Abstract
Unresolved hyperglycaemia, a hallmark of type 2 diabetes mellitus (T2DM), is a well characterised manifestation of altered fuel homeostasis and our understanding of its role in the pathologic activation of the inflammatory system continues to grow. Metabolic disorders like T2DM trigger changes in the regulation of key cellular processes such as cell trafficking and proliferation, and manifest as chronic inflammatory disorders with severe long-term consequences. Activation of inflammatory pathways has recently emerged as a critical link between T2DM and inflammation. A substantial body of evidence has suggested that this is due in part to increased flux through the hexosamine biosynthetic pathway (HBP). The HBP, a unique nutrient-sensing metabolic pathway, produces the activated amino sugar UDP-GlcNAc which is a critical substrate for protein O-GlcNAcylation, a dynamic, reversible post-translational glycosylation of serine and threonine residues in target proteins. Protein O-GlcNAcylation impacts a range of cellular processes, including inflammation, metabolism, trafficking, and cytoskeletal organisation. As increased HBP flux culminates in increased protein O-GlcNAcylation, we propose that targeting O-GlcNAcylation may be a viable therapeutic strategy for the prevention and management of glucose-dependent pathologies with inflammatory components.
1. Introduction
Sustained hyperglycaemia, which is the hallmark of type 2 diabetes mellitus (T2DM), comes with far reaching consequences such as increased risk of cardiovascular diseases (CVD) and kidney failure [1]. While some of the complications associated with hyperglycaemia are the result of acute metabolic derangements e.g., ketoacidosis, a majority of complications are due in part to chronically elevated blood glucose levels e.g., stroke, neuropathy, retinopathy and nephropathy [1]. Metabolic alterations associated with hyperglycaemia have been well characterised [2]. Specifically, the pathogenesis and progression of T2DM have been ascribed to four key mechanisms; increased polyol pathway flux, increased advanced glycation end product (AGE) formation, activation of protein kinase C (PKC) isoforms, and increased hexosamine pathway flux [2,3]. However, until recently, the contribution of increased hexosamine pathway flux to the development of metabolic disorders was unclear [2,4,5,6]. The final product of the hexosamine biosynthetic pathway (HBP) is the activated amino sugar UDP-GlcNAc, a critical substrate for protein glycosylation [6,7,8]. UDP-GlcNAc serves as the sugar donor for O-GlcNAcylation events which is a dynamic, reversible post-translational modification of serine and threonine residues in target proteins within cells [8,9]. This process is controlled by two enzymes: O-linked β-N-acetylglucosamine transferase (OGT) and O-linked β-N-acetylglucosaminidase (OGA) [8,9]. OGT is responsible for catalysing the addition of a O-GlcNAc moiety to either serine or threonine residues in target proteins [10,11]. Conversely, the enzyme OGA reverses this modification by catalysing the hydrolysis of O-GlcNAc from protein targets [10,11]. A large body of evidence has shown that increased hexosamine pathway flux results in increased OGT-mediated O-GlcNAcylation events [6,12,13,14,15]. Importantly, O-GlcNAcylation of inflammatory pathways, and trafficking and proliferation of cells have emerged as a critical link between T2DM and the chronic inflammation observed in pathologies that are a consequence of poor blood glucose control [2,4,5,16]. In recent years, a growing number of findings elucidating the role of O-GlcNAcylation as vital molecular mechanism in the pathogenesis of T2DM and its complications have emerged [2,4,5,12,13,14,15,16]. In this review, we will, therefore, evaluate the involvement of O-GlcNAcylation in the inflammatory events induced by T2DM and the potential therapeutic viability of targeting O-GlcNAcylation in preventing and managing disease progression.
2. Hexosamine Biosynthetic Pathway (HBP) and O-GlcNAcylation
2.1. Hexosamine Biosynthetic Pathway (HBP)
The HBP is a distinct nutrient-sensing metabolic pathway that produces the activated amino sugar UDP-N-acetyl-glucosamine (UDP-GlcNAc), a key substrate for protein glycosylation. This type of glycosylation is particularly sensitive to changes in UDP-GlcNAc concentration. However, HBP and O-GlcNAcylation are not only sensitive to changes in glucose concentrations but also changes in protein and lipid metabolism [17]. Over the years, there have been significant advances in our understanding of this dynamic pathway. An important breakthrough came when Marshall et al. [18] treated isolated rat adipocytes with glucosamine (GlcN) to demonstrate the contribution of glucose flux through the HBP to the development of insulin resistance. Given the likelihood that glucose and GlcN mediate desensitisation through the same mechanism and, since GlcN appears to be 40 times more potent than glucose, it was suggested that an estimated 2–3% of the incoming glucose that is converted to fructose-6-phosphate (Fru-6-P) enters the HBP [18]. However, recent findings by Olsen et al. [19] using a more reliable quantitative assay in an ex vivo mouse heart model showed that glucose metabolism through the HBP, as determined by the rates of glycolysis and UDP-GlcNAc synthesis, comprises only ~0.006% of the glycolytic efflux, which is much lower than that originally proposed [18]. The 2–3% estimate from Marshall et al. [18] was from a single study in rat adipocyte cultures and expressed as a representation of the percentage of total glucose uptake. However, it is unclear whether this value would be consistent between quiescent cells in culture versus metabolically active organs with high energy demand such as the heart. In view of these shortcomings, the estimate by Olsen et al. [19] may be more quantitatively reliable.
As shown in Figure 1, when glucose enters the cell, it is converted to fructose-6P (fructose-6-phosphate), following this, glutamine-fructose-6P amidotransferase 1 (GFAT), the rate-limiting enzyme in the HBP transfers an amino group to fructose-6-phosphate from glutamine to form glucosamine-6-phosphate (GlcN-6-P). GlcN-6P is then rapidly acetylated by glucosamine-phosphate N-acetyltransferase (GNPNAT, EMeg32) in the presence of Acetyl-CoA, to produce N-acetylglucosamine-6-phosphate (GlcNAc-6P) [20], which further undergoes isomerisation by GlcNAc phosphomutase (PGM3/AGM1) to produce N-acetylglucosamine-1-phosphate (GlcNAc-1P) [21]. Then the nucleoside is added to the sugar by UDP-N-acetylhexosamine pyrophosphorylase 1 (UAP/AGX1) to yield UDP-GlcNAc which is the amino sugar substrate. UDP-GlcNAc is then used as a substrate for N- and O-linked glycosylation reactions in the ER and Golgi and for O-GlcNAc modification of nuclear and cytoplasmic proteins by OGT (O-GlcNAc transferase). OGA (O-GlcNAcase) catalyses the removal of O-GlcNAc and adds back GlcNAc to the HBP pool for re-cycling through the salvage pathway. Studies have shown that unresolved and sustained hyperglycaemia increases glycolytic efflux and subsequent activation of HBP which then increases production of the amino sugar substrate UDP-GlcNAc for protein glycosylation [6,12,13,14,15].
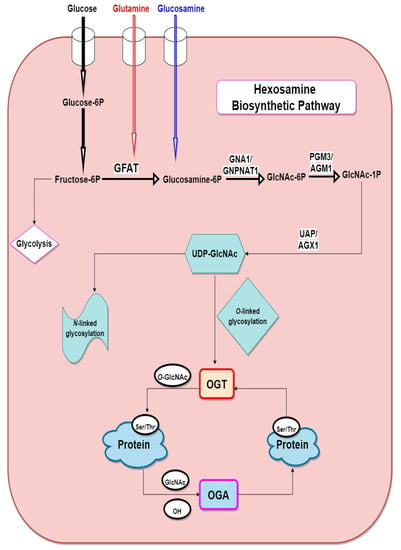
Figure 1.
Schematic of the hexosamine biosynthetic pathway. fructose-6P (fructose-6-phosphate), glucosamine-6P (glucosamine-6-phosphate), GFAT (glutamine:fructose-6-phosphate amidotransferase), OGT (O-GlcNAc transferase), OGA (O-GlcNAcase) GNA1/GNPNAT1 (glucosamine-6-phosphate N-acetyltransferase), GlcNAc-6P (N-acetylglucosamine-6-Phosphate), GlcNAc-1P (N-acetylglucosamine-1-phosphate), PGM3/AGM1 (phosphoglucomutase), UDP-GlcNAc (uridine diphosphate-N-acetylglucosamine), UAP/AGX1 (UDP-N-acetylhexosamine pyrophosphorylase).
2.2. Protein O-GlcNAcylation
O-GlcNAcylation is a dynamic post-translational glycosylation that links single GlcNAc molecules to target serine and threonine residueson target proteins by an O-linked β-glycosidic bond [21]. The O-GlcNAcome recently reached a milestone of 5000 human proteins identified [22]. Distinct from other protein glycosylation events, which are chiefly produced by the secretory pathways, O-GlcNAcylated proteins are predominantly localised in the nucleus and cytoplasm, with only approximately 7% of O-GlcNAc moieties detectable at the cell surface [7,21]. Furthermore, cellular distribution analysis has confirmed that O-GlcNAcylated proteins are chiefly concentrated in nuclear and cytoplasmic compartments (Figure 2) [22]. O-GlcNAcylation has been shown to be highly enriched on proteins in nuclear pore complexes and the nuclear envelope [21,23], as well as proteins that can interact with chromatin [24]. Cytoskeletal [25], and intrinsic membrane proteins in the Golgi apparatus and endoplasmic reticulum (ER) have also been shown to be O-GlcNAcylated [26]. Unlike phosphorylation and other post-translational modifications (PTMs), there is currently no consensus sequence that can accurately predict target protein modification by O-GlcNAcylation.

Figure 2.
Subcellular distribution of the O-GlcNAcome. Median confidence scores of human O-GlcNAcylated proteins (n = 4969) are from [22].
