
Targeting Epigenetic Modifiers of Tumor Plasticity and Cancer Stem Cell Behavior


 , and
, and
Abstract
:1. Introduction
2. Cancer Stem Cells
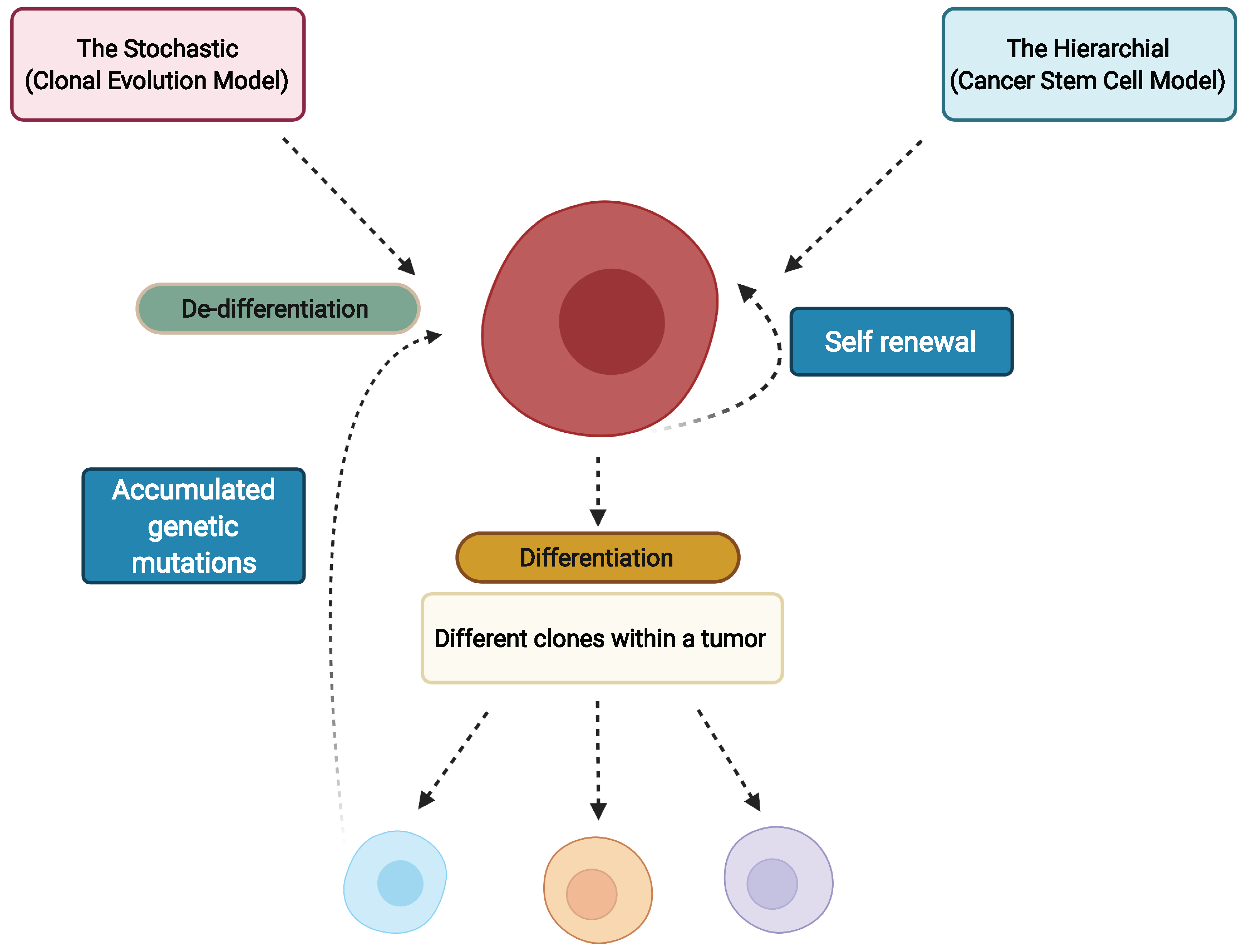
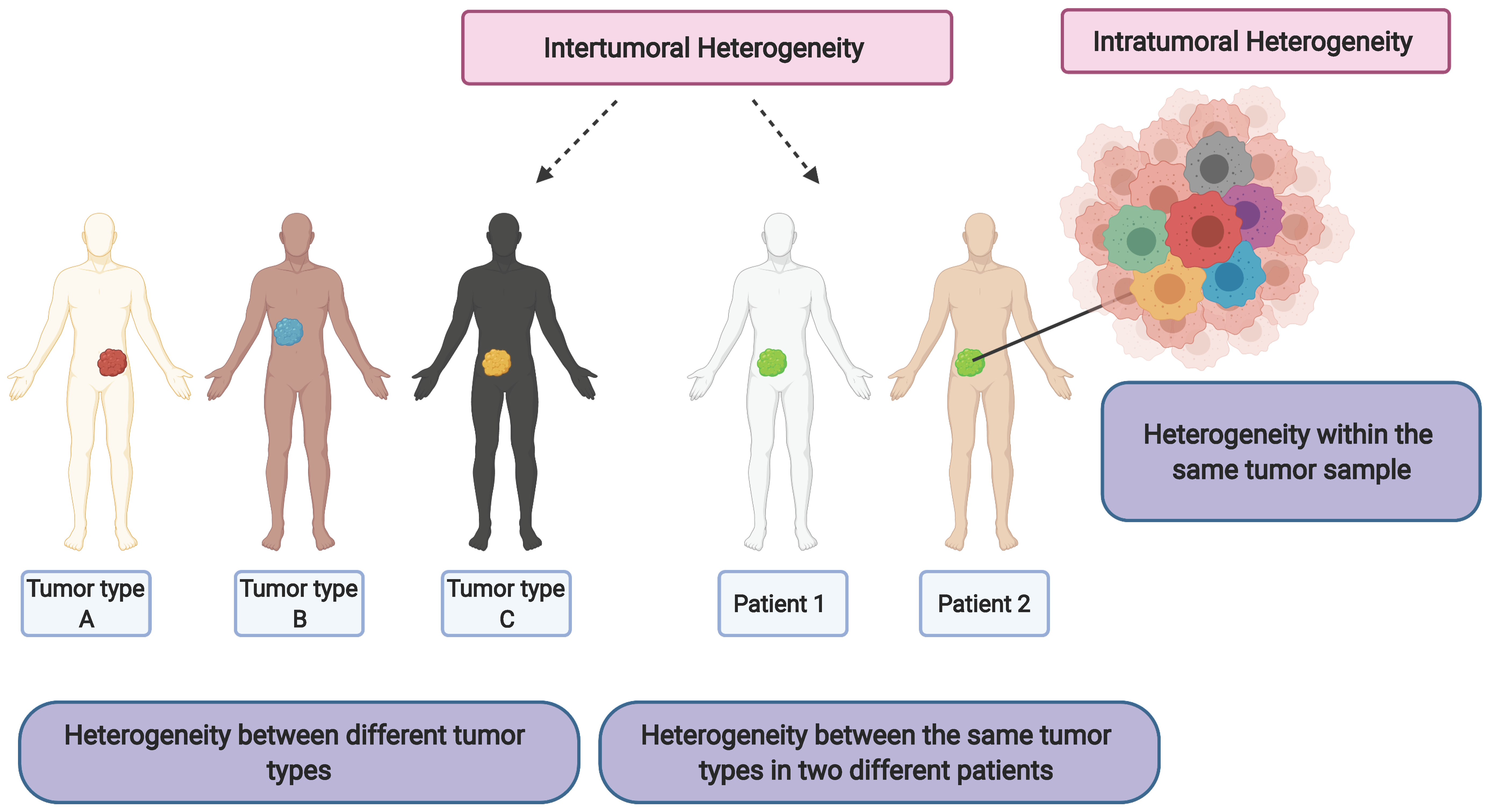
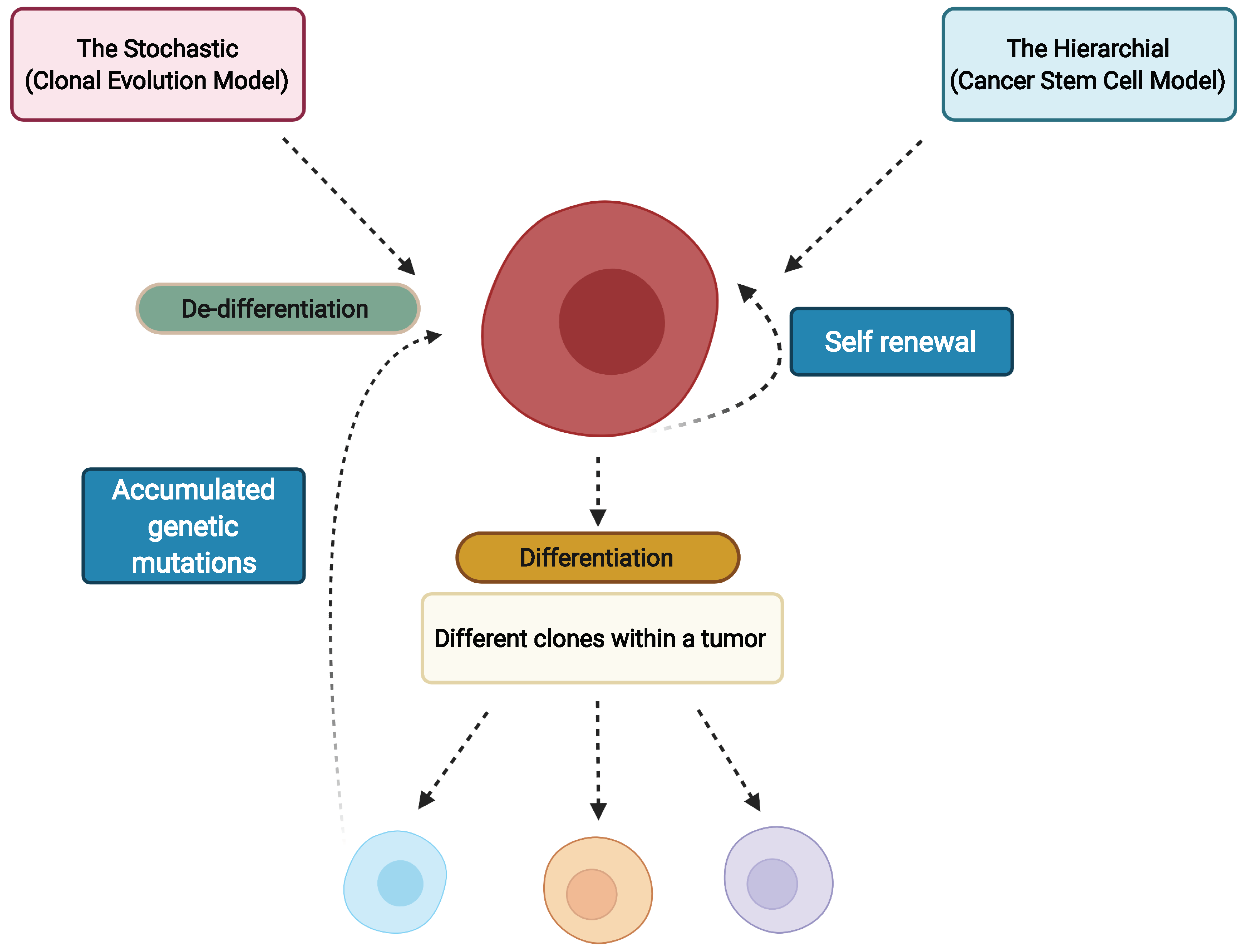
3. Cancer Stem Cells and Clonal Evolution as the Source of Intratumor Heterogeneity
4. Cancer Stem Cell Plasticity
5. Epigenetic Regulation/Dysregulation in Cancer Stem Cells
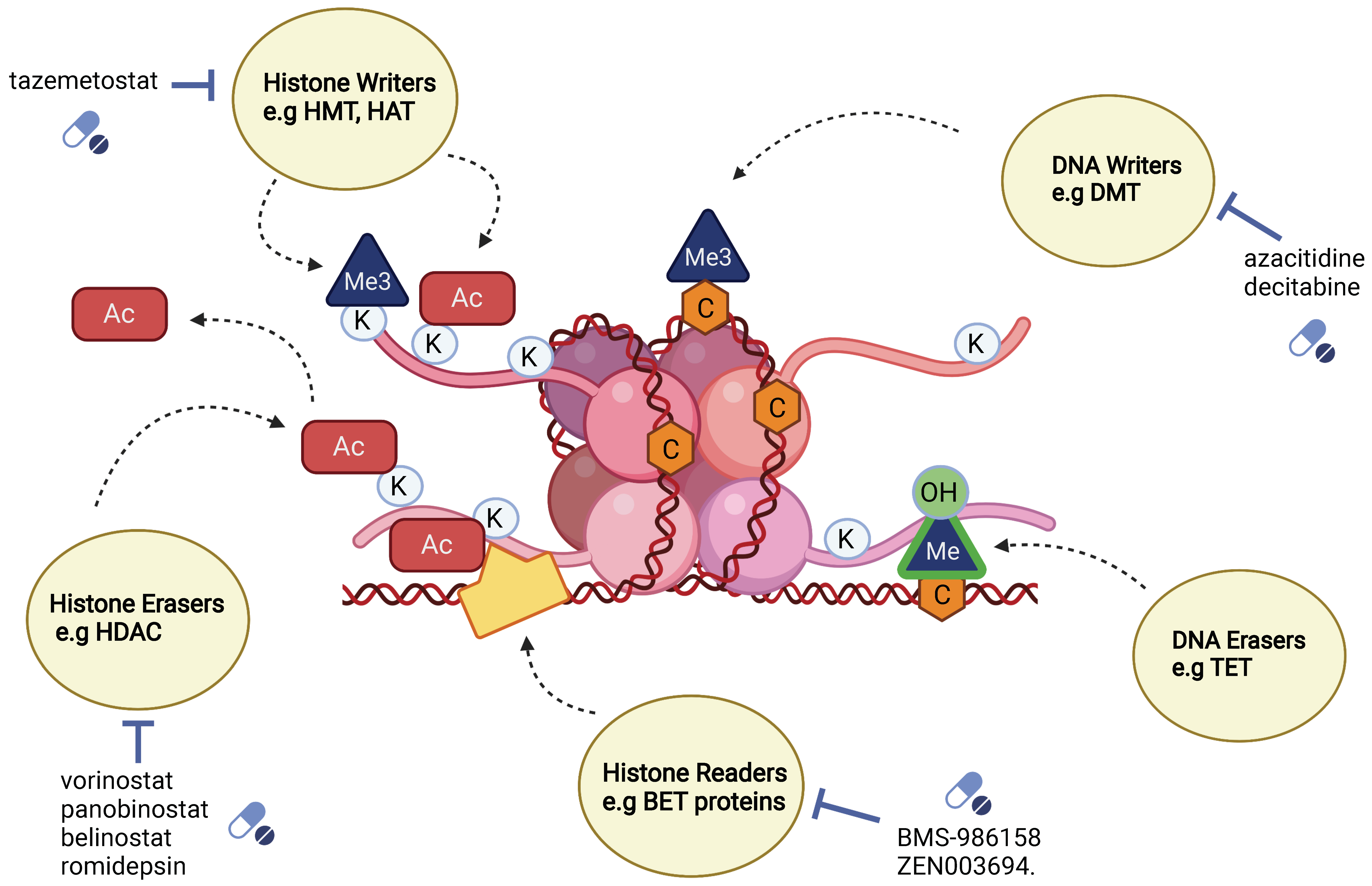
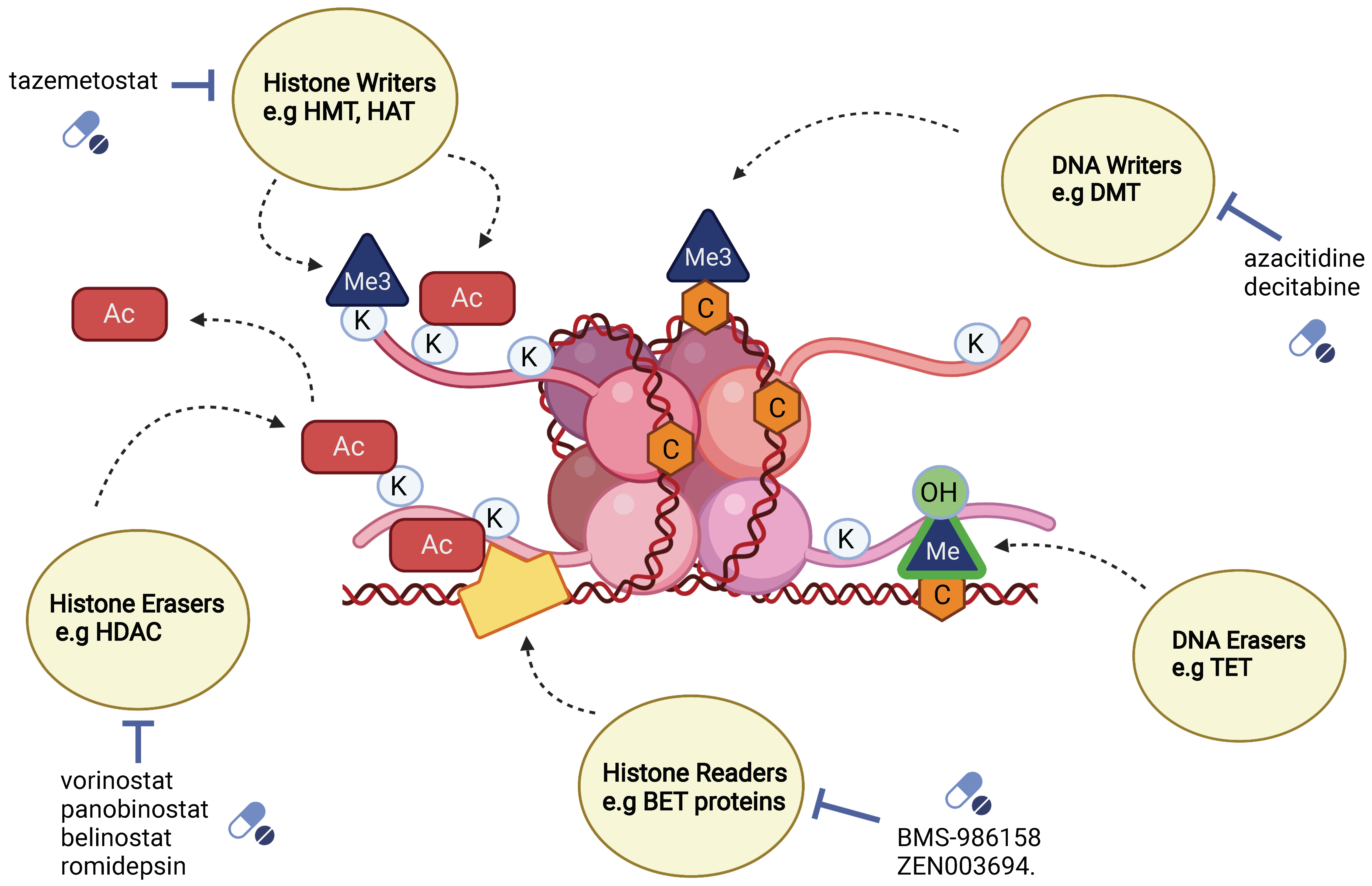
5.1. Classification of Epigenetic Mediators: Writers, Erasers and Readers
5.1.1. Writer Enzymes
5.1.2. Eraser Enzymes
5.1.3. Reader Enzymes
6. Other Regulators of Cancer Stem Cells
6.1. Long Non-Coding RNA
6.2. ATP-Dependent Chromatin-Remodeling Complexes
7. Hypoxia on Cancer Stem Cells
8. Cell Signaling Pathways Regulating Cancer Stem Cells
8.1. Wnt Signaling Pathways
8.2. Notch Signaling Pathway
8.3. Hedgehog Signaling Pathway
9. Epithelial–Mesenchymal Transition Is a Critical Regulator of the CSC Phenotype
10. Integrins Alter Cancer Stem Cells Behavior
11. Targeting Vulnerabilities in Cancer Stem Cells
11.1. Histone Methyltransferase Inhibitors
11.2. Histone Methylase Inhibitors
11.3. DNA Methyltransferase Inhibitors
11.4. Histone Deacetylase Inhibitors
11.5. BET Inhibitors
11.6. Inhibitors against Signaling Pathways
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Wahl, G.M.; Spike, B.T. Cell state plasticity, stem cells, EMT, and the generation of intra-tumoral heterogeneity. NPJ Breast Cancer 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Ailles, L.E.; Weissman, I.L. Cancer stem cells in solid tumors. Curr. Opin. Biotechnol. 2007, 18, 460–466. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H. Cancer stem cells and self-renewal. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, M.; Mortezaee, K.; Ahadi, R. Cancer stem cell (a)symmetry & plasticity: Tumorigenesis and therapy relevance. Life Sci. 2019, 231, 116520. [Google Scholar] [CrossRef]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Odoux, C.; Fohrer, H.; Hoppo, T.; Guzik, L.; Stolz, D.B.; Lewis, D.W.; Gollin, S.M.; Gamblin, T.C.; Geller, D.A.; Lagasse, E. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 2008, 68, 6932–6941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva-Diz, V.; Lorenzo-Sanz, L.; Bernat-Peguera, A.; Lopez-Cerda, M.; Munoz, P. Cancer cell plasticity: Impact on tumor progression and therapy response. Semin Cancer Biol. 2018, 53, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.S.; Heilmann, S.; Kansler, E.R.; Zhang, Y.; Zimmer, M.; Ratnakumar, K.; Bowman, R.L.; Simon-Vermot, T.; Fennell, M.; Garippa, R.; et al. Microenvironment-derived factors driving metastatic plasticity in melanoma. Nat. Commun. 2017, 8, 14343. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.G.; Zhu, L.C.; Wang, Y.J.; Li, Y.Y.; Wang, D. Current Advance of Therapeutic Agents in Clinical Trials Potentially Targeting Tumor Plasticity. Front. Oncol. 2019, 9, 887. [Google Scholar] [CrossRef]
- Merlo, A.; Herman, J.G.; Mao, L.; Lee, D.J.; Gabrielson, E.; Burger, P.C.; Baylin, S.B.; Sidransky, D. 5’ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med. 1995, 1, 686–692. [Google Scholar] [CrossRef]
- Ocker, M.; Bitar, S.A.; Monteiro, A.C.; Gali-Muhtasib, H.; Schneider-Stock, R. Epigenetic Regulation of p21(cip1/waf1) in Human Cancer. Cancers 2019, 11, 1343. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Shi, S.; Gong, T.; Zhang, Z.; Sun, X. Cancer stem cells: Therapeutic implications and perspectives in cancer therapy. Acta Pharm. Sin. B 2013, 3, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, E.N.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, T.B.; Lim, J.J.; Chow, E.K. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef] [Green Version]
- Roberti, A.; Valdes, A.F.; Torrecillas, R.; Fraga, M.F.; Fernandez, A.F. Epigenetics in cancer therapy and nanomedicine. Clin. Epigenetics 2019, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Asmar, F.; Søgaard, A.; Grønbæk, K. Chapter 2—DNA Methylation and Hydroxymethylation in Cancer. In Epigenetic Cancer Therapy; Gray, S.G., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 9–30. [Google Scholar]
- Peterson, C.L.; Laniel, M.A. Histones and histone modifications. Curr. Biol. 2004, 14, R546–R551. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, T.B.; Veland, N.; Chen, T. Chapter 3—Writers, Readers, and Erasers of Epigenetic Marks. In Epigenetic Cancer Therapy; Gray, S.G., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 31–66. [Google Scholar]
