

Analysis of Homologous Regions of Small RNAs MIR397 and MIR408 Reveals the Conservation of Microsynteny among Rice Crop-Wild Relatives


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval of MIR397 and MIR408 Homologs
2.2. MIRNA Precursor Sequence Analysis
2.3. Test of Neutrality
2.4. Microsynteny Analysis
2.5. Plant Material and Sequencing of Precursor MIR397 and MIR408 Sequences
2.6. Expression Analysis of Mature MIR397 and MIR408 via qRT-PCR
2.7. Phylogenetic Analysis of Precursor Sequences
3. Results
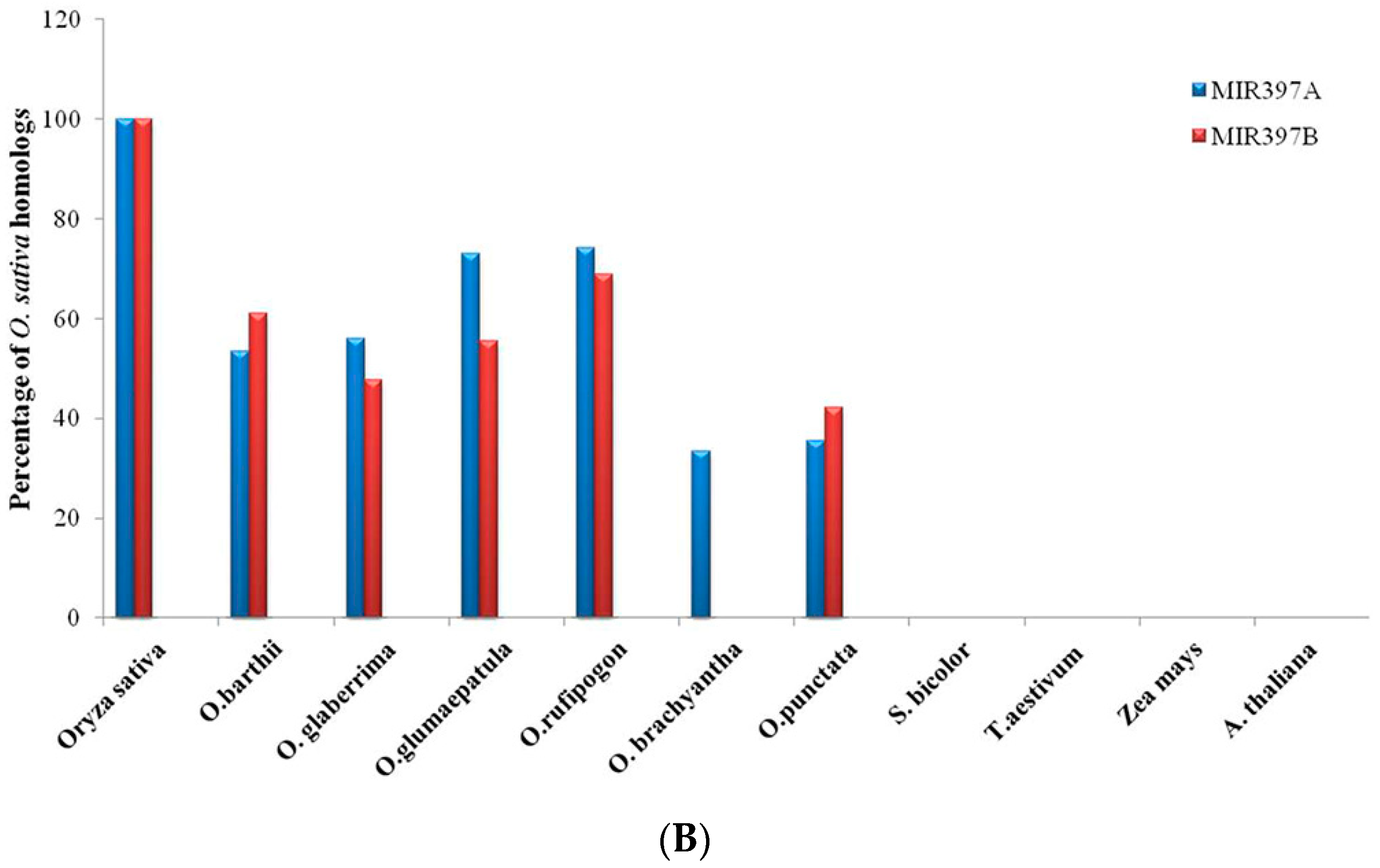
3.1. Identification of MIR397 and MIR408 Homologs in Oryza, Other Monocots and One Eudicot
3.2. Conservation and Divergence in Mature and Precursor MIR397 and MIR408
3.3. Selection Pattern of Sequence Variation
3.4. Gene Content and Gene Density Analysis
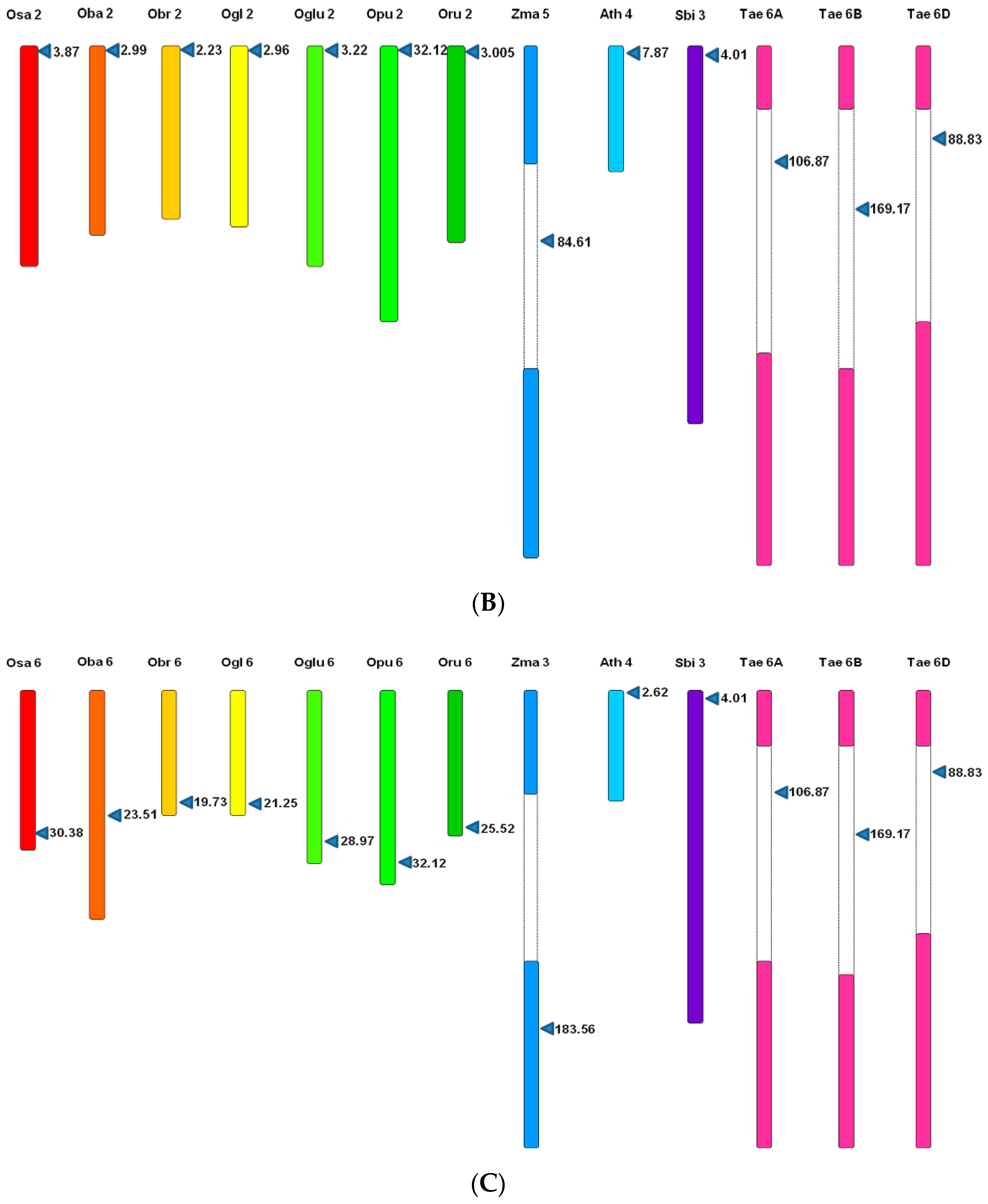
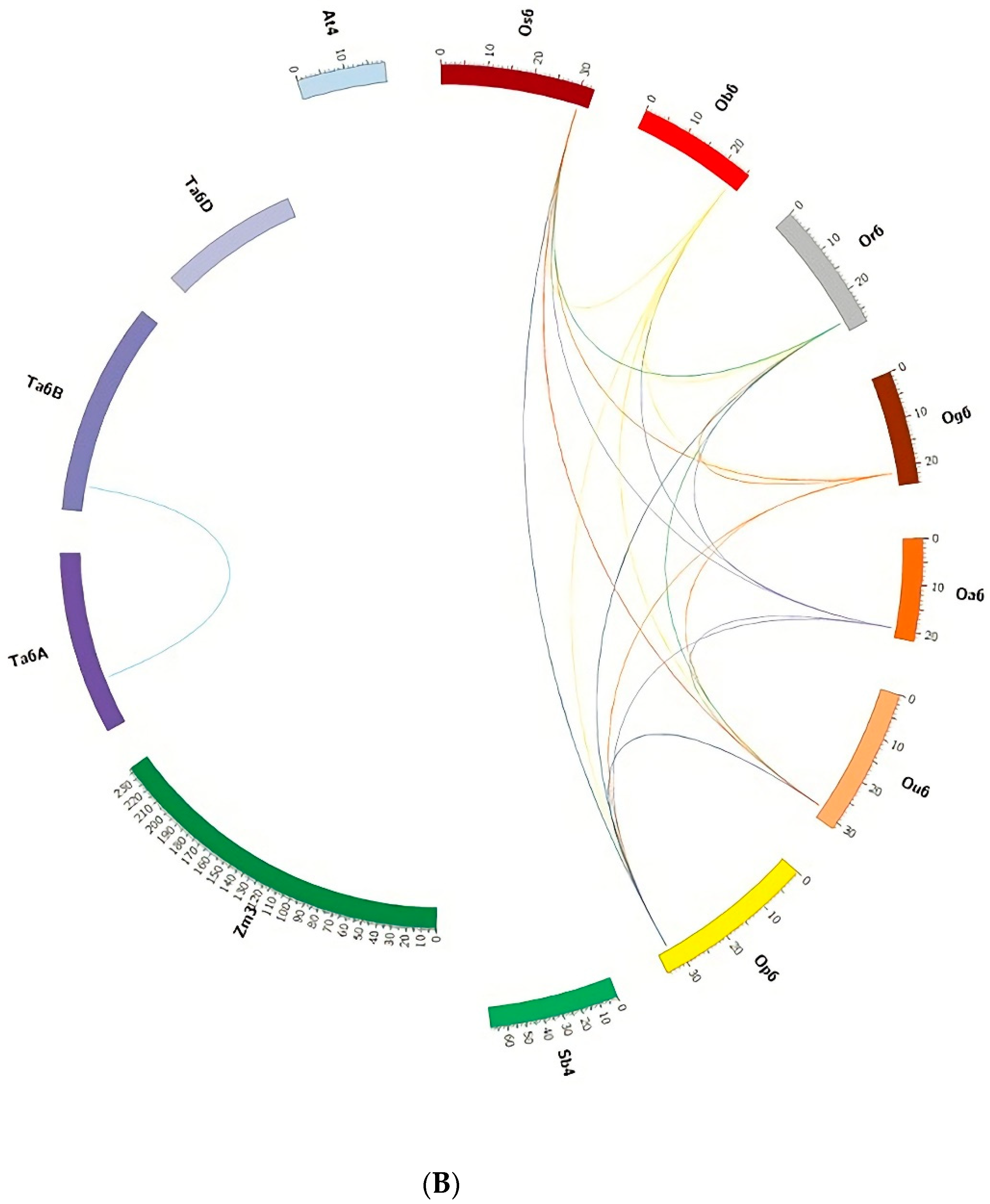
3.5. Microsynteny Analysis
3.5.1. (I) MIR397A
3.5.2. (II) MIR397B
3.5.3. (III) MIR408
3.6. Phylogenetic Analysis
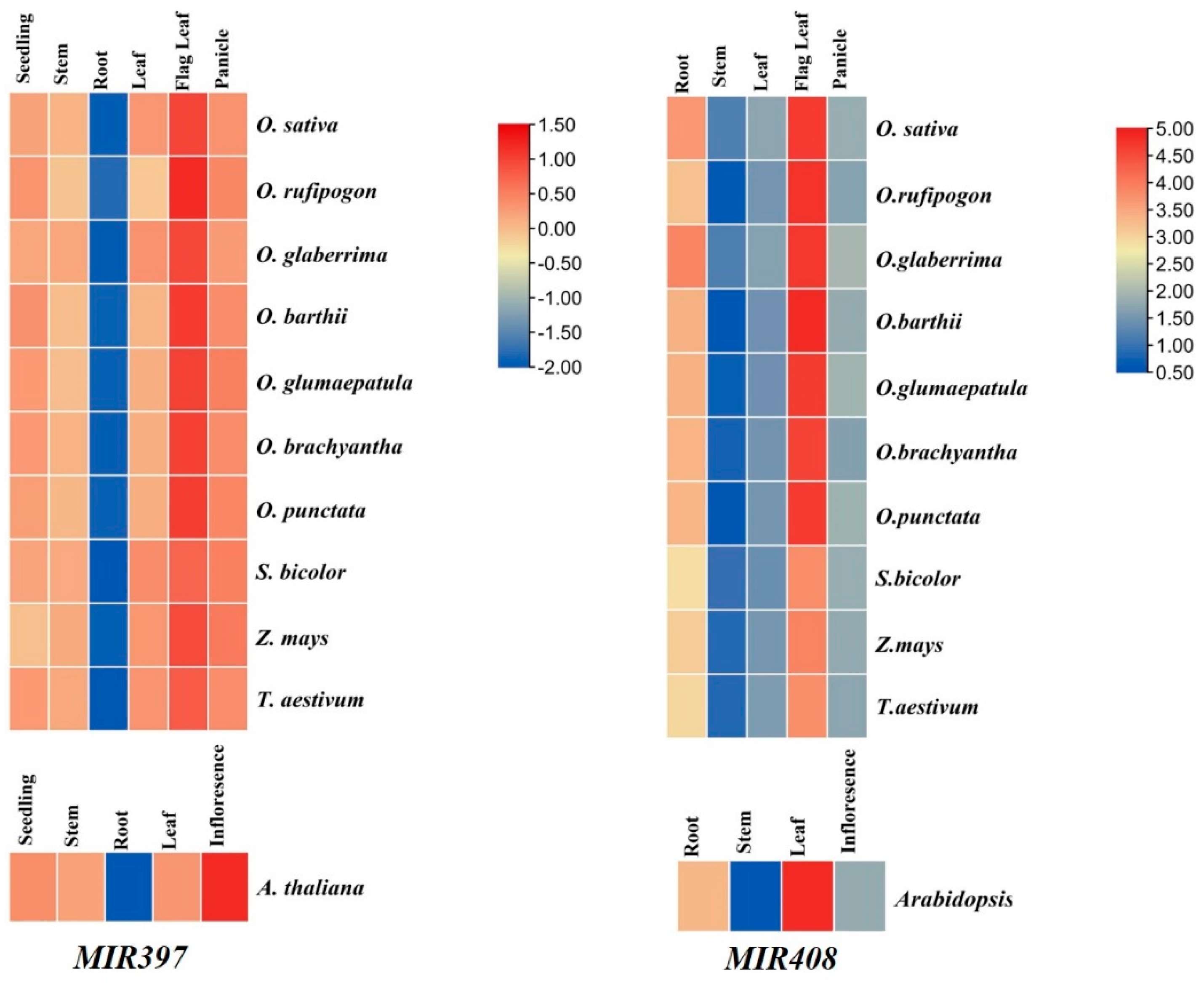
3.7. Expression Analysis of MIR397 and MIR408 in Tissue-Specific Manner
4. Discussion
4.1. Conservation in Mature MIRNAs
4.2. Evolution of MIR307 and MIR408
4.3. Selection Pattern of Sequence Polymorphism
4.4. Microsynteny Analysis across Poaceae
4.5. Conservation of Gene Organization around MIRNA Locus
4.6. Phylogenetic Analysis of Conserved and Non-Conserved MIRNAs across Poaceae
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kellogg, E.A. Evolutionary History of the Grasses. Plant Physiol. 2001, 125, 1198–1205. [Google Scholar] [CrossRef]
- Mammadov, J.; Buyyarapu, R.; Guttikonda, S.K.; Parliament, K.; Abdurakhmonov, I.Y.; Kumpatla, S.P. Wild Relatives of Maize, Rice, Cotton, and Soybean: Treasure Troves for Tolerance to Biotic and Abiotic Stresses. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef]