As stated earlier, a single pair of enzymes—OGT and OGA—controls this dynamic cycling [15,16]. OGT catalyses the attachment of a monosaccharide (GlcNAc) moiety to the free hydroxyl group of either serine or threonine residues in target proteins through an O-glycosidic linkage [10,11,27]. The human OGT gene produces three known isoforms of OGT namely the ncOGT, mOGT, and sOGT. The ncOGT which is the longest of the isoforms is localized to both the nucleus and cytoplasm. It contains a unique N-terminal sequence followed by 12 tetratricopeptide repeats (TPR) motifs, a linker region, and the catalytic domain. Furthermore, mOGT, the second isoform has a distinct N-terminal sequence which includes a mitochondrial targeting motif. The N-terminal sequence is then followed by 9 TPR motifs, a linker region, and the catalytic domain. sOGT, the shortest of the isoforms is ubiquitously expressed. Characteristically, it has only 2 TPR motifs, a linker region, and the catalytic domain. Importantly, all three isoforms have identical catalytic region which contained two sub-domains termed CD I and CD II [11,27].
Conversely, OGA, which was first purified from rat spleen cytosol, reverses O-GlcNAcylation by catalysing the hydrolysis of O-GlcNAc from protein targets [11]. OGA is largely enriched in the cytosol, unlike OGT, which accumulates in the cytoplasm and nucleus [28]. The OGA transcript encodes a protein with three distinct regions: a N-terminal catalytic domain, a stalk domain and a C-terminal pseudo-histone acetyltransferase domain [29,30]. The human OGA can exist as a homodimer with a unique interaction with other OGAs. During this interaction, it uses the substrate-recognition mode in which its stalk domain combines with the catalytic domain from other OGA monomer, and this interaction forms a cleft for substrate binding [30,31,32,33]. Also, hydrophobic residues are chiefly dominant in the inner surface of the substrate binding cleft, and are conserved in most eukaryotes. These hydrophobic interactions are important for protein binding and spatial constraints [30].
3. Involvement of O-GlcNAcylation in Inflammation
3.1. Impact of O-GlcNAcylation on Inflammatory Signalling Pathways
Activation of inflammatory signalling pathways by glucose-dependent metabolic stress is a major phenomenon in the pathogenesis and progression of T2DM [2,4] as well as other known pathologies such as cancers, autoimmune disorders and neurological disease [34,35,36,37,38]. Despite our incomplete understanding of the mechanisms responsible for nutrient regulation of inflammation, several studies suggest that O-GlcNAcylation is critical to the pathophysiology of inflammatory derangements [37,39,40,41,42,43]. Importantly, while some studies have indicated that O-GlcNAcylation of target proteins is a pro-inflammatory event in diabetes and is involved in the development of insulin resistance [44,45]. Conversely, others have shown that this dynamic post-translational modification can also confer protection against acute inflammatory stimuli [46,47,48]. These findings [44,45,46,47,48] strongly suggest that diverse forms of insults could result in dynamic changes to O-GlcNAcylation patterns to either promote or inhibit inflammation in response to fluctuations in cellular metabolism. This section of the review highlights core involvement of O-GlcNAc modification of target proteins in inflammatory pathways.
3.1.1. Pro-Inflammatory Role of O-GlcNAcylation
Emerging findings have continued to improve our understanding of the pro-inflammatory role of O-GlcNAcylation in the NF-κB signaling pathway. NF-κB is a chief regulator of innate and adaptive immune responses and plays a central role in chronic inflammation responsible for tumour development, rheumatoid arthritis, atherosclerosis, chronic obstructive pulmonary disease, asthma, multiple sclerosis, inflammatory bowel disease, and ulcerative colitis [49,50,51,52,53]. Yang et al. [54] have shown that the p65/RelA NF-κB subunit is O-GlcNAcylated in rat vascular smooth muscle cells. O-GlcNAc modification of NF-κB p65 on Thr352 inhibits the interaction between NF-κB p65 and IκB, thereby promoting translocation of NF-κB heterodimers to the nucleus and increasing transcription of target genes such as the gene for adhesion molecule VCAM-1 under hyperglycaemic conditions [50]. These findings strongly suggest that specific O-GlcNAcylation of p65/RelA on Thr352 may facilitate sustained activation of NF-κB-mediated chronic inflamamtion in response to prolonged hyperglycaemia and development of diabetes [50]. Although the mechanism of how O-GlcNAcylation activates NF-κB signaling is not fully understood, however, O-GlcNAcylation has been shown to increase the phosphorylation and acetylation of NF-κB subunit p65/RelA [51]. O-GlcNAc modification of p65 at Thr305 and Ser319 increased CREB-binding protein (CBP)/p300-dependent activating acetylation of p65 at Lys310, and this contributes to NF-κB transcriptional activation [51]. Also, elevation of O-GlcNAcylation by upregulating the expression of OGT increased the expression of p300, IKKα, and IKKβ and promoted IKK-mediated activating phosphorylation of p65 at Ser536, contributing to NF-κB activation (Figure 3) [51]. Similarly, it has been proposed that O-GlcNAcylation of IKKβ at Ser733 enhances NF-κB activity by increasing IKK activity, IκB phosphorylation and subsequent IκB degradation in both mouse and human fibroblasts [52]. These findings [49,50,52] suggest mechanisms through which O-GlcNAcylation regulates NF-κB signaling through interplay with phosphorylation and acetylation with consequent inflammatory outputs.
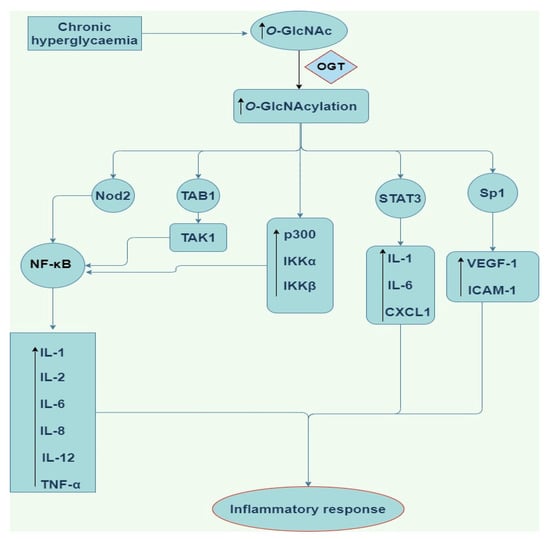
Figure 3.
Schematic of the pro-inflammatory downstream signalling modulation by O-GlcNAcylation. O-GlcNAc (O-linked β-N-acetylglucosamine), OGT (O-linked β-N-acetylglucosamine transferase), NF-κB (nuclear factor kappa light chain enhancer of activated B cells), STAT3 (signal transducer and activator of transcription3), TAK 1 (Transforming growth factor-β activated kinase 1), TAB1 (TAK1-binding protein 1), Nod2 (nucleotide-binding oligomerization domain-containing protein 2), IL-1,6,8, and 12 (Interleukin-1,6,8, and 12), Sp1 (substrate of Keap1), CXCL1 (chemokine ligand 1), VEGF-A (vascular endothelial growth factor), ICAM-1 (intercellular Adhesion Molecule 1), TNF-α (tumor necrosis factor alpha).
Furthermore, Sp1, a zinc finger transcription factor is also O-GlcNAcylated in response to high glucose concentrations and elevated Sp1 activity upon O-GlcNAcylation could play a role in hyperglycaemia-induced pro-fibrotic and pro-inflammatory factors involved in diabetic retinopathy [55,56]. Donovan et al. [55] showed that hyperglycaemia significantly increased Sp1 binding to the gene promoter of the pro-angiogenic mediator vascular endothelial growth factor (VEGF)-A, while shRNA-mediated knockdown of either OGT or Sp1 significantly inhibited glucose-induced increases in VEGF-A levels in ARPE-19 human retinal pigment epithelial cells and TR-iBRB rat retinal microvascular endothelial cells. These observations suggest that hyperglycaemia-induced O-GlcNAcylation of Sp1 drives VEGF-A production in both systems. Zhang et al. [56] have also proposed that hyperglycaemia also stimulates expression of intercellular adhesion molecule (ICAM)-1 by O-GlcNAcylation of Sp1 in both human umbilical vein endothelial cells and rat retinal capillary endothelial cells.
Alternatively, the transcriptional activity of Sp1 can be reduced when O-GlcNAc modified, and this may be due to the resultant distortion in its interaction with its cooperative factors such as Elf-1 [57], NF-Y [58], Ying-Yang 1 [59], and sterol regulatory element binding protein 2 [60]. Thus, O-GlcNAc modification of Sp1 could contribute to negative regulation of placental and embryonic expression of oncofetal protein gene (Pem), a gene which encodes a homeobox protein expressed in reproductive tissues and a wide range of transformed cells [57]. Furthermore, O-GlcNAcylation of Sp1 inhibits hyaluronan synthesis (an important component of the extracellular matrix that plays a crucial role for cumulus cell expansion which occurs during maturation of oocytes [59,60], and lipid synthesis [61]. Intriguingly, Suh et al. [62] demonstrated that O-GlcNAc modification of Sp1 protects primary cultured renal proximal tubule cells against hypoxia-induced dysfunction of Na/glucose cotransporter (SGLT). Similarly, Lee et al. [63] showed that O-GlcNAcylation of Sp1 protects mouse embryonic stem cells against hypoxia-induced apoptosis. A weakened association between Sp1 and its co-operative factors caused by O-GlcNAcylation is one potential mechanism which could explain these phenomena.
As a critical effector of cytokines which activate the Janus kinase (JAK)-STAT pathway, STAT3 function is tightly controlled [64]. However, our understanding of how metabolic changes regulate STAT3 function via specific PTMs that regulate STAT3 function, and their pathophysiological significance remain incomplete. Li et al. [46] demonstrated that there was an increased disease severity in azoxymethane-induced colitis and a colitis-associated cancer model in the bone marrow-derived macrophages isolated from CUL3-deficient mice. Li et al. [46] proposed that this was due to increased O-GlcNAcylation of STAT3. In addition, the expression levels of pro-inflammatory cytokines IL-1 and IL-6, and chemokines CXCL1 and CXCL2 were upregulated by O-GlcNAcylated STAT3 in BMMs from CUL3-deficient mice, thus contributing to azoxymethane-induced colitis and colitis-associated cancer.