- Li, S.; Han, Z.; Zhao, N.; Zhu, B.; Zhang, Q.; Yang, X.; Sheng, D.; Hou, J.; Guo, S.; Wei, L.; et al. Inhibition of DNMT suppresses the stemness of colorectal cancer cells through down-regulating Wnt signaling pathway. Cell Signal 2018, 47, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Morita, R.; Hirohashi, Y.; Suzuki, H.; Takahashi, A.; Tamura, Y.; Kanaseki, T.; Asanuma, H.; Inoda, S.; Kondo, T.; Hashino, S.; et al. DNA methyltransferase 1 is essential for initiation of the colon cancers. Exp. Mol. Pathol. 2013, 94, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Hon, G.C.; Hawkins, R.D.; Ren, B. Predictive chromatin signatures in the mammalian genome. Hum. Mol. Genet. 2009, 18, R195–R201. [Google Scholar] [CrossRef] [Green Version]
- Roca, M.S.; Di Gennaro, E.; Budillon, A. Implication for Cancer Stem Cells in Solid Cancer Chemo-Resistance: Promising Therapeutic Strategies Based on the Use of HDAC Inhibitors. J. Clin. Med. 2019, 8, 912. [Google Scholar] [CrossRef] [Green Version]
- Maiques-Diaz, A.; Somervaille, T.C. LSD1: Biologic roles and therapeutic targeting. Epigenomics 2016, 8, 1103–1116. [Google Scholar] [CrossRef] [Green Version]
- Adamo, A.; Sese, B.; Boue, S.; Castano, J.; Paramonov, I.; Barrero, M.J.; Izpisua Belmonte, J.C. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat. Cell Biol. 2011, 13, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Verigos, J.; Karakaidos, P.; Kordias, D.; Papoudou-Bai, A.; Evangelou, Z.; Harissis, H.V.; Klinakis, A.; Magklara, A. The Histone Demethylase LSD1/ΚDM1A Mediates Chemoresistance in Breast Cancer via Regulation of a Stem Cell Program. Cancers 2019, 11, 1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plch, J.; Hrabeta, J.; Eckschlager, T. KDM5 demethylases and their role in cancer cell chemoresistance. Int. J. Cancer. J. Int. Du Cancer 2019, 144, 221–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xhabija, B.; Kidder, B.L. KDM5B is a master regulator of the H3K4-methylome in stem cells, development and cancer. Semin Cancer Biol. 2019, 57, 79–85. [Google Scholar] [CrossRef] [PubMed]
- van den Beucken, T.; Koch, E.; Chu, K.; Rupaimoole, R.; Prickaerts, P.; Adriaens, M.; Voncken, J.W.; Harris, A.L.; Buffa, F.M.; Haider, S.; et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat. Commun. 2014, 5, 5203. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.A.; Laukka, T.; Myllykoski, M.; Ringel, A.E.; Booker, M.A.; Tolstorukov, M.Y.; Meng, Y.J.; Meier, S.R.; Jennings, R.B.; Creech, A.L.; et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 2019, 363, 1217–1222. [Google Scholar] [CrossRef] [Green Version]
- Caslini, C.; Hong, S.; Ban, Y.J.; Chen, X.S.; Ince, T.A. HDAC7 regulates histone 3 lysine 27 acetylation and transcriptional activity at super-enhancer-associated genes in breast cancer stem cells. Oncogene 2019, 38, 6599–6614. [Google Scholar] [CrossRef] [Green Version]
- Witt, A.E.; Lee, C.W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2017, 36, 1707–1720. [Google Scholar] [CrossRef]
- Bora-Singhal, N.; Mohankumar, D.; Saha, B.; Colin, C.M.; Lee, J.Y.; Martin, M.W.; Zheng, X.; Coppola, D.; Chellappan, S. Novel HDAC11 inhibitors suppress lung adenocarcinoma stem cell self-renewal and overcome drug resistance by suppressing Sox2. Sci. Rep. 2020, 10, 4722. [Google Scholar] [CrossRef]
- Cimmino, L.; Abdel-Wahab, O.; Levine, R.L.; Aifantis, I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell 2011, 9, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Thompson, E.C.; Hastie, R.; Tsangaratou, A.; Rajewsky, K.; Koralov, S.B.; Rao, A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 14566–14571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musselman, C.A.; Lalonde, M.E.; Cote, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkataraman, S.; Alimova, I.; Balakrishnan, I.; Harris, P.; Birks, D.K.; Griesinger, A.; Amani, V.; Cristiano, B.; Remke, M.; Taylor, M.D.; et al. Inhibition of BRD4 attenuates tumor cell self-renewal and suppresses stem cell signaling in MYC driven medulloblastoma. Oncotarget 2014, 5, 2355–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Xiao, R.; Pan, S.; Yang, X.; Yuan, W.; Tu, Z.; Xu, M.; Zhu, Y.; Yin, Q.; Wu, Y.; et al. Uncovering the roles of long non-coding RNAs in cancer stem cells. J. Hematol. Oncol. 