- Shanchez, P.; Wing, R.A.; Brar, D. Genetics and Genomics of Rice; Zhang, Q., Wing, R., Eds.; Springer Science+Business Media, LLC: Berlin, Germany, 2013. [Google Scholar]
- Shakiba, E.; Eizenga, G. Rice-Germplasm, Genetics and Improvement; Bao, J., Ed.; IntechOpen: London, UK, 2014. [Google Scholar]
- Levy, A.A.; Feldman, M. The Impact of Polyploidy on Grass Genome Evolution. Plant Physiol. 2002, 130, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Bowers, J.E.; Chapman, B.A. Ancient Polyploidization Predating Divergence of the Cereals, and Its Consequences for Comparative Genomics. Proc. Natl. Acad. Sci. USA 2004, 101, 9903–9908. [Google Scholar] [CrossRef] [PubMed]
- Dhaka, N.; Sharma, R. MicroRNA-Mediated Regulation of Agronomically Important Seed Traits: A Treasure Trove with Shades of Grey! Crit. Rev. Biotechnol. 2021, 41, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.; Chauhan, C.; Das, S. Microsynteny Analysis to Understand Evolution and Impact of Polyploidization on MIR319 Family within Brassicaceae. Dev. Genes Evol. 2018, 228, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Dhaka, N.; Sharma, S.; Vashisht, I.; Kandpal, M.; Sharma, M.K.; Sharma, R. Small RNA Profiling from Meiotic and Post-Meiotic Anthers Reveals Prospective MiRNA-Target Modules for Engineering Male Fertility in Sorghum. Genomics 2020, 112, 1598–1610. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Weinstein, E.; Rhoades, M.; Bartel, B.; Bartel, D. MicroRNAs in Plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef]
- Voinnet, O. Origin, Biogenesis, and Activity of Plant MicroRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef]
- Rogers, K.; Chen, X. Biogenesis, Turnover, and Mode of Action of Plant MicroRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhou, J.; Gao, L.; Tang, Y. Plant MiR397 and Its Functions. Funct Plant Biol. 2021, 48, 361. [Google Scholar] [CrossRef] [PubMed]