Apart from the transcription factors highlighted above, other proteins which control inflammation and immunity e.g., Transforming growth factor-β activated kinase (TAK) 1 are also O-GlcNAcylated. TAK1 is an important serine/threonine protein kinase that mediates signals transduced by multifunctional cytokines such as TGF-β, TNF-α and IL-1 [65]. Emerging data have suggested that O-GlcNAcylation of TAK1-binding protein 1 (TAB1) at Ser395 is needed for TAK1 activation [65]. More so, activated TAK1 is intricate to downstream activation of NF-κB and production of IL-6 and TNF-α in human embryonic kidney (HEK) 293 cells stably expressing the IL-1 receptor and reconstituted Tab1-deficient MEFs upon exposure to IL-1 or hyperosmotic stress [65]. Upon stimulation, O-GlcNAc modification of TAK1 at Ser427 is needed for the phosphorylation of Thr187/Ser192 and full activation of TAK1 in RAW264.7 cells [66]. Thus, O-GlcNAc modification of TAK1 has been shown to trigger downstream activation of JNK and NF-κB signaling pathways, facilitate M1 polarisation of macrophages, and increase production of pro-inflammatory cytokine, resulting in the development of acute inflammatory responses [67]. Also, a cytoplasmic human nucleotide-binding oligomerization domain-containing protein 2 (Nod2)-like receptor that recognises bacterial components, is O-GlcNAcylated and this modification causes it to produce various pro-inflammatory cytokines and chemokines via activation of NF-κB in HEK293T cells (Figure 3) [42].
3.1.2. Anti-Inflammatory Role of O-GlcNAcylation
There is evidence to support O-GlcNAc modification of target proteins in the vasculature as a novel anti-inflammatory and vasoprotective mechanism [47,48]. Xing et al. [47] acutely treated ovariectomized rats with GlcN and PUGNAc before balloon injury of the right carotid artery and found that GlcN and PUGNAc each upregulated protein O-GlcNAcylation and significantly ameliorated acute inflammation [47]. Also, increased levels of proinflammatory HIS48+ granulocytes and ED1+ monocytes in response to injury were significantly reduced by GlcN and PUGNAc treatment through reduced infiltration into injured arteries [47]. The authors were also able to demonstrate that chronic (14 days) treatment with GlcN significantly reduced neointima formation in injured arteries compared with vehicle controls [47]. A protective anti-inflammatory role for O-GlcNAcylation in the vasculature is also supported by Hilgers et al. [48], who demonstrated that acute increases in protein O-GlcNAcylation were associated with reduced TNF-α-induced hypocontractility of rat aortic rings. In this study, iNOS protein expression was increased in TNF-α-treated rings, and this was attenuated by pretreatment with either GlcN or Thiamet-G, suggesting that acute increases in protein O-GlcNAcylation ameliorated TNF-α induced vascular dysfunction at least in part by limiting iNOS expression (Figure 4) [48].
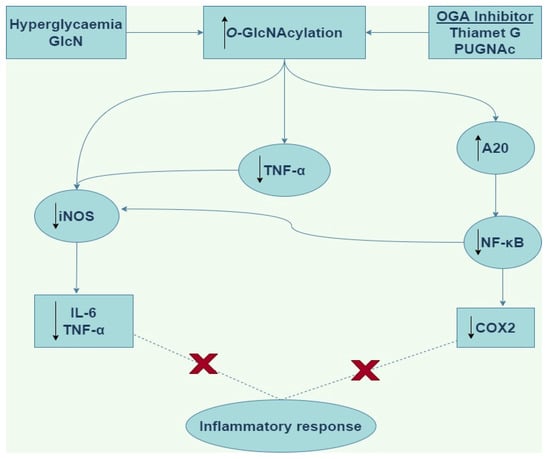
Figure 4.
Schematic of the anti-inflammatory downstream signaling by O-GlcNAcylation. GlcN (Glucosamine), OGA (O-linked β-N-acetylglucosaminidase), PUGNAc (O-(2-acetamido-2-deoxy-d-glucopyranosyliden)amino-N-phenylcarbamate), iNOS (inducible nitric oxide synthase), IL-6 (interleukin-6), TNF-α (tumor necrosis factor alpha), NF-κB (nuclear factor kappa light chain enhancer of activated B cells), COX2 (cyclooxygenase-2).
Furthermore, Hwang et al. [68] proposed that GlcN could be a novel neuroprotective or anti-inflammatory agent. In this study [68], administration of GlcN was found to reduce infarct volume and ameliorates motor impairment and neurological deficits in a rat middle cerebral artery occlusion model of ischaemic stroke. Also, GlcN suppressed LPS-induced upregulation of pro-inflammatory mediators in BV2 microglial cells and RAW264.7 macrophages [68]. GlcN is a substrate for GFAT, the rate-limiting enzyme in the HBP (Figure 1). Increase in the influx of glucose and glutamine would result in increased UDP-GlcNAc levels and upregulation of O-GlcNAcylation. However, in this case, Hwang et al. [68] suggest that GlcN inhibits the O-GlcNAcylation of NF-κB, probably by disturbing the association between OGT and NF-κB.
In addition, Zou et al. [69] showed that increased O-GlcNAcylation by PUGNAc administration following induction of trauma-haemorrhage (TH) in fasted rats improves recovery of organ perfusion and function. In this study [69], they demonstrated that PUGNAc attenuated the TH-induced increase in plasma IL-6 levels, and suggested that PUGNAc, like GlcN, improves cardiac function and organ perfusion by reducing the level of circulating IL-6 and TNF-α after TH. The same researchers have also shown that treatment of fasted rats with GlcN attenuated TH-induced increases in NF-κB activation and reduced cardiac levels of TNF-α and IL-6 mRNAs, ICAM-1 protein and myeloperoxidase activity (Figure 4) [70]. These results demonstrate that the modulation of O-GlcNAc levels regulates activation of the NF-κB pathway in the heart, which may contribute to the observed GlcN-mediated improvement in cardiac function following haemorrhagic shock.
Also, sepsis leading to multiple organ damage is mainly caused by an uncontrolled systemic inflammatory response. However, the mechanisms responsible for this are unclear and our understanding of its pathogenesis remains incompletely understood [71,72]. As it shares common major blood lineages [73] and strikingly similar immune system architecture with humans, the zebrafish has been proposed as a tractable model to investigate the underlying pathophysiology of infectious diseases as well as immune and inflammatory disorders [73,74,75]. In addition, Hwang et al. [76] have demonstrated that pre-treatment of mice with GlcN upregulated protein O-GlcNAcylation and improved survival in the caecal ligation and puncture (CLP)-induced sepsis, and attenuated LPS-induced septic lung injury and systemic inflammation. In this study [76], they demonstrated that LPS reduced O-GlcNAcylation of nucleocytoplasmic proteins in liver, lung, and spleen. Also, LPS-induced downregulation O-GlcNAcylation in mouse lung was inhibited by GlcN, and the OGA level increased by LPS was suppressed by GlcN [76].
Another study has provided evidence that GlcN stimulates the O-GlcNAcylation of both nuclear and cytosolic forms of c-Rel [77]. Furthermore, this study demonstrated that upon stimulation with LPS, GlcN inhibits the binding of c-Rel to the NF-κB site in the iNOS promoter [77]. Although the mechanism through which GlcN exacts these effects is not fully understood, however, it is suggested that GlcN is impairs the interaction between OGT and c-Rel [77]. It has been observed in N9 microglial cells treated with LPS, the enhanced expression of iNOS, NO and ROS is mediated via the downregulation of OGT and protein O-GlcNAcylation, or via the upregulation of MAPKs phosphorylation and NF-κB translocation [78]. Also, overexpression of OGT inhibits LPS-induced activation of NF-κB and iNOS through modulation of histone acetylation either directly or indirectly [79]. Furthermore, Thiamet G improved clinical outcomes and ameliorated the neurological deficits when used to treat mice either before or after middle cerebral artery occlusion (MCAO) [80]. Additionally, the number of Iba1+ cells in MCAO mice were reduced when treated with Thiamet G [80]. Also, expression of MI markers was decreased by Thiamet G, conversely, the expression of the M2 markers increased in vivo [80]. Also, Thiamet G decreased the expression of iNOS and COX2 mainly by suppressing NF-κB p65 signaling (Figure 4) [80]. These results suggest that Thiamet G exerts a neuroprotective effect which suggest its usefulness as a potential anti-inflammatory agent for ischemic stroke therapy.
Furthermore, the deubiquitinase A20 has been identified as suppressor of NF-κB pathway activation [81]. Our understanding of the downstream regulation of the activities of A20 is still unclear. However, it has been shown that GlcN and Thiamet G which significantly increased O-GlcNAc modification of A20 enhanced its binding to TAB1, a key regulator of A20 activity [81]. These studies [81,82] suggest that O-GlcNAcylation is a critical regulatory modulator of the activities of A20, which consequently downregulates NF-κB signaling, including in vascular smooth muscle cells. Furthermore, it has been shown that the activities of Glutathione peroxidase 1 (GPX1) is induced by hyperglycaemia [83]. GPX1 is an anti-oxidant enzyme that is critical for cell survival. Hyperglycaemia enhances the O-GlcNAc modification of GPX1 and subsequently increases the association between non-receptor tyrosine kinase c-Abl and Arg in rat vascular smooth muscle cells [83]. Also, 1,2-dideoxy-2′-propyl-alpha-d-glucopyranoso-[2,1-d]-Delta2′-thiazoline (an OGA inhibitor) was shown to cause the activation of GPX1 in the mouse liver [83]. Furthermore, Hwang et al. [84] after stimulating with LPS, discovered that GlcN relieves the basal transcription activity of RNA polymerase II (RNAPII), a multiprotein complex that catalyzes the transcription of all protein-coding genes and several non-coding RNAs. They suggest that this observation was due to increased O-GlcNAcylation of RNAPII and DNA binding upon treatment with GlcN, which are inhibited by LPS [84].