2017, 10, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, C.M. Chapter 5—Long Noncoding RNAs and Cancer. In Epigenetic Cancer Therapy; Gray, S.G., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 91–114. [Google Scholar]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; He, L.; Du, Y.; Zhu, P.; Huang, G.; Luo, J.; Yan, X.; Ye, B.; Li, C.; Xia, P.; et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell 2015, 16, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Zhu, X.; Chen, F.; Huang, C.; Ai, K.; Wu, H.; Zhang, L.; Zhao, X. LncRNA XIST/miR-200c regulates the stemness properties and tumourigenicity of human bladder cancer stem cell-like cells. Cancer Cell Int. 2018, 18, 41. [Google Scholar] [CrossRef]
- Flaus, A.; Owen-Hughes, T. Mechanisms for ATP-dependent chromatin remodelling: Farewell to the tuna-can octamer? Curr. Opin. Genet. Dev. 2004, 14, 165–173. [Google Scholar] [CrossRef]
- Saha, A.; Wittmeyer, J.; Cairns, B.R. Chromatin remodelling: The industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006, 7, 437–447. [Google Scholar] [CrossRef]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Allis, C.D.; Chi, P. Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling. Trends Mol. Med. 2007, 13, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, L.; Wu, T.; Zhang, H.W.; Lu, N.; Hu, R.; Wang, Y.J.; Zhao, L.; Chen, F.H.; Wang, X.T.; You, Q.D.; et al. HIF-1alpha is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Okuda, H.; Watabe, M.; Kobayashi, A.; Pai, S.K.; Liu, W.; Pandey, P.R.; Fukuda, K.; Hirota, S.; Sugai, T.; et al. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene 2011, 30, 4075–4086. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.W.; Zhang, Z.G.; Hao, Y.X.; Zhao, Y.L.; Qian, F.; Shi, Y.; Li, P.A.; Liu, C.Y.; Yu, P.W. HIF-1alpha induces the epithelial-mesenchymal transition in gastric cancer stem cells through the Snail pathway. Oncotarget 2017, 8, 9535–9545. [Google Scholar] [CrossRef] [Green Version]
- Peng, F.; Wang, J.H.; Fan, W.J.; Meng, Y.T.; Li, M.M.; Li, T.T.; Cui, B.; Wang, H.F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef] [Green Version]
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95, S8–S19. [Google Scholar] [CrossRef]
- Wend, P.; Holland, J.D.; Ziebold, U.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Semin. Cell Dev. Biol. 2010, 21, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, C.; Mendoza-Topaz, C.; Mieszczanek, J.; Bienz, M. Stability elements in the LRP6 cytoplasmic tail confer efficient signalling upon DIX-dependent polymerization. J. Cell Sci. 2010, 123, 1588–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Z.; Zhao, J.; Zhang, Q.; Cao, C.; Tian, S.; Zhang, K.; Liu, L.; Shi, L.; Yu, N.; Yang, S. USP9X-mediated deubiquitination of B-cell CLL/lymphoma 9 potentiates Wnt signaling and promotes breast carcinogenesis. J. Biol. Chem. 2019, 294, 9844–9857. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fang, R.; Yue, C.; Chang, G.; Li, P.; Guo, Q.; Wang, J.; Zhou, A.; Zhang, S.; Fuller, G.N.; et al. Wnt-Induced Stabilization of KDM4C Is Required for Wnt/beta-Catenin Target Gene Expression and Glioblastoma Tumorigenesis. Cancer Res. 2020, 80, 1049–1063. [Google Scholar] [CrossRef]
- Regel, I.; Eichenmuller, M.; Mahajan, U.M.; Hagl, B.; Benitz, S.; Haberle, B.; Vokuhl, C.; von Schweinitz, D.; Kappler, R. Downregulation of SFRP1 is a protumorigenic event in hepatoblastoma and correlates with beta-catenin mutations. J. Cancer Res. Clin. Oncol. 2020, 146, 1153–1167. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Karkhanis, V.; Baiocchi, R.A.; Sif, S. Protein arginine methyltransferase 5 (PRMT5) promotes survival of lymphoma cells via activation of WNT/beta-catenin and AKT/GSK3beta proliferative signaling. J. Biol. Chem. 2019, 294, 7692–7710. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Andersson, E.R.; Lendahl, U. Therapeutic modulation of Notch signalling--are we there yet? Nat. Rev. 2014, 13, 357–378. [Google Scholar] [CrossRef]