- Swetha, C.; Basu, D.; Pachamuthu, K.; Tirumalai, V.; Nair, A.; Prasad, M.; Shivaprasad, P.V. Major Domestication-Related Phenotypes in Indica Rice Are Due to Loss of MiRNA-Mediated Laccase Silencing. Plant Cell 2018, 30, 2649–2662. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Annotating High Confidence MicroRNAs Using Deep Sequencing Data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Zaman, F.; Zhang, M.; Liu, Y.; Wang, Z.; Xu, L.; Guo, D.; Luo, Z.; Zhang, Q. DkmiR397 Regulates Proanthocyanidin Biosynthesis via Negative Modulating DkLAC2 in Chinese PCNA Persimmon. Int. J. Mol. Sci. 2022, 23, 3200. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Yu, Y.; Wang, C.Y.; Li, Z.Y.; Liu, Q.; Xu, J.; Liao, J.Y.; Wang, X.J.; Qu, L.H.; Chen, F.; et al. Overexpression of MicroRNA OsmiR397 Improves Rice Yield by Increasing Grain Size and Promoting Panicle Branching. Nat. Biotechnol. 2013, 31, 848–852. [Google Scholar] [CrossRef]
- Wang, C.Y.; Zhang, S.; Yu, Y.; Luo, Y.C.; Liu, Q.; Ju, C.; Zhang, Y.C.; Qu, L.H.; Lucas, W.J.; Wang, X.; et al. MiR397b Regulates Both Lignin Content and Seed Number in Arabidopsis via Modulating a Laccase Involved in Lignin Biosynthesis. Plant Biotechnol. J. 2014, 12, 1132–1142. [Google Scholar] [CrossRef]
- Feng, Y.-Z.; Yu, Y.; Zhou, Y.-F.; Yang, Y.-W.; Lei, M.-Q.; Lian, J.-P.; He, H.; Zhang, Y.-C.; Huang, W.; Chen, Y.-Q. A Natural Variant of MiR397 Mediates a Feedback Loop in Circadian Rhythm. Plant Physiol. 2020, 182, 204–214. [Google Scholar] [CrossRef]
- Gupta, O.P.; Meena, N.L.; Sharma, I.; Sharma, P. Differential Regulation of MicroRNAs in Response to Osmotic, Salt and Cold Stresses in Wheat. Mol. Biol. Rep. 2014, 41, 4623–4629. [Google Scholar] [CrossRef]
- Patel, P.; Yadav, K.; Srivastava, A.K.; Suprasanna, P.; Ganapathi, T.R. Overexpression of Native Musa-MiR397 Enhances Plant Biomass without Compromising Abiotic Stress Tolerance in Banana. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Axtell, M.J.; Bowman, J.L. Evolution of Plant MicroRNAs and Their Targets. Trends Plant Sci. 2008, 13, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Yang, Y.; Du, J.; Deng, X.W.; Li, L. The PCY-SAG14 Phytocyanin Module Regulated by PIFs and MiR408 Promotes Dark-Induced Leaf Senescence in Arabidopsis. Proc. Natl. Acad. Sci. USA 2022, 119. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Integrating MicroRNA Annotation and Deep-Sequencing Data. Nucleic. Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhang, L.; Wang, Y.; Li, H.; Li, S.; Zhao, H.; Zhang, H. Constitutive Expression of Mir408 Improves Biomass and Seed Yield in Arabidopsis. Front. Plant Sci. 2018, 8, 2114. [Google Scholar] [CrossRef]
- Feng, H.; Zhang, Q.; Wang, Q.; Wang, X.; Liu, J.; Li, M.; Huang, L.; Kang, Z. Target of Tae-MiR408, a Chemocyanin-like Protein Gene (TaCLP1), Plays Positive Roles in Wheat Response to High-Salinity, Heavy Cupric Stress and Stripe Rust. Plant Mol. Biol. 2013, 83, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Feng, B.; Gao, C.; Zhang, H.; Wen, F.; Tao, L.; Fu, G.; Xiong, J. The Evolution and Functional Roles of MiR408 and Its Targets in Plants. Int. J. Mol. Sci. 2022, 23, 530. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Hong, P.; Wu, J.Y.; Chen, X.B.; Ye, X.G.; Pan, Y.Y.; Wang, J.; Zhang, X.S. The Tae-MiR408-Mediated Control of TaTOC1 Genes Transcription Is Required for the Regulation of Heading Time in Wheat. Plant Physiol. 2016, 170, 1578–1594. [Google Scholar] [CrossRef]
- Yamasaki, H.; Hayashi, M.; Fukazawa, M.; Kobayashi, Y.; Shikanai, T. SQUAMOSA Promoter Binding Protein-Like7 Is a Central Regulator for Copper Homeostasis in Arabidopsis. Plant Cell 2009, 21, 347–361. [Google Scholar] [CrossRef]
- Zhang, H.; Li, L. SQUAMOSA Promoter Binding Protein-Like7 Regulated MicroRNA408 Is Required for Vegetative Development in Arabidopsis. Plant J. 2013, 74, 98–109. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, X.; Li, J.; Cai, H.; Deng, X.W.; Li, L. MicroRNA408 Is Critical for the HY5-SPl7 Gene Network That Mediates the Coordinated Response to Light and Copper. Plant Cell 2014, 26, 4933–4953. [Google Scholar] [CrossRef]