3.2. Intrinsic Role of OGT and OGA in Inflammatory Pathways
Findings showing the intrinsic involvement of the key modulating enzymes of O-GlcNAcylation in inflammation are beginning to emerge [46,85,86,87]. Mutual and balanced interaction between OGT and OGA is critical for normal O-GlcNAc homeostasis. However, sustained alteration in form of enhancement or inhibition of these key enzymes initiates the development of O-GlcNAcylation mediated pathologies with inflammatory components. Recent findings have demonstrated that OGT promotes inflammatory responses in macrophages [46,49,85,86,88]. NF-κB which is a critical regulator of pro-inflammatory responses in macrophages has been shown to be O-GlcNAc modified [49]. Allison et al. [49] demonstrated that OGT could co-localise to NF-κB–regulated promoters causing the modification of Thr305 residue of the NF-κB subunit RelA. This was shown to promote acetylation on K310 of RelA, and further enhanced NF-κB transcriptional activity after stimulation of tumour necrosis factor (TNF) [49]. Allison et al. [49] further showed that a knockdown of OGT abolishes p300-mediated acetylation of RelA on Lys310, a posttranslational mark required for full NF-κB transcription. Similarly, Ramakrishnan et al. [85] identified Ser350 as the site of modification and activation of NF-κB subunit c-Rel. They also showed that preventing the OGT mediated O-GlcNAc modification of this residue repealed c-Rel–mediated expression of the cytokine-encoding genes IL2, IFNG, and CSF2 in response to TCR activation, whereas increasing the extent of O-GlcNAcylation of cellular proteins promoted the expression of these genes [85].
Microglial cells are the brain resident macrophage [89]. In BV2 microglia cells, c-Rel has been shown to interact with OGT upon lipopolysaccharide (LPS) treatment, which promotes c-Rel O-GlcNAcylation and formation of a c-Rel-p50/p105 heterodimeric complex [77]. More so, this study [88] demonstrated that the S-nitrosylation of OGT inhibits its catalytic activity in resting RAW 264.7 murine macrophage cells [88]. However, stimulation of OGT with LPS results in de-nitrosylation of OGT which then enhanced OGT-mediated O-GlcNAc modification of p65/RelA [88]. Furthermore, attenuation of O-GlcNAcylation negatively modulates p65 nuclear translocation, resulting in diminished production of nitric oxide (NO) and IL-1β [88]. Also, the role of STAT3, a critical transcription factor in promoting inflammation and tissue repair has been well enumerated [90,91]. More so, STAT3 can induce IL-10 production in macrophages to suppress inflammation [92]. It has been previously demonstrated that OGT modulated O-GlcNAc modification of STAT3 at transactivation domain Thr717 competitively downregulates STAT3 phosphorylation which results in reduced production of IL-10 [46]. Furthermore, in macrophages derived from the bone-marrow, OGT expression can be transcriptionally downregulated by myeloid-derived cullin 3 (CUL3), a process dependent on nuclear factor-2 (Nrf2) which binds to the Ogt promoter to increase gene transcription [46,93,94]. Therefore, CUL3 counteracts STAT3 O-GlcNAcylation to elevate STAT3 phosphorylation and inhibit inflammation [46].
In contrast, many studies have demonstrated that increased O-GlcNAcylation correlates with suppression of inflammation in sepsis and ischemia [76,95,96,97,98,99]. Upon GlcN treatment, which bypasses the GFAT rate-limiting step to induce hyper-O-GlcNAcylation [95], the transcriptional activity of c-Rel is inhibited, thereby reducing NF-κB-mediated expression of inducible nitric oxide synthase (iNOS) in LPS stimulated BV2 microglial cells [68,77]. Furthermore, it has recently been shown that receptor-interacting serine/threonine-protein kinase 3 (RIPK3) can be O-GlcNAc modified [96]. RIPK3 is a member of the necrosome complex, a complex consisting of RIP1, RIP3, and Fas-associated protein with death domain causing rapid plasma membrane rupture and induction of an inflammatory response. This is done through the release of damage-associated molecular patterns and cytokines [97,98]. Also, RIPK3 reduces inflammation by diminishing the production of pro-inflammatory cytokines resulting from necroptosis of macrophages [100]. Interestingly, O-GlcNAc modification of RIPK3 at Thr467 is believed to suppress RIPK3-RIPK1 and RIPK3-RIPK3 complex formation, therefore preventing necroptosis of macrophages [96]. Ogtf/f Lyz2-cre conditional knockout mice, in which Ogt is specifically deleted in macrophages, revealed significantly increased activation of RIPK3, elevated production of inflammatory cytokine, and more severe mortality in experimental sepsis when compared with control mice [96]. Furthermore, Yang et al. [87] have shown that OGT prevents macrophage mediated inflammation and metabolic disturbance by suppressing the phosphorylation of S6 kinase beta-1 (S6K1). Phosphorylation of S6K1, a ribosomal protein is critical for macrophage proinflammatory activation [87]. More so, OGT mediated O-GlcNAcylation is downregulated during macrophage proinflammatory activation, and knockout of OGT in high fat-diet induced obese mice enhances macrophage proinflammatory polarization and promotes adipose tissue inflammation [87].
On the other hand, acute upregulation of O-GlcNAcylation by inhibiting OGA have been shown to prevent inflammation-induced vascular dysfunction, hence, confer a protection on the heart and vasculature [48]. Hilgers et al. [48] demonstrated that acute increase in O-GlcNAcylation by inhibiting OGA prevented TNF-α-induced vascular dysfunction. Furthermore, O-GlcNAcylation can possibly modulate the polarisation of M2 macrophages in a pattern that contribute to resolution of inflammation and tissue repair [76,80,99]. This study [76] demonstrated that treatment with GlcN reduced M1 macrophage-specific gene expression profiles in macrophages in an LPS-induced septic lung injury animal model. More so, when Thiamet-G, a selective OGA inhibitor [80] was administered to a mouse model of middle cerebral artery occlusion, there was an increase in the expression M2-specific markers in microglia [80]. Also, there was suppression of NF-κB p65 signaling which resulted in diminished expression of iNOS and cyclooxygenase-2 (COX-2) expression [80]. This strongly suggests that alteration O-GlcNAc homeostasis can vary the differentiation of M2 versus M1 macrophages in tissues.
4. Targeting O-GlcNAcylation in Inflammatory Derangements: Perspectives
O-GlcNAcylation is currently an unexplored therapeutic target in the management of pathologies with inflammatory derangements; this may be due to the fact that the involvement of this dynamic PTM is just beginning to evolve. Also, our current understanding of the involvement of O-GlcNAcylation in inflammatory pathways is unclear. More so, that O-GlcNAcylation seem to be a dual-directional regulator of inflammatory system. Therefore, further characterisation would help improve our current knowledge which could pave way for the development of new therapeutics that can find their way into mainstay clinical applications. Over 5000 proteins have now been identified as targets for O-GlcNAcylation and this will continue to increase due to technological advances in the mapping of protein O-GlcNAcylation sites [22,100,101]. As we have described in detail, the role of O-GlcNAcylation in the activation of pro-inflammatory and immunomodulating pathways has gained more attention. For example, findings have shown that O-GlcNAcylation activates NF-κB and NFAT pathway [39,50,51,52,102]. Activation of NF-κB initiates the transcription of relevant downstream target genes with pro-inflammatory tendencies and on the other hand, activation of the NFAT pathway has been linked with modification of proteins in human bronchial epithelial cells by fibroblast growth factor 23 resulting in upregulation of IL-6 and regulation of airway inflammation [103].
Furthermore, recurring and/ prolonged inflammation, a common and characteristic feature of a variety of disorders such as autoimmune disease, diabetes, and cancer have been attributed to aberrant regulation of NF-κB activity [104,105,106]. Therefore, a clearer understanding of the mechanisms that underpin the involvement of NF-κB in the inflammatory process may have great biological and clinical significance. Increased cellular O-GlcNAc level which is typical in diabetes and insulin resistance is usually accompanied by NF-κB activation, and treatments that upregulate O-GlcNAcylation appear to have anti-inflammatory and pro-survival effects during acute injuries like myocardial infarction, burns, trauma and sepsis [69,70,107,108]. For instance, GlcN and PUGNAc improved organ perfusion and function after trauma-hemorrhage in fasted male rats [69]. Also, Xing et al. [109] showed that treatment of rat aortic smooth muscle cells with GlcN and PUGNAc prevented TNF-α induced inflammatory stress by upregulation of O-GlcNAc modification of p65 on Ser536. Xing et al. [109] further suggested that GlcN and PUGNAc inhibited TNF-α induced phosphorylation of NF-κB p65, thus inhibiting NF-κB signaling in rat aortic smooth muscle cells. In vivo, dextran sodium sulfate-induced phosphorylation of NF-κB p65 and IL-1β mRNA expression are significantly lower in Ogt-transgenic when compared wild type mice. This suggests that acute colitis could be prevented by upregulating O-GlcNAcylation which result in diminished acute inflammation [110].
Also, alteration in the metabolic pathways of effector T cells is core to the progress of insulin resistance and atherosclerosis and this may lead to enhanced supply of metabolites to the HBP, thereby promoting O-GlcNAc modification. More so, effector T cells such as Th1 and Th17 cells are crucial for several autoimmune diseases, including inflammatory bowel disease, rheumatoid arthritis, multiple sclerosis (MS), and systemic lupus erythematosus (SLE) [111]. It has been suggested that the progression of SLE is enhanced by the reactivation of the silenced X-chromosome due to CD4+ T cell DNA demethylation and diet [112]. Hence, it is proposed that the magnitude of overexpression of OGT in CD4+ T cells could be a critical factor that contributes to the progression of SLE in women [112]. Additionally, it has been demonstrated that miR-15b suppressed the differentiation of Th17 cells which are likely the most critical pathogenic factor of human MS [113]. It is suggested that this results in suppression of the pathogenesis of MS by decreasing the expression of OGT in an NF-κB p65- and c-Rel-dependent manner [113]. Taken together, these observations should lead to the investigation of the links between protein O-GlcNAcylation and the activation of T cells in metabolic and autoimmune diseases. We believe that further characterisation of the role of OGT and O-GlcNAc modification in autoimmunity may yield new therapeutic targets for autoimmune diseases with inflammatory components.