- Munoz, P.; Iliou, M.S.; Esteller, M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol. Oncol. 2012, 6, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Bhagat, T.D.; Zou, Y.; Huang, S.; Park, J.; Palmer, M.B.; Hu, C.; Li, W.; Shenoy, N.; Giricz, O.; Choudhary, G.; et al. Notch Pathway Is Activated via Genetic and Epigenetic Alterations and Is a Therapeutic Target in Clear Cell Renal Cancer. J. Biol. Chem. 2017, 292, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, B.; He, Y.; He, J.; Wang, L.; Liu, Z.; Yang, J.; Gao, Z.; Lu, G.; Zou, C.; Zhao, W. Epigenetic Profiling Identifies LIF as a Super-enhancer-Controlled Regulator of Stem Cell-like Properties in Osteosarcoma. Mol. Cancer Res. MCR 2020, 18, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohnken, R.; Wen, J.; Mundy-Bosse, B.; McConnell, K.; Keiter, A.; Grinshpun, L.; Hartlage, A.; Yano, M.; McNeil, B.; Chakravarti, N.; et al. Diminished microRNA-29b level is associated with BRD4-mediated activation of oncogenes in cutaneous T-cell lymphoma. Blood 2018, 131, 771–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima-Fernandes, E.; Murison, A.; da Silva Medina, T.; Wang, Y.; Ma, A.; Leung, C.; Luciani, G.M.; Haynes, J.; Pollett, A.; Zeller, C.; et al. Targeting bivalency de-represses Indian Hedgehog and inhibits self-renewal of colorectal cancer-initiating cells. Nat. Commun. 2019, 10, 1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samadani, A.A.; Nikbakhsh, N.; Taheri, H.; Shafaee, S.; Fattahi, S.; Pilehchian Langroudi, M.; Hajian, K.; Akhavan-Niaki, H. CDX1/2 and KLF5 Expression and Epigenetic Modulation of Sonic Hedgehog Signaling in Gastric Adenocarcinoma. Pathol. Oncol. Res. 2019, 25, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Pak, E.; Segal, R.A. Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev. Cell 2016, 38, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, Y.; Kong, J.; Kim, E.; Choi, J.H.; Yuk, H.D.; Lee, H.; Kim, H.R.; Lee, K.H.; Kang, M.; et al. Epigenetic regulation of mammalian Hedgehog signaling to the stroma determines the molecular subtype of bladder cancer. eLife 2019, 8, e43024. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [Green Version]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manshouri, R.; Coyaud, E.; Kundu, S.T.; Peng, D.H.; Stratton, S.A.; Alton, K.; Bajaj, R.; Fradette, J.J.; Minelli, R.; Peoples, M.D.; et al. ZEB1/NuRD complex suppresses TBC1D2b to stimulate E-cadherin internalization and promote metastasis in lung cancer. Nat. Commun. 2019, 10, 5125. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Liu, Y.; Wu, F.; Liu, R.; Xie, Y.; Yang, Q.; Li, Y.; Liu, M.; Li, S.; Tang, H. miR-639 Expression Is Silenced by DNMT3A-Mediated Hypermethylation and Functions as a Tumor Suppressor in Liver Cancer Cells. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 587–598. [Google Scholar] [CrossRef]
- Bow, Y.D.; Wang, Y.Y.; Chen, Y.K.; Su, C.W.; Hsu, C.W.; Xiao, L.Y.; Yuan, S.S.; Li, R.N. Silencing of FOXA2 decreases E-cadherin expression and is associated with lymph node metastasis in oral cancer. Oral Dis. 2020, 26, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Yu, X.; Cong, P.; Zhou, Y.; Xu, Y.; Jiang, Y. Six2 promotes non-small cell lung cancer cell stemness via transcriptionally and epigenetically regulating E-cadherin. Cell Prolif. 2019, 52, e12617. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.H.; Sarkissian, M.; Hu, G.Q.; Wang, Z.; Bhattacharjee, A.; Gordon, D.B.; Gonzales, M.; Lan, F.; Ongusaha, P.P.; Huarte, M.; et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 2010, 466, 503–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Tanasa, B.; Tyurina, O.V.; Zhou, T.Y.; Gassmann, R.; Liu, W.T.; Ohgi, K.A.; Benner, C.; Garcia-Bassets, I.; Aggarwal, A.K.; et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 2010, 466, 508–512. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Gong, L.; Wu, Q.; Xing, C.; Wei, B.; Chen, T.; Zhou, Y.; Yin, S.; Jiang, B.; Xie, H.; et al. PHF8 upregulation contributes to autophagic degradation of E-cadherin, epithelial-mesenchymal transition and metastasis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 215. [Google Scholar] [CrossRef]
- Hu, Y.; Zheng, Y.; Dai, M.; Wang, X.; Wu, J.; Yu, B.; Zhang, H.; Cui, Y.; Kong, W.; Wu, H.; et al. G9a and histone deacetylases are crucial for Snail2-mediated E-cadherin repression and metastasis in hepatocellular carcinoma. Cancer Sci. 2019, 110, 3442–3452. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Lorton, B.; Gupta, V.; Shechter, D. A TGFbeta-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene 2017, 36, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Richter, A.M.; Kuster, M.M.; Woods, M.L.; Walesch, S.K.; Gokyildirim, M.Y.; Krueger, M.; Dammann, R.H. RASSF10 Is a TGFbeta-Target That Regulates ASPP2 and E-Cadherin Expression and Acts as Tumor Suppressor That Is Epigenetically Downregulated in Advanced Cancer. Cancers 2019, 11, 1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinchure, O.S.; Sharma, V.; Tabasum, S.; Ghosh, S.; Singh, R.P.; Sarkar, C.; Kulshreshtha, R. Polycomb complex mediated epigenetic reprogramming alters TGF-beta signaling via a novel EZH2/miR-490/TGIF2 axis thereby inducing migration and EMT potential in glioblastomas. Int. J. Cancer. J. Int. Du Cancer 2019, 145, 1254–1269. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.A.; Policastro, R.A.; Savant, S.S.; Sriramkumar, S.; Ding, N.; Lu, X.; Mohammad, H.P.; Cao, S.; Kalin, J.H.; Cole, P.A.; et al. Lysine-Specific Demethylase 1 Mediates AKT Activity and Promotes Epithelial-to-Mesenchymal Transition in PIK3CA-Mutant Colorectal Cancer. Mol. Cancer Res. MCR 2020, 18, 264–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.S.; Liu, H.Y.; Zhou, Z.; Sun, H.L.; Liu, M.Y. TSPAN8 promotes colorectal cancer cell growth and migration in LSD1-dependent manner. Life Sci. 2020, 241, 117114. [Google Scholar] [CrossRef] [PubMed]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and cancer: Regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Rinkenbaugh, A.L.; Baldwin, A.S. The NF-kappaB Pathway and Cancer Stem Cells. Cells 2016, 5, 16. [Google Scholar] [CrossRef]
- Xiao, W.; Ma, W.; Wei, S.; Li, Q.; Liu, R.; Carney, R.P.; Yang, K.; Lee, J.; Nyugen, A.; Yoneda, K.Y.; et al. High-affinity peptide ligand LXY30 for targeting alpha3beta1 integrin in non-small cell lung cancer. J. Hematol. Oncol. 2019, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiao, W.; Zhang, Y.; Meza, L.; Tseng, H.; Takada, Y.; Ames, J.B.; Lam, K.S. Optimization of RGD-Containing Cyclic Peptides against alphavbeta3 Integrin. Mol. Cancer Ther. 2016, 15, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin beta(3)-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; Macswords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebsbach, P.H.; Villa-Diaz, L.G. The Role of Integrin alpha6 (CD49f) in Stem Cells: More than a Conserved Biomarker. Stem Cells Dev. 2017, 26, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.R.; Yang, S.R.; Jung, J.W.; Kim, H.; Ko, K.; Han, D.W.; Park, S.B.; Choi, S.W.; Kang, S.K.; Scholer, H.; et al. CD49f enhances multipotency and maintains stemness through the direct regulation of OCT4 and SOX2. Stem Cells 2012, 30, 876–887. [Google Scholar] [CrossRef]
- Lamar, J.M.; Xiao, Y.; Norton, E.; Jiang, Z.G.; Gerhard, G.M.; Kooner, S.; Warren, J.S.A.; Hynes, R.O. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J. Biol. Chem. 2019, 294, 2302–2317. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Goel, H.L.; Gao, H.; Pursell, B.; Shultz, L.D.; Greiner, D.L.; Ingerpuu, S.; Patarroyo, M.; Cao, S.; Lim, E.; et al. A laminin 511 matrix is regulated by TAZ and functions as the ligand for the alpha6Bbeta1 integrin to sustain breast cancer stem cells. Genes Dev. 2015, 29, 1–6. [Google Scholar] [CrossRef]
- Sougawa, N.; Miyagawa, S.; Fukushima, S.; Yokoyama, J.; Kitahara, M.; Harada, A.; Mochizuki-Oda, N.; Sato-Nishiuchi, R.; Sekiguchi, K.; Sawa, Y. Laminin-511 Supplementation Enhances Stem Cell Localization With Suppression in the Decline of Cardiac Function in Acute Infarct Rats. Transplantation 2019, 103, e119–e127. [Google Scholar] [CrossRef]
- Goel, H.L.; Gritsko, T.; Pursell, B.; Chang, C.; Shultz, L.D.; Greiner, D.L.; Norum, J.H.; Toftgard, R.; Shaw, L.M.; Mercurio, A.M. Regulated splicing of the alpha6 integrin cytoplasmic domain determines the fate of breast cancer stem cells. Cell Rep. 2014, 7, 747–761. [Google Scholar] [CrossRef] [Green Version]
- Varzavand, A.; Hacker, W.; Ma, D.; Gibson-Corley, K.; Hawayek, M.; Tayh, O.J.; Brown, J.A.; Henry, M.D.; Stipp, C.S. alpha3beta1 Integrin Suppresses Prostate Cancer Metastasis via Regulation of the Hippo Pathway. Cancer Res. 2016, 76, 6577–6587. [Google Scholar] [CrossRef] [Green Version]
- Nakada, M.; Nambu, E.; Furuyama, N.; Yoshida, Y.; Takino, T.; Hayashi, Y.; Sato, H.; Sai, Y.; Tsuji, T.; Miyamoto, K.I.; et al. Integrin alpha3 is overexpressed in glioma stem-like cells and promotes invasion. Br. J. Cancer 2013, 108, 2516–2524. [Google Scholar] [CrossRef]
- Ellis, S.J.; Tanentzapf, G. Integrin-mediated adhesion and stem-cell-niche interactions. Cell Tissue Res. 2010, 339, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ouyang, G. Periostin: A bridge between cancer stem cells and their metastatic niche. Cell Stem Cell 2012, 10, 111–112. [Google Scholar] [CrossRef] [Green Version]
