP3 Inflammasome. Front. Physiol. 2019, 10, 866. [Google Scholar] [CrossRef] [PubMed]
- Cañadas-Lozano, D.; Marín-Aguilar, F.; Castejón-Vega, B.; Ryffel, B.; Navarro-Pando, J.M.; Ruiz-Cabello, J.; Alcocer-Gómez, E.; Bullón, P.; Cordero, M.D. Blockade of the NLRP3 inflammasome improves metabolic health and lifespan in obese mice. Geroscience 2020, 42, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Stokman, G.; Kors, L.; Bakker, P.J.; Rampanelli, E.; Claessen, N.; Teske, G.J.D.; Butter, L.; van Andel, H.; van den Bergh Weerman, M.A.; Larsen, P.W.B.; et al. NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J. Exp. Med. 2017, 214, 2405–2420. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G. A common signature of cellular senescence; does it exist? Ageing Res. Rev. 2021, 71, 101458. [Google Scholar] [CrossRef]
- Tang, X.; Li, P.H.; Chen, H.Z. Cardiomyocyte Senescence and Cellular Communications Within Myocardial Microenvironments. Front. Endocrinol. 2020, 11, 280. [Google Scholar] [CrossRef]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech. Ageing Dev. 2018, 170, 30–36. [Google Scholar] [CrossRef]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.A.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes. Dev. 2020, 34, 428–445. [Google Scholar] [CrossRef]
- Victorelli, S.; Salmonowicz, H.; Chapman, J.; Martini, H.; Vizioli, M.G.; Riley, J.S.; Cloix, C.; Hall-Younger, E.; Machado Espindola-Netto, J.; Jurk, D.; et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature 2023, 622, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Passos, J.F. Targeting the SASP to combat ageing: Mitochondria as possible intracellular allies? Bioessays 2017, 39, 1600235. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, H.; Hoek, J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 2017, 16, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef]
- Agius, L.; Ford, B.E.; Chachra, S.S. The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective. Int. J. Mol. Sci. 2020, 21, 3240. [Google Scholar] [CrossRef]
- Ma, T.; Tian, X.; Zhang, B.; Li, M.; Wang, Y.; Yang, C.; Wu, J.; Wei, X.; Qu, Q.; Yu, Y.; et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature 2022, 603, 159–165. [Google Scholar] [CrossRef]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.H. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes 2017, 66, 193–205. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Ducommun, S.; Deak, M.; Sumpton, D.; Ford, R.J.; Núñez Galindo, A.; Kussmann, M.; Viollet, B.; Steinberg, G.R.; Foretz, M.; Dayon, L.; et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: Identification of mitochondrial fission factor as a new AMPK substrate. Cell. Signal 2015, 27, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Abdelgawad, I.Y.; Agostinucci, K.; Sadaf, B.; Grant, M.K.O.; Zordoky, B.N. Metformin mitigates SASP secretion and LPS-triggered hyper-inflammation in Doxorubicin-induced senescent endothelial cells. Front. Aging 2023, 4, 1170434. [Google Scholar] [CrossRef]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Yang, J.; Wu, X.; Zhang, G.; Li, T.; Wang, X.; Zhang, H.; Wang, C.C.; Liu, G.H.; Wang, L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 2018, 17, e12765. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Campbell, J.M.; Bellman, S.M.; Stephenson, M.D.; Lisy, K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: A systematic review and meta-analysis. Ageing Res. Rev. 2017, 40, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Peng, M.; Tang, X.; Xu, X.; Wu, Y.; Chen, A.F.; Yang, X. Protective effects of metformin in various cardiovascular diseases: Clinical evidence and AMPK-dependent mechanisms. J. Cell. Mol. Med. 2022, 26, 4886–4903. [Google Scholar] [CrossRef]