Furthermore, the involvement of O-GlcNAcylation in pro- versus anti-inflammatory processes is dependent on the distinct GlcN stimulus which regulates inflammation by sensing both a healthy nutritional status and overnutrition. At normoglycaemic concentrations, GlcN enhances LPS-triggered inflammation in macrophages in a dose-dependent manner, conversely, in high glucose cell culture the inflammation of macrophages was suppressed [114]. Also, it was observed that LPS-stimulated induction of iNOS as well as an increase in DNA binding of c-Rel to the iNOS promoter in macrophage cells is increased by GlcN in normoglycaemic conditions and decreased in high glucose conditions [114]. In addition, there is a high risk in pregnancy due to hyperglycaemia which could have negative effects on the fetus. Considering that O-GlcNAcylation is a nutritionally responsive modification; hence, excess flux of glucose may alter the O-GlcNAc homeostasis in the intrauterine environment resulting in metabolic deregulation of the offspring [115]. Furthermore, inhibition of TNF-α and IL-8 gene expression in osteoarthritis is mediated in part by GlcN-induced O-GlcNAcylation [116]. Therefore, these findings suggest that depending on the cell state, the nature of insult, and the cellular nutrition state, HBP may swiftly adapt the management pattern to regulate inflammation, resulting in either pro- or anti-inflammatory outcomes [114]. Thus, proper modulation of O-GlcNAc homeostasis presents a viable therapeutic strategy for combating inflammatory diseases and metabolic dysregulation such as sepsis, diabetes, and osteoarthritis.
Pharmacological modulation of the critical enzymes (OGT and OGA) that regulate this dynamic PTM might be a critical lead way in treating inflammatory derangements. Drugs like alloxan [117,118], benzoxazolinones [119], BADGP (Benzyl-2-acetymido-2-deoxy-α-d-galactopyranoside) [120,121,122] are used as OGT inhibitors but due to off-target effects and toxicities, their role over the years has been limited to experimental and in vitro studies [122,123]. However, more recently developed OGT inhibitors such as Ac-5SGlcNAc [124,125], OSMI 1 [126], OSMI 2-4 [127], and L01 [128] have not only helped improve the understanding and characterisation of this dynamic PTM but are more promising as potential pharmacological agents as they have shown improved specificity and encouraging pharmacodynamic profiles [122]. On the other hand, development of therapeutic agents used clinically to increase O-GlcNAcylation level has been more successful [122]. These agents include OGA inhibitors such as Thiamet-G (5H-Pyrano[3,2-d]thiazole-6,7-diol2-(ethylamino)-3a,6,7,7a-tetrahydro-5-(hydroxymethyl)-(3aR,5R,6S,7R,7aR)), PUGNAc (O-(2-acetamido-2-deoxy-d-glucopyranosyliden)amino-N-phenylcarbamate), NButGT (1,2-dideoxy-2′-propyl-α-d-glucopyranoso-[2,1-d]-Δ2′-thiazoline), and more recently developed GlcNAcstatin [122,129,130]. Also, glucosamine, which increases the UDP-GlcNAc concentrations by bypassing GFAT [95,122].
However, considering that OGA and OGT are ubiquitously expressed, modulation of their expression with drugs could come with numerous adverse drug reactions. Also, considering that the use of OGA and OGT global knockout mice is not viable as this usually result in high mortality [131,132,133,134]. Therefore, cell-specific knock out of these key enzymes to fully validate the role of O-GlcNAc modification in inflammatory responses in key cell types in animal models of disease would inform studies in human. Also, assessment of the expression of OGT/OGA change in target cell types in disease and determining if targeting OGA/OGT is limited to catalytic inhibitors targeted at their active sites could be characteristic. In view of this, we believe it is viable to target OGT/OGA-target protein interaction with protein-protein interaction inhibitors [135,136]. Having identified the sites of interaction, it is possible to design and optimize a blocking peptide and use this as a basis to screen for drug-like compounds that function as peptidomimetics [135,136].
5. Conclusions
Findings highlighted in this review have shown that the involvement of O-GlcNAcylation in the modulation of inflammatory pathways is complex, wide-ranging and substantial, yet it is not a populartarget for drug development in the management of inflammatory pathologies. Therefore, we propose that targeting O-GlcNAcylation is a viable therapeutic target for future management of pathologies characterised by dysregulation of inflammatory pathways.
Author Contributions
Paper was written by I.O.B., and it was revised and edited by T.M.P. All authors have read and agreed to the published version of the manuscript.
Funding
Work in TMP’s laboratory is supported by the Hull and East Riding Cardiac Trust Fund. IOB is supported by scholarships from the Tertiary Education Trust Fund (TETFund) and University of Benin, Nigeria, and Hull York Medical School, University of Hull, UK.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Hameed, I.; Masoodi, S.R.; Mir, S.A.; Nabi, M.; Ghazanfar, K.; Ganai, B.A. Type 2 diabetes mellitus: From a metabolic disorder to an inflammatory condition. World J. Diabetes 2015, 6, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C.; Crook, M.A. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 1998, 41, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef]
- Chang, Y.H.; Weng, C.L.; Lin, K.I. O-GlcNAcylation and its role in the immune system. J. Biomed. Sci. 2020, 27, 57. [Google Scholar] [CrossRef]
- Torres, C.R.; Hart, G.W. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [CrossRef]
- Haltiwanger, R.S.; Holt, G.D.; Hart, G.W. Enzymatic addition of O-GlcNAc to nuclear and cytoplasmic proteins. Identification of a uridine diphospho-N-acetylglucosamine:peptide beta-N-acetylglucosaminyltransferase. J. Biol. Chem. 1990, 265, 2563–2568. [Google Scholar] [CrossRef]
- Dong, D.L.; Hart, G.W. Purification and characterization of an O-GlcNAc selective N-acetyl-beta-d-glucosaminidase from rat spleen cytosol. J. Biol. Chem. 1994, 269, 19321–19330. [Google Scholar] [CrossRef]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef]
- Lazarus, B.D.; Love, D.C.; Hanover, J.A. Recombinant O-heyGlcNAc transferase isoforms: Identification of O-GlcNAcase, yes tyrosine kinase, and tau as isoform-specific substrates. Glycobiology 2006, 16, 415–421. [Google Scholar] [CrossRef]
- Wellen, K.E.; Lu, C.; Mancuso, A.; Lemons, J.M.; Ryczko, M.; Dennis, J.W.; Rabinowitz, J.D.; Coller, H.A.; Thompson, C.B. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010, 24, 2784–2799. [Google Scholar] [CrossRef]
- Swamy, M.; Pathak, S.; Grzes, K.M.; Damerow, S.; Sinclair, L.V.; van Aalten, D.M.; Cantrell, D.A. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat. Immunol. 2016, 17, 712–720. [Google Scholar] [CrossRef]
- Taylor, R.P.; Geisler, T.S.; Chambers, J.H.; McClain, D.A. Up-regulation of O-GlcNAc transferase with glucose deprivation in HepG2 cells is mediated by decreased hexosamine pathway flux. J. Biol. Chem. 2009, 284, 3425–3432. [Google Scholar] [CrossRef]
- Weigert, C.; Klopfer, K.; Kausch, C.; Brodbeck, K.; Stumvoll, M.; Haring, H.U.; Schleicher, E.D. Palmitate-induced activation of the hexosamine pathway in human myotubes: Increased expression of glutamine:fructose-6-phosphate aminotransferase. Diabetes 2003, 52, 650–656. [Google Scholar] [CrossRef]
- Donath, M.Y.; Dalmas, É.; Sauter, N.S.; Böni-Schnetzler, M. Inflammation in obesity and diabetes: Islet dysfunction and therapeutic opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the fire: Emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019, 17, 52. [Google Scholar] [CrossRef]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991, 266, 4706–4712. [Google Scholar] [CrossRef]
- Olson, A.K.; Bouchard, B.; Zhu, W.Z.; Chatham, J.C.; Des Rosiers, C. First characterization of glucose flux through the hexosamine biosynthesis pathway (HBP) in ex vivo mouse heart. J. Biol. Chem. 2020, 295, 2018–2033. [Google Scholar] [CrossRef]
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [Google Scholar] [CrossRef]
- Holt, G.D.; Hart, G.W. The subcellular distribution of terminal N-acetylglucosamine moieties. Localization of a novel protein-saccharide linkage, O-linked GlcNAc. J. Biol. Chem. 1986, 261, 8049–8057. [Google Scholar] [CrossRef]
- Wulff-Fuentes, E.; Berendt, R.R.; Massman, L.; Danner, L.; Malard, F.; Vora, J.; Kahsay, R.; Stichelen, O.V. The human O-GlcNAcome database and meta-analysis. Sci. Data 2021, 8, 25. [Google Scholar] [CrossRef]