- Gailhouste, L.; Liew, L.C.; Yasukawa, K.; Hatada, I.; Tanaka, Y.; Nakagama, H.; Ochiya, T. Differentiation Therapy by Epigenetic Reconditioning Exerts Antitumor Effects on Liver Cancer Cells. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1840–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. 2020, 19, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Schoffski, P.; Jones, R.; Agulnik, M.; Villalobos, V.M.; Jahan, T.M.; Chen, T.W.-W.; Italiano, A.; Demetri, G.D.; Cote, G.M.; et al. Safety and efficacy of tazemetostat, a first-in-class EZH2 inhibitor, in patients (pts) with epithelioid sarcoma (ES) (NCT02601950). J. Clin. Oncol. 2019, 37, 11003. [Google Scholar] [CrossRef]
- Dai, G.; Deng, S.; Guo, W.; Yu, L.; Yang, J.; Zhou, S.; Gao, T. Notch pathway inhibition using DAPT, a gamma-secretase inhibitor (GSI), enhances the antitumor effect of cisplatin in resistant osteosarcoma. Mol. Carcinog 2019, 58, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Du, F.Y.; Zhou, Q.F.; Sun, W.J.; Chen, G.L. Targeting cancer stem cells in drug discovery: Current state and future perspectives. World J. Stem Cells 2019, 11, 398–420. [Google Scholar] [CrossRef]
- Girard, N.; Bazille, C.; Lhuissier, E.; Benateau, H.; Llombart-Bosch, A.; Boumediene, K.; Bauge, C. 3-Deazaneplanocin A (DZNep), an inhibitor of the histone methyltransferase EZH2, induces apoptosis and reduces cell migration in chondrosarcoma cells. PLoS ONE 2014, 9, e98176. [Google Scholar] [CrossRef] [Green Version]
- Hoy, S.M. Tazemetostat: First Approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef]
- Ribrag, V.; Soria, J.-C.; Michot, J.-M.; Schmitt, A.; Postel-Vinay, S.; Bijou, F.; Thomson, B.; Keilhack, H.; Blakemore, S.J.; Reyderman, L.; et al. Phase 1 Study of Tazemetostat (EPZ-6438), an Inhibitor of Enhancer of Zeste-Homolog 2 (EZH2): Preliminary Safety and Activity in Relapsed or Refractory Non-Hodgkin Lymphoma (NHL) Patients. Blood 2015, 126, 473. [Google Scholar] [CrossRef]
- Yap, T.A.; Winter, J.N.; Giulino-Roth, L.; Longley, J.; Lopez, J.; Michot, J.-M.; Leonard, J.P.; Ribrag, V.; McCabe, M.T.; Creasy, C.L.; et al. Phase I Study of the Novel Enhancer of Zeste Homolog 2 (EZH2) Inhibitor GSK2816126 in Patients with Advanced Hematologic and Solid Tumors. Clin. Cancer Res. 2019, 25, 7331–7339. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Wang, Z.; Zhou, J.; Huang, J.; Zhou, L.; Luo, J.; Wan, Y.Y.; Long, H.; Zhu, B. EZH2 Inhibitor GSK126 Suppresses Antitumor Immunity by Driving Production of Myeloid-Derived Suppressor Cells. Cancer Res. 2019, 79, 2009–2020. [Google Scholar] [CrossRef]
- Magliulo, D.; Bernardi, R.; Messina, S. Lysine-Specific Demethylase 1A as a Promising Target in Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 255. [Google Scholar] [CrossRef] [Green Version]
- Shorstova, T.; Marques, M.; Su, J.; Johnston, J.; Kleinman, C.L.; Hamel, N.; Huang, S.; Alaoui-Jamali, M.A.; Foulkes, W.D.; Witcher, M. SWI/SNF-Compromised Cancers Are Susceptible to Bromodomain Inhibitors. Cancer Res. 2019, 79, 2761–2774. [Google Scholar] [CrossRef] [Green Version]
- Diesch, J.; Zwick, A.; Garz, A.-K.; Palau, A.; Buschbeck, M.; Götze, K.S. A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin. Epigenetics 2016, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Qian, J. Chapter 23—Clinical Trials. In Epigenetic Cancer Therapy; Gray, S.G., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 525–568. [Google Scholar]
- Notari, R.E.; Deyoung, J.L. Kinetics and Mechanisms of Degradation of the Antileukemic Agent 5-Azacytidine in Aqueous Solutions. J. Pharm. Sci. 1975, 64, 1148–1157. [Google Scholar] [CrossRef]
- Agrawal, K.; Das, V.; Vyas, P.; Hajdúch, M. Nucleosidic DNA demethylating epigenetic drugs—A comprehensive review from discovery to clinic. Pharmacol. Ther. 2018, 188, 45–79. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Zahnow, C.A.; Ahuja, N.; Baylin, S.B. Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer Res. 2016, 76, 1683–1689. [Google Scholar] [CrossRef] [Green Version]