- Petrie, J.R.; Chaturvedi, N.; Ford, I.; Brouwers, M.; Greenlaw, N.; Tillin, T.; Hramiak, I.; Hughes, A.D.; Jenkins, A.J.; Klein, B.E.K.; et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (REMOVAL): A double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 597–609. [Google Scholar] [CrossRef]
- De Jager, J.; Kooy, A.; Lehert, P.; Bets, D.; Wulffelé, M.G.; Teerlink, T.; Scheffer, P.G.; Schalkwijk, C.G.; Donker, A.J.; Stehouwer, C.D. Effects of short-term treatment with metformin on markers of endothelial function and inflammatory activity in type 2 diabetes mellitus: A randomized, placebo-controlled trial. J. Intern. Med. 2005, 257, 100–109. [Google Scholar] [CrossRef]
- Jadhav, S.; Ferrell, W.; Greer, I.A.; Petrie, J.R.; Cobbe, S.M.; Sattar, N. Effects of metformin on microvascular function and exercise tolerance in women with angina and normal coronary arteries: A randomized, double-blind, placebo-controlled study. J. Am. Coll. Cardiol. 2006, 48, 956–963. [Google Scholar] [CrossRef]
- Preiss, D.; Lloyd, S.M.; Ford, I.; McMurray, J.J.; Holman, R.R.; Welsh, P.; Fisher, M.; Packard, C.J.; Sattar, N. Metformin for non-diabetic patients with coronary heart disease (the CAMERA study): A randomised controlled trial. Lancet Diabetes Endocrinol. 2014, 2, 116–124. [Google Scholar] [CrossRef]
- Xu, W.; Deng, Y.Y.; Yang, L.; Zhao, S.; Liu, J.; Zhao, Z.; Wang, L.; Maharjan, P.; Gao, S.; Tian, Y.; et al. Metformin ameliorates the proinflammatory state in patients with carotid artery atherosclerosis through sirtuin 1 induction. Transl. Res. 2015, 166, 451–458. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2023, 290, 1362–1383. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D.; Alehagen, U.; Steurer, G.; Littarru, G.P. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: Results from Q-SYMBIO: A randomized double-blind trial. JACC Heart Fail. 2014, 2, 641–649. [Google Scholar] [CrossRef]
- Zeb, I.; Ahmadi, N.; Nasir, K.; Kadakia, J.; Larijani, V.N.; Flores, F.; Li, D.; Budoff, M.J. Aged garlic extract and coenzyme Q10 have favorable effect on inflammatory markers and coronary atherosclerosis progression: A randomized clinical trial. J. Cardiovasc. Dis. Res. 2012, 3, 185–190. [Google Scholar] [CrossRef]
- MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 23–33. [CrossRef]
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2008, 300, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Mariscal, F.M.; de la Cruz-Ares, S.; Torres-Peña, J.D.; Alcalá-Diaz, J.F.; Yubero-Serrano, E.M.; López-Miranda, J. Coenzyme Q(10) and Cardiovascular Diseases. Antioxidants 2021, 10, 906. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Liu, Y. Efficacy of coenzyme Q10 in patients with cardiac failure: A meta-analysis of clinical trials. BMC Cardiovasc. Disord. 2017, 17, 196. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.S.; Nauduri, D.; Anders, M.W. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta 2006, 1762, 256–265. [Google Scholar] [CrossRef]
- Reily, C.; Mitchell, T.; Chacko, B.K.; Benavides, G.; Murphy, M.P.; Darley-Usmar, V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013, 1, 86–93. [Google Scholar] [CrossRef]
- Kulkarni, C.A.; Fink, B.D.; Gibbs, B.E.; Chheda, P.R.; Wu, M.; Sivitz, W.I.; Kerns, R.J. A Novel Triphenylphosphonium Carrier to Target Mitochondria without Uncoupling Oxidative Phosphorylation. J. Med. Chem. 2021, 64, 662–676. [Google Scholar] [CrossRef]
- Ribeiro Junior, R.F.; Dabkowski, E.R.; Shekar, K.C.; KA, O.C.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef]
- Dare, A.J.; Logan, A.; Prime, T.A.; Rogatti, S.; Goddard, M.; Bolton, E.M.; Bradley, J.A.; Pettigrew, G.J.; Murphy, M.P.; Saeb-Parsy, K. The mitochondria-targeted anti-oxidant MitoQ decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model. J. Heart Lung Transplant. 2015, 34, 1471–1480. [Google Scholar] [CrossRef]
- Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cochemé, H.M.; Murphy, M.P.; Dominiczak, A.F. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 2009, 54, 322–328. [Google Scholar] [CrossRef]
- Goh, K.Y.; He, L.; Song, J.; Jinno, M.; Rogers, A.J.; Sethu, P.; Halade, G.V.; Rajasekaran, N.S.; Liu, X.; Prabhu, S.D.; et al. Mitoquinone ameliorates pressure overload-induced cardiac fibrosis and left ventricular dysfunction in mice. Redox Biol. 2019, 21, 101100. [Google Scholar] [CrossRef]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef]
- Varesi, A.; Chirumbolo, S.; Campagnoli, L.I.M.; Pierella, E.; Piccini, G.B.; Carrara, A.; Ricevuti, G.; Scassellati, C.; Bonvicini, C.; Pascale, A. The Role of Antioxidants in the Interplay between Oxidative Stress and Senescence. Antioxidants 2022, 11, 1224. [Google Scholar] [CrossRef]
- Breccia, M.; Alimena, G. Activity and safety of dasatinib as second-line treatment or in newly diagnosed chronic phase chronic myeloid leukemia patients. BioDrugs 2011, 25, 147–157. [Google Scholar] [CrossRef]
- Chan, C.M.; Jing, X.; Pike, L.A.; Zhou, Q.; Lim, D.J.; Sams, S.B.; Lund, G.S.; Sharma, V.; Haugen, B.R.; Schweppe, R.E. Targeted inhibition of Src kinase with dasatinib blocks thyroid cancer growth and metastasis. Clin. Cancer Res. 2012, 18, 3580–3591. [Google Scholar] [CrossRef]
- Bruning, A. Inhibition of mTOR signaling by quercetin in cancer treatment and prevention. Anti-Cancer Agents Med. Chem. 2013, 13, 1025–1031. [Google Scholar] [CrossRef]
- Olave, N.C.; Grenett, M.H.; Cadeiras, M.; Grenett, H.E.; Higgins, P.J. Upstream stimulatory factor-2 mediates quercetin-induced suppression of PAI-1 gene expression in human endothelial cells. J. Cell. Biochem. 2010, 111, 720–726. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Lérida-Viso, A.; Estepa-Fernández, A.; Morellá-Aucejo, Á.; Lozano-Torres, B.; Alfonso, M.; Blandez, J.F.; Bisbal, V.; Sepúlveda, P.; García-Fernández, A.; Orzáez, M.; et al. Pharmacological senolysis reduces doxorubicin-induced cardiotoxicity and improves cardiac function in mice. Pharmacol. Res. 2022, 183, 106356. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.M.; Kaistha, A.; Uryga, A.K.; Oc, S.; Foote, K.; Shah, A.; Finigan, A.; Figg, N.; Dobnikar, L.; Jørgensen, H.; et al. Efficacy and limitations of senolysis in atherosclerosis. Cardiovasc. Res. 2022, 118, 1713–1727. [Google Scholar] [CrossRef] [PubMed]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Robbins, P.D. Senotherapeutics for healthy ageing. Nat. Rev. Drug Discov. 2018, 17, 377. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camacho-Encina, M.; Booth, L.K.; Redgrave, R.E.; Folaranmi, O.; Spyridopoulos, I.; Richardson, G.D. Cellular Senescence, Mitochondrial Dysfunction, and Their Link to Cardiovascular Disease. Cells 2024, 13, 353. https://doi.org/10.3390/cells13040353
Camacho-Encina M, Booth LK, Redgrave RE, Folaranmi O, Spyridopoulos I, Richardson GD. Cellular Senescence, Mitochondrial Dysfunction, and Their Link to Cardiovascular Disease. Cells. 2024; 13(4):353. https://doi.org/10.3390/cells13040353
Chicago/Turabian StyleCamacho-Encina, Maria, Laura K. Booth, Rachael E. Redgrave, Omowumi Folaranmi, Ioakim Spyridopoulos, and Gavin D. Richardson. 2024. "Cellular Senescence, Mitochondrial Dysfunction, and Their Link to Cardiovascular Disease" Cells 13, no. 4: 353. https://doi.org/10.3390/cells13040353
APA StyleCamacho-Encina, M., Booth, L. K., Redgrave, R. E., Folaranmi, O., Spyridopoulos, I., & Richardson, G. D. (2024). Cellular Senescence, Mitochondrial Dysfunction, and Their Link to Cardiovascular Disease. Cells, 13(4), 353. https://doi.org/10.3390/cells13040353