- Dash Prasanta, K.; Rai, R. Green Revolution to Grain Revolution: Florigen in the Frontiers. J. Biotechnol. 2021, 343, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Burgarella, C.; Berger, A.; Glémin, S.; David, J.; Terrier, N.; Deu, M.; Pot, D. The Road to Sorghum Domestication: Evidence from Nucleotide Diversity and Gene Expression Patterns. Front. Plant Sci. 2021, 12, 66075. [Google Scholar] [CrossRef] [PubMed]
- Jena, K.K. The Species of the Genus Oryza and Transfer of Useful Genes from Wild Species into Cultivated Rice, O. Sativa. Breed. Sci. 2010, 60, 518–523. [Google Scholar] [CrossRef]
- Dash, P.K.; Cao, Y.; Jailani, A.K.; Gupta, P.; Venglat, P.; Xiang, D.; Rai, R.; Sharma, R.; Thirunavukkarasu, N.; Abdin, M.Z.; et al. Genome-Wide Analysis of Drought Induced Gene Expression Changes in Flax (Linum usitatissimum). GM Crops Food 2014, 5, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The Genome of Flax (Linum usitatissimum) Assembled de Novo from Short Shotgun Sequence Reads. Plant J. 2012, 72, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Shivaraj, S.M.; Deshmukh, R.K.; Rai, R.; Bélanger, R.; Agrawal, P.K.; Dash, P.K. Genome-Wide Identification, Characterization, and Expression Profile of Aquaporin Gene Family in Flax (Linum usitatissimum). Sci. Rep. 2017, 7, srep46137. [Google Scholar] [CrossRef]
- Dash, P.K.; Rai, R.; Mahato, A.K.; Gaikwad, K.; Singh, N.K. Transcriptome Landscape at Different Developmental Stages of a Drought Tolerant Cultivar of Flax (Linum usitatissimum). Front. Chem. 2017, 5, 82. [Google Scholar] [CrossRef]
- Dash, P.K.; Gupta, P.; Rai, R. Hydroponic Method of Halophobic Response Elicitation in Flax (Linum usitatissimum) for Precise down- Stream Gene Expression Studies. Int. J. Trop. Agric. 2015, 33, 1079–1085. [Google Scholar]