- Unnikrishnan, A.; Papaemmanuil, E.; Beck, D.; Deshpande, N.P.; Verma, A.; Kumari, A.; Woll, P.S.; Richards, L.A.; Knezevic, K.; Chandrakanthan, V.; et al. Integrative Genomics Identifies the Molecular Basis of Resistance to Azacitidine Therapy in Myelodysplastic Syndromes. Cell Rep. 2017, 20, 572–585. [Google Scholar] [CrossRef] [Green Version]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination Epigenetic Therapy Has Efficacy in Patients with Refractory Advanced Non–Small Cell Lung Cancer. Cancer Discov. 2011, 1, 598–607. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.H.; Kang, K.W.; Jeon, M.J.; Yu, E.S.; Kim, D.S.; Choi, H.; Lee, S.R.; Sung, H.J.; Kim, B.S.; Choi, C.W.; et al. Comparison between 5-day decitabine and 7-day azacitidine for lower-risk myelodysplastic syndromes with poor prognostic features: A retrospective multicentre cohort study. Sci. Rep. 2020, 10, 39. [Google Scholar] [CrossRef]
- La, X.; Zhang, L.; Li, Z.; Li, H.; Yang, Y. (-)-Epigallocatechin Gallate (EGCG) Enhances the Sensitivity of Colorectal Cancer Cells to 5-FU by Inhibiting GRP78/NF-kappaB/miR-155-5p/MDR1 Pathway. J. Agric. Food Chem 2019, 67, 2510–2518. [Google Scholar] [CrossRef]
- Trudel, D.; Labbé, D.P.; Araya-Farias, M.; Doyen, A.; Bazinet, L.; Duchesne, T.; Plante, M.; Grégoire, J.; Renaud, M.-C.; Bachvarov, D.; et al. A two-stage, single-arm, phase II study of EGCG-enriched green tea drink as a maintenance therapy in women with advanced stage ovarian cancer. Gynecol. Oncol. 2013, 131, 357–361. [Google Scholar] [CrossRef]
- Candelaria, M.; Gallardo-Rincón, D.; Arce, C.; Cetina, L.; Aguilar-Ponce, J.L.; Arrieta, O.; Serrano, A.; Perez-Plasencia, C.; Gonzalez-Fierro, A.; de la Cruz-Hernandez, E.; et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. BMC Cancer 2007, 7, A27. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Cárdenas, E.; Taja-Chayeb, L.; Trejo-Becerril, C.; Chanona-Vilchis, J.; Chávez-Blanco, A.; Domínguez-Gómez, G.; Langley, E.; García-Carrancá, A.; Dueñas-González, A. Antimetastatic effect of epigenetic drugs, hydralazine and valproic acid, in Ras-transformed NIH 3T3 cells. OncoTargets Ther. 2018, 11, 8823–8833. [Google Scholar] [CrossRef] [Green Version]
- Kirschbaum, M.H.; Goldman, B.H.; Zain, J.M.; Cook, J.R.; Rimsza, L.M.; Forman, S.J.; Fisher, R.I. A phase 2 study of vorinostat for treatment of relapsed or refractory Hodgkin lymphoma: Southwest Oncology Group Study S0517. Leuk Lymphoma 2012, 53, 259–262. [Google Scholar] [CrossRef]
- De Souza, C.; Chatterji, B.P. HDAC Inhibitors as Novel Anti-Cancer Therapeutics. Recent Pat. Anticancer Drug Discov. 2015, 10, 145–162. [Google Scholar] [CrossRef]
- Gao, X.; Shen, L.; Li, X.; Liu, J. Efficacy and toxicity of histone deacetylase inhibitors in relapsed/refractory multiple myeloma: Systematic review and meta-analysis of clinical trials. Exp. Med. 2019, 18, 1057–1068. [Google Scholar] [CrossRef] [Green Version]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, L.; Yang, X.B. Harnessing the HDAC–histone deacetylase enzymes, inhibitors and how these can be utilised in tissue engineering. Int. J. Oral Sci. 2019, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, F.U.; Rehman, M.S.; Khan, M.S.; Ali, M.A.; Javed, A.; Nawaz, A.; Yang, C. Curcumin as an Alternative Epigenetic Modulator: Mechanism of Action and Potential Effects. Front. Genet. 2019, 10, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, M.I.; Iwuji, C.; Irving, G.; Karmokar, A.; Higgins, J.A.; Griffin-Teal, N.; Thomas, A.; Greaves, P.; Cai, H.; Patel, S.R.; et al. Curcumin inhibits cancer stem cell phenotypes in ex vivo models of colorectal liver metastases, and is clinically safe and tolerable in combination with FOLFOX chemotherapy. Cancer Lett. 2015, 364, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct Target. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, Y.; Zhu, H.; Lee, J.H.; Kossenkov, A.V.; Wu, S.Y.; Wickramasinghe, J.M.; Yin, X.; Palozola, K.C.; Gardini, A.; Showe, L.C.; et al. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res. 2016, 76, 6320–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piya, S.; Mu, H.; Bhattacharya, S.; Lorenzi, P.L.; Davis, R.E.; McQueen, T.; Ruvolo, V.; Baran, N.; Wang, Z.; Qian, Y.; et al. BETP degradation simultaneously targets acute myelogenous leukemic stem cells and the microenvironment. J. Clin. Investig. 2019, 129, 1878–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.; Paradise, B.D.; Ma, W.W.; Fernandez-Zapico, M.E. Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer. Cells 2019, 8, 394. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, C.; Cortes, J.E.; Oehler, V.; Baccarani, M.; Kantarjian, H.M.; Papayannidis, C.; Rice, K.N.; Zhang, X.; Shaik, N.; Courtney, R.; et al. Phase 1 Dose-Escalation Study of PF-04449913, An Oral Hedgehog (Hh) Inhibitor, in Patients with Select Hematologic Malignancies. Blood 2011, 118, 424. [Google Scholar] [CrossRef]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.