- Holt, G.D.; Snow, C.M.; Senior, A.; Haltiwanger, R.S.; Gerace, L.; Hart, G.W. Nuclear pore complex glycoproteins contain cytoplasmically disposed O-linked N-acetylglucosamine. J. Cell Biol. 1987, 104, 1157–1164. [Google Scholar] [CrossRef]
- Kelly, W.G.; Hart, G.W. Glycosylation of chromosomal proteins: Localization of O-linked N-acetylglucosamine in Drosophila chromatin. Cell 1989, 57, 243–251. [Google Scholar] [CrossRef]
- Tarbet, H.J.; Dolat, L.; Smith, T.J.; Condon, B.M.; O’Brien, E.T., 3rd; Valdivia, R.H.; Boyce, M. Site-specific glycosylation regulates the form and function of the intermediate filament cytoskeleton. eLife 2018, 7, e31807. [Google Scholar] [CrossRef]
- Cho, H.J.; Mook-Jung, I. O-GlcNAcylation regulates endoplasmic reticulum exit sites through Sec31A modification in conventional secretory pathway. FASEB J. 2018, 32, 4641–4657. [Google Scholar] [CrossRef]
- Bolanle, I.O.; Riches-Suman, K.; Loubani, M.; Williamson, R.; Palmer, T.M. Revascularization of type 2 diabetics with coronary artery disease: Insights and therapeutic targeting of O-GlcNAcylation. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 1349–1356. [Google Scholar] [CrossRef]
- Gao, Y.; Wells, L.; Comer, F.I.; Parker, G.J.; Hart, G.W. Dynamic O-glycosylation of nuclear and cytosolic proteins: Cloning and characterization of a neutral, cytosolic β-N-acetylglucosaminidase from human brain. J. Biol. Chem. 2001, 276, 9838–9845. [Google Scholar] [CrossRef]
- Schimpl, M.; Schuttelkopf, A.W.; Borodkin, V.S.; van Aalten, D.M.F. Human OGA binds substrates in a conserved peptide recognition groove. Biochem. J. 2010, 432, 1–7. [Google Scholar] [CrossRef]
- Li, B.B.; Li, H.; Lu, L.; Jiang, J.Y. Structures of human O-GlcNAcase and its complexes reveal a new substrate recognition mode. Nat. Struct. Mol. Biol. 2017, 24, 362. [Google Scholar] [CrossRef]
- Li, B.B.; Li, H.; Hu, C.W.; Jiang, J.Y. Structural insights into the substrate binding adaptability and specificity of human O-GlcNAcase. Nat. Commun. 2017, 8, 666. [Google Scholar] [CrossRef]
- Roth, C.; Chan, S.; Offen, W.A.; Hemsworth, G.R.; Willems, L.I.; King, D.T.; Varghese, V.; Britton, R.; Vocadlo, D.J.; Davies, G.J. Structural and functional insight into human O-GlcNAcase. Nat. Chem. Biol. 2017, 13, 610–612. [Google Scholar] [CrossRef]
- Elsen, N.L.; Patel, S.B.; Ford, R.E.; Hall, D.L.; Hess, F.; Kandula, H.; Kornienko, M.; Reid, J.; Selnick, H.; Shipman, J.M.; et al. Insights into activity and inhibition from the crystal structure of human O-GlcNAcase. Nat. Chem. Biol. 2017, 13, 613–615. [Google Scholar] [CrossRef]
- Tseng, C.H. The Relationship between Diabetes Mellitus and Gastric Cancer and the Potential Benefits of Metformin: An Extensive Review of the Literature. Biomolecules 2021, 11, 1022. [Google Scholar] [CrossRef]
- Tseng, C.H.; Tseng, F.H. Diabetes and gastric cancer: The potential links. World J. Gastroenterol. 2014, 20, 1701–1711. [Google Scholar] [CrossRef]
- Subauste, A.; Gianani, R.; Chang, A.M.; Plunkett, C.; Pietropaolo, S.L.; Zhang, Y.J.; Barinas-Mitchell, E.; Kuller, L.H.; Galecki, A.; Halter, J.B.; et al. Islet autoimmunity identifies a unique pattern of impaired pancreatic beta-cell function, markedly reduced pancreatic beta cell mass and insulin resistance in clinically diagnosed type 2 diabetes. PLoS ONE 2014, 9, e106537. [Google Scholar] [CrossRef][Green Version]
- Liu, J.; Ting, J.P.; Al-Azzam, S.; Ding, Y.; Afshar, S. Therapeutic Advances in Diabetes, Autoimmune, and Neurological Diseases. Int. J. Mol. Sci. 2021, 22, 2805. [Google Scholar] [CrossRef]
- Schedin-Weiss, S.; Winblad, B.; Tjernberg, L.O. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014, 281, 46–62. [Google Scholar] [CrossRef]
- Li, Y.; Xie, M.; Men, L.; Du, J. O-GlcNAcylation in immunity and inflammation: An intricate system (Review). Int. J. Mol. Sci. 2019, 44, 363–374. [Google Scholar] [CrossRef]
- Machacek, M.; Slawson, C.; Fields, P.E. O-GlcNAc: A novel regulator of immunometabolism. J. Bioenerg. Biomembr. 2018, 50, 223–229. [Google Scholar] [CrossRef]
- Woo, C.M.; Lund, P.J.; Huang, A.C.; Davis, M.M.; Bertozzi, C.R.; Pitteri, S.J. Mapping and quantification of over 2000 O-linked glycopeptides in activated human T cells with isotope-targeted glycoproteomics (IsoTaG). Mol. Cell. Proteom. 2018, 17, 764–775. [Google Scholar] [CrossRef]
- Hou, C.W.; Mohanan, V.; Zachara, N.E.; Grimes, C.L. Identification and biological consequences of the O-GlcNAc modification of the human innate immune receptor, Nod2. Glycobiology 2016, 26, 13–18. [Google Scholar] [CrossRef]
- Zhang, D.; Cai, Y.; Chen, M.; Gao, L.; Shen, Y.; Huang, Z. OGT-mediated O-GlcNAcylation promotes NF-κB activation and inflammation in acute pancreatitis. Inflamm. Res. 2015, 64, 943–952. [Google Scholar] [CrossRef]
- Li, Y.; Liu, H.; Xu, Q.S.; Du, Y.G.; Xu, J. Chitosan oligosaccharides block LPS-induced O-GlcNAcylation of NF-κB and endothelial inflammatory response. Carbohydr. Polym. 2014, 99, 568–578. [Google Scholar] [CrossRef]
- James, L.R.; Tang, D.; Ingram, A.; Ly, H.; Thai, K.; Cai, L.; Scholey, J.W. Flux through the hexosamine pathway is a determinant of nuclear factor κB- dependent promoter activation. Diabetes 2002, 51, 1146–1156. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Z.; Li, L.; Gong, W.; Lazenby, A.J.; Swanson, B.J.; Herring, L.E.; Asara, J.M.; Singer, J.D.; Wen, H. Myeloid-derived cullin 3 promotes STAT3 phosphorylation by inhibiting OGT expression and protects against intestinal inflammation. J. Exp. Med. 2017, 214, 1093–1109. [Google Scholar] [CrossRef]
- Xing, D.; Feng, W.; Not, L.G.; Miller, A.P.; Zhang, Y.; Chen, Y.F.; Majid-Hassan, E.; Chatham, J.C.; Oparil, S. Increased protein O-GlcNAc modification inhibits inflammatory and neointimal responses to acute endoluminal arterial injury. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H335–H342. [Google Scholar] [CrossRef]
- Hilgers, R.H.; Xing, D.; Gong, K.; Chen, Y.F.; Chatham, J.C.; Oparil, S. Acute O-GlcNAcylation prevents inflammation-induced vascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H513–H522. [Google Scholar] [CrossRef]
- Allison, D.F.; Wamsley, J.J.; Kumar, M.; Li, D.; Gray, L.G.; Hart, G.W.; Jones, D.R.; Mayo, M.W. Modification of RelA by O-linked N-acetylglucosamine links glucose metabolism to NF-κB acetylation and transcription. Proc. Natl. Acad. Sci. USA 2012, 109, 16888–16893. [Google Scholar] [CrossRef]
- Yang, W.H.; Park, S.Y.; Nam, H.W.; Kim, D.H.; Kang, J.G.; Kang, E.S.; Kim, Y.S.; Lee, H.C.; Kim, K.S.; Cho, J.W. NFkappaB activation is associated with its O-GlcNAcylation state under hyperglycemic conditions. Proc. Natl. Acad. Sci. USA 2008, 105, 17345–17350. [Google Scholar] [CrossRef]
- Ma, Z.; Chalkley, R.J.; Vosseller, K. Hyper-O-GlcNAcylation activates nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) signaling through interplay with phosphorylation and acetylation. J. Biol. Chem. 2017, 292, 9150–9163. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-N-acetyl glucosamine modification. Proc. Natl. Acad. Sci. USA 2009, 106, 3431–3436. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Pantarelli, C.; Welch, H.C.E. Rac-GTPases and Rac-GEFs in neutrophil adhesion, migration and recruitment. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), e12939. [Google Scholar] [CrossRef]
- Donovan, K.; Alekseev, O.; Qi, X.; Cho, W.; Azizkhan-Clifford, J. O-GlcNAc modification of transcription factor Sp1 mediates hyperglycemia-induced VEGF-A upregulation in retinal cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7862–7873. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, Y.; Niu, T.; Wang, H.; Liu, K. O-GlcNAc modification of Sp1 mediates hyperglycaemia-induced ICAM-1 up-regulation in endothelial cells. Biochem. Biophys. Res. Commun. 2017, 484, 79–84. [Google Scholar] [CrossRef]
- Lim, K.; Chang, H.I. O-GlcNAc inhibits interaction between Sp1 and Elf-1 transcription factors. Biochem. Biophys. Res. Commun. 2009, 380, 569–574. [Google Scholar] [CrossRef]
- Lim, K.; Chang, H.I. O-GlcNAcylation of Sp1 interrupts Sp1 interaction with NF-Y. Biochem. Biophys. Res. Commun. 2009, 382, 593–597. [Google Scholar] [CrossRef]
- Jokela, T.A.; Makkonen, K.M.; Oikari, S.; Kärnä, R.; Koli, E.; Hart, G.W.; Tammi, R.H.; Carlberg, C.; Tammi, M.I. Cellular content of UDP-N-acetylhexosamines controls hyaluronan synthase 2 expression and correlates with O-linked N-acetylglucosamine modification of transcription factors YY1 and SP1. J. Biol. Chem. 2011, 286, 33632–33640. [Google Scholar] [CrossRef]
- Marei, W.F.; Ghafari, F.; Fouladi-Nashta, A.A. Role of hyaluronic acid in maturation and further early embryo development of bovine oocytes. Theriogenology 2012, 78, 670–677. [Google Scholar] [CrossRef]