- Gupta, P.; Dash, P.K. Molecular Details of Secretory Phospholipase A2 from Flax (Linum usitatissimum L.) Provide Insight into Its Structure and Function. Sci. Rep. 2017, 7, 11080. [Google Scholar] [CrossRef]
- Gupta, P.; Dash, P.K. Precise Method of in Situ Drought Stress Induction in Flax (Linum usitatissimum) for RNA Isolation towards down-Stream Analysis. Anals Agric. Res. 2015, 36, 10–17. [Google Scholar]
- Dash, P.K.; Gupta, P.; Jailani, A.K.; Rai, R. Hydropenia Induces Expression of Drought Responsive Genes (DRGs) erd1, hat, pld-δ, and zfa in Linum usitatissimum L. Ind. J. Exp. Biol. 2018, 56, 743–749. [Google Scholar]
- Gupta, P.; Rai, R.; Vasudev, S.; Yadava, D.; Dash, P.K. Ex-Foliar Application of Glycine Betaine and Its Impact on Protein, Carbohydrates and Induction of ROS Scavenging System during Drought Stress in Flax (Linum usitatissimum). J. Biotechnol. 2021, 337, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Duong, H.; Repalli, S.; Sreevathsa, R.; Yadava, D.; Dash, P. Cloning and Structural Elucidation of a Brassinosteroids Biosynthetic Gene (Atdwarf4) and Genetic Transformation of Indian Mustard (Brassica juncea L.). Indian J. Biochem. Biophys. 2022, 59, 320–330. [Google Scholar]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. MiRBase: MicroRNA Sequences, Targets and Gene Nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res. 2014, 42, 320–324. [Google Scholar] [CrossRef]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.X.; Li, W.H. Statistical Tests of Neutrality of Mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Solovyev, V.; Fokin, O.; Seledtsov, I.; Salamov, A.; Molodtsov, V.; Okhalin, N.; Bachinskii, A.; Kosarev, P.; Bakulina, N.; Genaev, M.; et al. MolQuest-Bioinformatics Toolbox for Analysis of Biomedical Data. 2021. Available online: https://molquest.com/molquest.phtml?topic=downloads (accessed on 22 September 2022).