-C.; Peaceman, D.; Özdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Long, B.; Wang, L.-X.; Zheng, F.-M.; Lai, S.-P.; Xu, D.-R.; Hu, Y.; Lin, D.-J.; Zhang, X.-Z.; Dong, L.; Long, Z.-J.; et al. Targeting GLI1 Suppresses Cell Growth and Enhances Chemosensitivity in CD34+ Enriched Acute Myeloid Leukemia Progenitor Cells. Cell. Physiol. Biochem. 2016, 38, 1288–1302. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; González-Ramírez, M.; Alcaine-Colet, A.; Aranda, S.; Di Croce, L. The Bivalent Genome: Characterization, Structure, and Regulation. Trends Genet. 2020, 36, 118–131. [Google Scholar] [CrossRef]
- Hall, A.W.; Battenhouse, A.M.; Shivram, H.; Morris, A.R.; Cowperthwaite, M.C.; Shpak, M.; Iyer, V.R. Bivalent Chromatin Domains in Glioblastoma Reveal a Subtype-Specific Signature of Glioma Stem Cells. Cancer Res. 2018, 78, 2463–2474. [Google Scholar] [CrossRef] [Green Version]
- Curry, E.; Zeller, C.; Masrour, N.; Patten, D.K.; Gallon, J.; Wilhelm-Benartzi, C.S.; Ghaem-Maghami, S.; Bowtell, D.D.; Brown, R. Genes Predisposed to DNA Hypermethylation during Acquired Resistance to Chemotherapy Are Identified in Ovarian Tumors by Bivalent Chromatin Domains at Initial Diagnosis. Cancer Res. 2018, 78, 1383–1391. [Google Scholar] [CrossRef] [Green Version]
- Alarcón, T.; Sardanyés, J.; Guillamon, A.; Menendez, J.A. Bivalent chromatin as a therapeutic target in cancer: An in silico predictive approach for combining epigenetic drugs. PLoS Comput. Biol. 2021, 17, e1008408. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Study | Phase/ Randomization | Drug | Disease | Combination with |
|---|---|---|---|---|
| NCT01897571 | Phase I/II | Tazemetostat | Epithelial sarcoma | Single agent |
| NCT02082977 | Phase I | GSK126 | Relapsed/refractory diffuse large B cell lymphoma, transformed follicular lymphoma, other non-Hodgkin’s lymphomas, solid tumors and multiple myeloma | Single agent |
| Study | Phase/ Randomization | Drug | Disease | Combination with |
|---|---|---|---|---|
| NCT02913443 | Non-randomized | ORY-1001 | ED SCLC * | Single agent |
| NCT02034123 | Non-randomized | GSK2879552 | Relapsed/refractory small cell lung carcinoma | Single agent |
| Study | Phase/ Randomization | Drug | Disease | Combination with |
|---|---|---|---|---|
| NCT00748553 | Not available | Azacitidine | Breast cancer and metastatic solid tumors | Abraxane |
| NCT00404508 | Non-randomized | Hydralazine | Refractory solid tumors | Magnesium valproate |
| Study | Phase/ Randomization | Drug | Disease | Combination with |
|---|---|---|---|---|
| NCT01583283 | Not available | Ricolinonstat (ACY-1215) | Multiple myeloma | Lenalidomide and dexamethasone |
| Study | Phase/ Randomization | Drug | Disease | Combination with |
|---|---|---|---|---|
| NCT03936465 | Phase I | BMS-986158 | Pediatric solid tumors, lymphomas, brain tumor | Monotherapy |
| NCT03901469 | Phase II | ZEN003694 | Triple Negative Breast Cancer | Talazoparib |
| Study | Phase/ Randomization | Drug | Disease | Combination with |
|---|---|---|---|---|
| NCT01981551 | Phase I/II | Nirogacestat | Desmoid tumors | Monotherapy |
| NCT02753127 | Phase III | Napabucasin (BBI-608) | Metastatic colorectal cancer | 5-Fluorouracil, Leucovorin, Irinotecan |
| NCT04065269 | Phase II | AZD6738 | Gynecological cancers | Olaparib |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, V.E.; Nambiar, R.; De Souza, C.; Nguyen, A.; Chien, J.; Lam, K.S. Targeting Epigenetic Modifiers of Tumor Plasticity and Cancer Stem Cell Behavior. Cells 2022, 11, 1403. https://doi.org/10.3390/cells11091403
Kumar VE, Nambiar R, De Souza C, Nguyen A, Chien J, Lam KS. Targeting Epigenetic Modifiers of Tumor Plasticity and Cancer Stem Cell Behavior. Cells. 2022; 11(9):1403. https://doi.org/10.3390/cells11091403
Chicago/Turabian StyleKumar, Vigneshwari Easwar, Roshni Nambiar, Cristabelle De Souza, Audrey Nguyen, Jeremy Chien, and Kit S. Lam. 2022. "Targeting Epigenetic Modifiers of Tumor Plasticity and Cancer Stem Cell Behavior" Cells 11, no. 9: 1403. https://doi.org/10.3390/cells11091403
APA StyleKumar, V. E., Nambiar, R., De Souza, C., Nguyen, A., Chien, J., & Lam, K. S. (2022). Targeting Epigenetic Modifiers of Tumor Plasticity and Cancer Stem Cell Behavior. Cells, 11(9), 1403. https://doi.org/10.3390/cells11091403