- Lim, K.; Chang, H.I. O-GlcNAc inhibits interaction between Sp1 and sterol regulatory element binding protein 2. Biochem. Biophys. Res. Commun. 2010, 393, 314–318. [Google Scholar] [CrossRef]
- Suh, H.N.; Lee, Y.J.; Kim, M.O.; Ryu, J.M.; Han, H.J. Glucosamine- induced Sp1 O-GlcNAcylation ameliorates hypoxia-induced SGLT dysfunction in primary cultured renal proximal tubule cells. J. Cell. Physiol. 2014, 229, 1557–1568. [Google Scholar] [CrossRef]
- Lee, H.J.; Ryu, J.M.; Jung, Y.H.; Lee, K.H.; Kim, D.I.; Han, H.J. Glycerol-3-phosphate acyltransferase-1 upregulation by O-GlcNAcylation of Sp1 protects against hypoxia-induced mouse embryonic stem cell apoptosis via mTOR activation. Cell Death Dis. 2016, 7, e2158. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- Pathak, S.; Borodkin, V.S.; Albarbarawi, O.; Campbell, D.G.; Ibrahim, A.; van Aalten, D.M. O-GlcNAcylation of TAB1 modulates TAK1-mediated cytokine release. EMBO J. 2012, 31, 1394–1404. [Google Scholar] [CrossRef]
- Hirata, Y.; Takahashi, M.; Morishita, T.; Noguchi, T.; Matsuzawa, A. Post-translational modifications of the TAK1-TAB complex. Int. J. Mol. Sci. 2017, 18, 205. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, Z.; Tao, T.; Liu, X.; Sun, X.; Ji, Y.; Han, L.; Qiu, H.; Zhu, G.; Shen, Y.; et al. Modification of TAK1 by O-linked N-acetylglucosamine facilitates TAK1 activation and promotes M1 macrophage polarization. Cell. Signal. 2016, 28, 1742–1752. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Shin, J.H.; Hwang, J.S.; Kim, S.Y.; Shin, J.A.; Oh, E.S.; Oh, S.; Kim, J.B.; Lee, J.K.; Han, I.O. Glucosamine exerts a neuroprotective effect via suppression of inflammation in rat brain ischemia/reperfusion injury. Glia 2010, 58, 1881–1892. [Google Scholar] [CrossRef]
- Zou, L.; Yang, S.; Hu, S.; Chaudry, I.H.; Marchase, R.B.; Chatham, J.C. The protective effects of PUGNAc on cardiac function after trauma-hemorrhage are mediated via increased protein O-GlcNAc levels. Shock 2007, 27, 402–408. [Google Scholar] [CrossRef]
- Zou, L.; Yang, S.; Champattanachai, V.; Hu, S.; Chaudry, I.H.; Marchase, R.B.; Chatham, J.C. Glucosamine improves cardiac function following trauma-hemorrhage by increased protein O-GlcNAcylation and attenuation of NF-{kappa}B signaling. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H515–H523. [Google Scholar] [CrossRef]
- Fleischmann, C.; Scherag, A.; Adhikari, N.K.; Hartog, C.S.; Tsaganos, T.; Schlattmann, P.; Angus, D.C.; Reinhart, K.; International Forum of Acute Care Trialists. Assessment of global incidence and mortality of hospital-treated sepsis: Current estimates and limitations. Am. J. Respir. Crit. Care Med. 2016, 193, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Sunyer, J.O. Fishing for mammalian paradigms in the teleost immune system. Nat. Immunol. 2013, 14, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Stachura, D.L.; Svoboda, O.; Campbell, C.A.; Espín-Palazon, R.; Lau, R.P.; Zon, L.I.; Bartunek, P.; Traver, D. The zebrafish granulocyte colony-stimulating factors (Gcsfs): 2 paralogous cytokines and their roles in hematopoietic development and maintenance. Blood 2013, 122, 3918–3928. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Trede, N.S. Fish immunology. Curr. Biol. 2009, 19, R678–R682. [Google Scholar] [CrossRef]
- Trede, N.S.; Langenau, D.M.; Traver, D.; Look, A.T.; Zon, L.I. The use of zebrafish to understand immunity. Immunity 2004, 20, 367–379. [Google Scholar] [CrossRef]
- Hwang, J.S.; Kim, K.H.; Park, J.; Kim, S.M.; Cho, H.; Lee, Y.; Han, I.J. Glucosamine improves survival in a mouse model of sepsis and attenuates sepsis-induced lung injury and inflammation. J. Biol. Chem. 2019, 294, 608–622. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Hwang, J.S.; Kim, S.Y.; Han, I.O. O-GlcNAcylation and p50/p105 binding of c-Rel are dynamically regulated by LPS and glucosamine in BV2 microglia cells. Br. J. Pharmacol. 2013, 169, 1551–1560. [Google Scholar] [CrossRef]
- Zheng, G.M.; Yu, C.; Yang, Z. Puerarin suppresses production of nitric oxide and inducible nitric oxide synthase in lipopolysac-charide-induced N9 microglial cells through regulating MAPK phosphorylation, O-GlcNAcylation and NF-κB translocation. Int. J. Oncol. 2012, 40, 1610–1618. [Google Scholar]
- Hwang, S.Y.; Hwang, J.S.; Kim, S.Y.; Han, I.O. O-GlcNAc transferase inhibits LPS-mediated expression of inducible nitric oxide synthase through an increased interaction with mSin3A in RAW264.7 cells. Am. J. Physiol. Cell Physiol. 2013, 305, C601–C608. [Google Scholar] [CrossRef]
- He, Y.; Ma, X.; Li, D.; Hao, J. Thiamet G mediates neuroprotection in experimental stroke by modulating microglia/macrophage polarization and inhibiting NF-κB p65 signaling. J. Cereb. Blood Flow Metab. 2017, 37, 2938–2951. [Google Scholar] [CrossRef]
- Coornaert, B.; Carpentier, I.; Beyaert, R. A20: Central gatekeeper in inflammation and immunity. J. Biol. Chem. 2009, 284, 8217–8221. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Xu, L.; Xu, O.; Li, R.; Chen, M.; Shen, H.; Zhu, H.; Zhang, F.; Yao, D.; Chen, Y.F.; et al. O-Linked β-N-acetylglucosamine modification of A20 enhances the inhibition of NF-κB (nuclear factor-κB) activation and elicits vascular protection after acute endoluminal arterial injury. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Park, S.Y.; Ji, S.; Kang, J.G.; Kim, J.E.; Song, H.; Mook-Jung, I.; Choe, K.M.; Cho, J.W. O-GlcNAcylation regulates hyperglycemia-induced GPX1 activation. Biochem. Biophys. Res. Commun. 2010, 391, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Hwang, S.Y.; Han, I.O. Basal transcription is regulated by lipopolysaccharide and glucosamine via the regulation of DNA binding of RNA polymerase II in RAW264.7 cells. Life Sci. 2014, 110, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, P.; Clark, P.M.; Mason, D.E.; Peters, E.C.; Hsieh-Wilson, L.C.; Baltimore, D. Activation of the transcriptional function of the NF-κB protein c-Rel by O-GlcNAc glycosylation. Sci. Signal. 2013, 6, ra75. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, X.; Attri, K.S.; Liu, C.; Li, L.; Herring, L.E.; Asara, J.M.; Lei, Y.L.; Singh, P.K.; Gao, C.; et al. O-GlcNAc transferase links glucose metabolism to MAVS-mediated antiviral innate immunity. Cell Host Microbe 2018, 24, 791–803. [Google Scholar] [CrossRef]
- Yang, Y.; Li, X.; Luan, H.H.; Zhang, B.; Zhang, K.; Nam, J.H.; Li, Z.; Fu, M.; Munk, A.; Zhang, D.; et al. OGT suppresses S6K1-mediated macrophage inflammation and metabolic disturbance. Proc. Natl. Acad. Sci. USA 2020, 117, 16616–16625. [Google Scholar] [CrossRef]
- Ryu, I.H.; Do, S.I. Denitrosylation of S-nitrosylated OGT is triggered in LPS-stimulated innate immune response. Biochem. Biophys. Res. Commun. 2011, 408, 52–57. [Google Scholar] [CrossRef]
- Sierra, A.; de Castro, F.; Del Rio-Hortega, J.; Rafael Iglesias-Rozas, J.; Garrosa, M.; Kettenmann, H. The “big-bang” for modern glial biology: Translation and comments on Pio del Rio-Hortega 1919 series of papers on microglia. Glia 2016, 64, 1801–1840. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—Using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Staples, K.J.; Smallie, T.; Williams, L.M.; Foey, A.; Burke, B.; Foxwell, B.M.; Ziegler-Heitbrock, L. IL-10 induces IL-10 in primary human monocyte-derived macrophages via the transcription factor Stat3. J. Immunol. 2007, 178, 4779–4785. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef]
- Marshall, S.; Nadeau, O.; Yamasaki, K. Dynamic actions of glucose and glucosamine on hexosamine biosynthesis in isolated adipocytes: Differential effects on glucosamine 6-phosphate, UDP-N-acetylglucosamine, and ATP levels. J. Biol. Chem. 2004, 279, 35313–35319. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gong, W.; Wang, H.; Li, T.; Attri, K.S.; Lewis, R.E.; Kalil, A.C.; Bhinderwala, F.; Powers, R.; Yin, G.; et al. O-GlcNAc transferase suppresses inflammation and necroptosis by targeting receptor-interacting serine/threonine-protein kinase 3. Immunity 2019, 50, 576–590. [Google Scholar] [CrossRef]
- Newton, K.; Manning, G. Necroptosis and inflammation. Annu. Rev. Biochem. 2016, 85, 743–763. [Google Scholar] [CrossRef]
- Seifert, L.; Miller, G. Molecular Pathways: The Necrosome—A Target for Cancer Therapy. Clin. Cancer Res. 2017, 23, 1132–1136. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef]
- Ma, J.; Hart, G.W. O-GlcNAc profiling: From proteins to proteomes. Clin. Proteom. 2014, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Golks, A.; Tran, T.T.; Goetschy, J.F.; Guerini, D. Requirement for O-linked N-acetylglucosaminyltransferase in lymphocytes activation. EMBO J. 2007, 26, 4368–4379. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Helton, E.S.; Hutcheson, S.B.; Blumhof, S.; Garth, J.M.; Denson, R.S.; Zaharias, R.S.; Wickham, H.; Barnes, J.W. FGF23 Induction of O-Linked N-Acetylglucosamine Regulates IL-6 Secretion in Human Bronchial Epithelial Cells. Front. Endocrinol. 2018, 9, 708. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Gaynor, R.B. IκB kinases: Key regulators of the NF-κB pathway. Trends Biochem. Sci. 2004, 29, 72–79. [Google Scholar] [CrossRef]