- Götz, S.; Arnold, R.; Sebastián-León, P.; Martín-Rodríguez, S.; Tischler, P.; Jehl, M.A.; Dopazo, J.; Rattei, T.; Conesa, A. B2G-FAR, a Species-Centered GO Annotation Repository. Bioinformatics 2011, 27, 919–924. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Ghiurcuta, C.G.; Moret, B.M.E. Evaluating Synteny for Improved Comparative Studies. Bioinformatics 2014, 30, i9–i18. [Google Scholar] [CrossRef]
- Tang, H.; Bowers, J.E.; Wang, X.; Ming, R.; Alam, M.; Paterson, A.H. Synteny and Collinearity in Plant Genomes. Science 2008, 320, 486–488. [Google Scholar] [CrossRef]
- Lallemand, T.; Leduc, M.; Landès, C.; Rizzon, C.; Lerat, E. An Overview of Duplicated Gene Detection Methods: Why the Duplication Mechanism Has to Be Accounted for in Their Choice. Genes 2020, 11, 1046. [Google Scholar] [CrossRef]
- Casneuf, T.; De Bodt, S.; Raes, J.; Maere, S.; Van de Peer, Y. Nonrandom Divergence of Gene Expression Following Gene and Genome Duplications in the Flowering Plant Arabidopsis Thaliana. Genome Biol. 2006, 7, R13. [Google Scholar] [CrossRef]
- Restrepo-Montoya, D.; McClean, P.E.; Osorno, J.M. Orthology and Synteny Analysis of Receptor-like Kinases “RLK” and Receptor-like Proteins “RLP” in Legumes. BMC Genom. 2021, 22, 1–17. [Google Scholar] [CrossRef]
- Jain, A.; Das, S. Synteny and Comparative Analysis of MiRNA Retention, Conservation, and Structure across Brassicaceae Reveals Lineage- and Sub-Genome-Specific Changes. Funct. Integr. Genom. 2016, 16, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Allaby, R.G. Domestication Syndrome in Plants. In Encyclopedia of Global Archaeology; Springer: New York, NY, USA, 2014. [Google Scholar]
- Allaby, R.G.; Ware, R.L.; Kistler, L. A Re-evaluation of the Domestication Bottleneck from Archaeogenomic Evidence. Evol. Appl. 2019, 12, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Sang, T.; Lu, B.R.; Hong, D.Y. Phylogeny of Rice Genomes with Emphasis on Origins of Allotetraploid Species. Proc. Natl. Acad. Sci. USA 1999, 96, 14400–14405. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.H.; Zhang, F.M.; Zhang, J.G.; Zang, L.L.; Tang, L.; Wang, J.; Sang, T.; Ge, S. Analysis of 142 Genes Resolves the Rapid Diversification of the Rice Genus. Genome Biol. 2008, 9, R49. [Google Scholar] [CrossRef]
- Zhang, Q.-J.; Zhu, T.; Xia, E.-H.; Shi, C.; Liu, Y.-L.; Zhang, Y.; Liu, Y.; Jiang, W.-K.; Zhao, Y.-J.; Mao, S.-Y.; et al. Rapid Diversification of Five Oryza AA Genomes Associated with Rice Adaptation. Proc. Natl. Acad. Sci. USA 2014, 111, E4954–E4962. [Google Scholar] [CrossRef]
- Ammiraju, J.S.S.; Fan, C.; Yu, Y.; Song, X.; Cranston, K.A.; Pontaroli, A.C.; Lu, F.; Sanyal, A.; Jiang, N.; Rambo, T.; et al. Spatio-Temporal Patterns of Genome Evolution in Allotetraploid Species of the Genus Oryza. Plant J. 2010, 63, 430–442. [Google Scholar] [CrossRef]
- Axtell, M.J.; Bartel, D.P. Antiquity of MicroRNAs and Their Targets in Land Plants. Plant Cell 2005, 17, 1658–1673. [Google Scholar] [CrossRef]
- Cammaerts, S.; Strazisar, M.; de Rijk, P.; del Favero, J. Genetic Variants in MicroRNA Genes: Impact on MicroRNA Expression, Function, and Disease. Front. Genet. 2015, 6, 186. [Google Scholar] [CrossRef]
- Han, M.; Zheng, Y. Comprehensive Analysis of Single Nucleotide Polymorphisms in Human MicroRNAs. PLoS ONE 2013, 8, e78028. [Google Scholar] [CrossRef]