- Meirow, Y.; Baniyash, M. Immune biomarkers for chronic inflammation related complications in non-cancerous and cancerous diseases. Cancer Immunol. Immunother. 2017, 66, 1089–1101. [Google Scholar] [CrossRef]
- Holmdahl, R.; Sareila, O.; Olsson, L.M.; Backdahl, L.; Wing, K. Ncf1 polymorphism reveals oxidative regulation of autoimmune chronic inflammation. Immunol. Rev. 2016, 269, 228–247. [Google Scholar] [CrossRef]
- Pietropaolo, M.; Barinas-Mitchell, E.; Kuller, L.H. The heterogeneity of diabetes: Unraveling a dispute: Is systemic inflammation related to islet autoimmunity? Diabetes 2007, 56, 1189–1197. [Google Scholar] [CrossRef]
- Vaidyanathan, K.; Wells, L. Multiple tissue-specific roles for the O-GlcNAc post-translational modification in the induction of and complications arising from type II diabetes. J. Biol. Chem. 2014, 289, 34466–34471. [Google Scholar] [CrossRef]
- Xing, D.; Gong, K.; Feng, W.; Nozell, S.E.; Chen, Y.F.; Chatham, J.C.; Oparil, S. O-GlcNAc modification of NFκB p65 inhibits TNF-α-induced inflammatory mediator expression in rat aortic smooth muscle cells. PLoS ONE 2011, 6, e24021. [Google Scholar] [CrossRef]
- Hirata, Y.; Nakagawa, T.; Moriwaki, K.; Koubayashi, E.; Kakimoto, K.; Takeuchi, T.; Inoue, T.; Higuchi, K.; Asahi, M. Augmented O-GlcNAcylation alleviates inflammation-mediated colon carcinogenesis via suppression of acute inflammation. J. Clin. Biochem. Nutr. 2018, 62, 221–229. [Google Scholar] [CrossRef]
- Carbo, A.; Hontecillas, R.; Kronsteiner, B.; Viladomiu, M.; Pedragosa, M.; Lu, P.; Philipson, C.W.; Hoops, S.; Marathe, M.; Eubank, S.; et al. Systems modeling of molecular mechanisms controlling cytokine-driven CD4+ T cell differentiation and phenotype plasticity. PLoS Comput. Biol. 2013, 9, e1003027. [Google Scholar] [CrossRef] [PubMed]
- Hewagama, A.; Gorelik, G.; Patel, D.; Liyanarachchi, P.; McCune, W.J.; Somers, E.; Gonzalez-Rivera, T.; The Michigan Lupus Cohort; Strickland, F.; Richardson, B. Overexpression of X-linked genes in T cells from women with lupus. J. Autoimmun. 2013, 41, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Ma, X.; Chen, L.; Yang, Y.; Zeng, Y.; Gao, J.; Jiang, W.; Zhang, F.; Li, D.; Han, B.; et al. MicroRNA-15b suppresses Th17 differentiation and is associated with pathogenesis of multiple sclerosis by targeting O-GlcNAc transferase. J. Immunol. 2017, 198, 2626–2639. [Google Scholar] [CrossRef]
- Hwang, J.S.; Kwon, M.Y.; Kim, K.H.; Lee, Y.; Lyoo, I.K.; Kim, J.E.; Oh, E.S.; Han, I.O. Lipopolysaccharide (LPS)-stimulated iNOS induction is increased by glucosamine under normal glucose conditions but is inhibited by glucosamine under high glucose conditions in macrophage cells. J. Biol. Chem. 2017, 292, 1724–1736. [Google Scholar] [CrossRef] [PubMed]
- Olivier-Van Stichelen, S.; Hanover, J.A. You are what you eat: O-linked N-acetylglucosamine in disease, development and epigenetics. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Someya, A.; Ikegami, T.; Sakamoto, K.; Nagaoka, I. Glucosamine downregulates the IL-1β-induced expression of proinflammatory cytokine genes in human synovial MH7A cells by O-GlcNAc modification-dependent and -independent mechanisms. PLoS ONE 2016, 11, e0165158. [Google Scholar] [CrossRef]
- Lenzen, S.; Panten, U. Alloxan: History and mechanism of action. Diabetologia 1988, 31, 337–342. [Google Scholar] [CrossRef]
- Konrad, R.J.; Zhang, F.; Hale, J.E.; Knierman, M.D.; Becker, G.W.; Kudlow, J.E. Alloxan is an inhibitor of the enzyme O-linked N-acetylglucosamine transferase. Biochem. Biophys. Res. Commun. 2002, 293, 207–212. [Google Scholar] [CrossRef]
- Jiang, J.; Lazarus, M.B.; Pasquina, L.; Sliz, P.; Walker, S. A neutral diphosphate mimic crosslinks the active site of human O-GlcNAc transferase. Nat. Chem. Biol. 2011, 8, 72–77. [Google Scholar] [CrossRef]
- Celiberto, L.S.; Chan, J.Y.; Law, H.T.; Bhullar, K.; Xia, L.; Cavallini, D.C.; Vallance, B. A10 core-1 derived o-glycosylation of the mucin muc2 plays a key role in host defense against enteric citrobacter rodentium infection. Can J. Gastroenterol. Hepatol. 2018, 1, 15–16. [Google Scholar] [CrossRef][Green Version]
- Hennebicq-Reig, S.; Lesuffleur, T.; Capon, C.; De Bolos, C.; Kim, I.; Moreau, O.; Richet, C.; Hémon, B.; Recchi, M.A.; Maës, E.; et al. Permanent exposure of mucin-secreting HT-29 cells to benzyl-N-acetylalpha-d-galactosaminide induces abnormal O-glycosylation of mucins and inhibits constitutive and stimulated MUC5AC secretion. Biochem. J. 1998, 334, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Bolanle, I.O.; Riches-Suman, K.; Williamson, R.; Palmer, T.M. Emerging roles of protein O-GlcNAcylation in cardiovascular diseases: Insights and novel therapeutic targets. Pharmacol. Res. 2021, 165, 105467. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Denis, M.; Persello, A.; Rathagirishnan, R.; Lauzier, B. Protein O-GlcNAcylation in Cardiac Pathologies: Past, Present, Future. Front. Endocrinol. 2019, 9, 819. [Google Scholar] [CrossRef] [PubMed]
- Gloster, T.M.; Zandberg, W.F.; Heinonen, J.E.; Shen, D.L.; Deng, L.; Vocadlo, D.J. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat. Chem. Biol. 2011, 7, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.W.; Zandberg, W.F.; Gloster, T.M.; Deng, L.; Murray, K.D.; Shan, X.; Vocadlo, D.J. Metabolic Inhibitors of O-GlcNAc Transferase That Act In Vivo Implicate Decreased O-GlcNAc Levels in Leptin-Mediated Nutrient Sensing. Angew. Chem. Int. Ed. 2018, 57, 7644–7648. [Google Scholar] [CrossRef]
- Ortiz-Meoz, R.F.; Jiang, J.; Lazarus, M.B.; Orman, M.; Janetzko, J.; Fan, C.; Duveau, D.Y.; Tan, Z.W.; Thomas, C.J.; Walker, S. A small molecule that inhibits OGT activity in cells. ACS Chem. Biol. 2015, 10, 1392–1397. [Google Scholar] [CrossRef]
- Martin, S.; Tan, Z.W.; Itkonen, H.M.; Duveau, D.Y.; Paulo, J.A.; Janetzko, J.; Boutz, P.L.; Törk, L.; Moss, F.A.; Thomas, C.J.; et al. Structure-Based Evolution of Low Nanomolar O-GlcNAc Transferase Inhibitors. J. Am. Chem. Soc. 2018, 140, 13542–13545. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, Y.; Cao, Y.; Huang, H.; Wu, Q.; Li, W.; Wu, S.; Zhang, J. Discovery of a Low Toxicity O-GlcNAc Transferase (OGT) Inhibitor by Structure-based Virtual Screening of Natural Products. Sci. Rep. 2017, 7, 12334. [Google Scholar] [CrossRef]
- Dorfmueller, H.C.; Borodkin, V.S.; Schimpl, M.; van Aalten, D.M.F. GlcNAcstatins are nanomolar inhibitors of human O-GlcNAcase inducing cellular hyper-O-GlcNAcylation. Biochem. J. 2009, 420, 221–227. [Google Scholar] [CrossRef]
- Dorfmueller, H.C.; Borodkin, V.S.; Schimpl, M.; Shepherd, S.M.; Shpiro, N.A.; van Aalten, D.M.F. GlcNAcstatin: A picomolar, selective O-GlcNAcase inhibitor that modulates intracellular O-GlcNAcylation levels. J. Am. Chem. Soc. 2006, 128, 16484–16485. [Google Scholar] [CrossRef]
- Keembiyehetty, C.; Love, D.C.; Harwood, K.R.; Gavrilova, O.; Comly, M.E.; Hanover, J.A. Conditional knock-out reveals a requirement for O-linked N-Acetylglucosaminase (O-GlcNAcase) in metabolic homeostasis. J. Biol. Chem. 2015, 290, 7097–7113. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.R.; Song, M.; Lee, H.; Jeon, Y.; Choi, E.J.; Jang, H.J.; Moon, H.Y.; Byun, H.Y.; Kim, E.K.; Kim, D.H.; et al. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell. 2012, 11, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.R.; Jang, H.J.; Yoon, S.; Lee, Y.H.; Nam, D.; Kim, I.S.; Lee, H.; Kim, H.; Choi, J.H.; Kang, B.H.; et al. OGA heterozygosity suppresses intestinal tumorigenesis in Apc(min/+) mice. Oncogenesis 2014, 3, e109. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Murakami, K.; Kurotaki, D.; Kawase, W.; Soma, S.; Fukuchi, Y.; Kunimoto, H.; Yoshimi, R.; Koide, S.; Oshima, M.; Hishiki, T.; et al. OGT Regulates Hematopoietic Stem Cell Maintenance via PINK1-Dependent Mitophagy. Cell Rep. 2021, 34, 108579. [Google Scholar] [CrossRef]
- Tarbet, H.J.; Toleman, C.A.; Boyce, M. A Sweet Embrace: Control of Protein-Protein Interactions by O-Linked β-N-Acetylglucosamine. Biochemistry 2018, 57, 13–21. [Google Scholar] [CrossRef]
- Ma, J.; Hou, C.; Li, Y.; Chen, S.; Wu, C. OGT Protein Interaction Network (OGT-PIN): A Curated Database of Experimentally Identified Interaction Proteins of OGT. Int. J. Mol. Sci. 2021, 22, 9620. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).