- Gong, J.; Tong, Y.; Zhang, H.M.; Wang, K.; Hu, T.; Shan, G.; Sun, J.; Guo, A.Y. Genome-Wide Identification of SNPs in MicroRNA Genes and the SNP Effects on MicroRNA Target Binding and Biogenesis. Hum. Mutat. 2012, 33, 254–263. [Google Scholar] [CrossRef]
- Salse, J. In Silico Archeogenomics Unveils Modern Plant Genome Organisation, Regulation and Evolution. Curr. Opin. Plant Biol. 2012, 15, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Hilu, K.W. Phylogenetics and Chromosomal Evolution in the Poaceae (Grasses). Aust. J. Bot. 2004, 52, 13–22. [Google Scholar] [CrossRef]
- Sun, S.; Wang, J.; Yu, J.; Meng, F.; Xia, R.; Wang, L.; Wang, Z.; Ge, W.; Liu, X.; Li, Y.; et al. Alignment of Common Wheat and Other Grass Genomes Establishes a Comparative Genomics Research Platform. Front. Plant Sci. 2017, 8, 1480. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Murat, F.; Li, A.; Pont, C.; Mao, L.; Salsea, J. Grass MicroRNA Gene Paleohistory Unveils New Insights into Gene Dosage Balance in Subgenome Partitioning after Whole-Genome Duplication. Plant Cell 2012, 24, 1776–1792. [Google Scholar] [CrossRef]
- Rathore, P.; Geeta, R.; Das, S. Microsynteny and Phylogenetic Analysis of Tandemly Organised MiRNA Families across Five Members of Brassicaceae Reveals Complex Retention and Loss History. Plant Sci. 2016, 247, 35–48. [Google Scholar] [CrossRef]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and Functional Diversification of MIRNA Genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef]
- Ehrenreich, I.M.; Purugganan, M.D. Sequence Variation of MicroRNAs and Their Binding Sites in Arabidopsis. Plant Physiol. 2008, 146, 1974–1982. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, D.; Bo, S.; Chen, H.; Zheng, J.; Zhu, Q.-H.; Cai, D.; Helliwell, C.; Fan, L. Sequence Variation and Selection of Small RNAs in Domesticated Rice. BMC Evol. Biol. 2010, 10, 119. [Google Scholar] [CrossRef]
- Ganie, S.A.; Debnath, A.B.; Gumi, A.M.; Mondal, T.K. Comprehensive Survey and Evolutionary Analysis of Genome-Wide MiRNA Genes from Ten Diploid Oryza Species. BMC Genom. 2017, 18, 711. [Google Scholar] [CrossRef]
- Yang, L.; Liu, T.; Li, B.; Sui, Y.; Chen, J.; Shi, J.; Wing, R.A.; Chen, M. Comparative Sequence Analysis of the Ghd7 Orthologous Regions Revealed Movement of Ghd7 in the Grass Genomes. PLoS ONE 2012, 7, e50236. [Google Scholar] [CrossRef]
- Sanyal, A.; Ammiraju, J.S.S.; Lu, F.; Yu, Y.; Rambo, T.; Currie, J.; Kollura, K.; Kim, H.R.; Chen, J.; Ma, J.; et al. Orthologous Comparisons of the Hd1 Region across Genera Reveal Hd1 Gene Lability within Diploid Oryza Species and Disruptions to Microsynteny in Sorghum. Mol. Biol. Evol. 2010, 27, 2487–2506. [Google Scholar] [CrossRef][Green Version]
- Jiao, Y.; Li, J.; Tang, H.; Paterson, A.H. Integrated Syntenic and Phylogenomic Analyses Reveal an Ancient Genome Duplication in Monocots. Plant Cell 2014, 26, 2792–2802. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Jiao, Y.; Tan, X.; Wang, X.; Huang, X.; Jin, H.; Paterson, A.H. Gene Duplication and Genetic Innovation in Cereal Genomes. Genome Res. 2019, 29, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.G.; Xiong, Y.Q.; Liu, T.Y.; Sun, S.H.; Chen, L.B.; Chen, M.S. Evidence for an Ancient Whole-Genome Duplication Event in Rice and Other Cereals. Acta Genet. Sin. 2005, 32, 519–527. [Google Scholar] [PubMed]
- Pont, C.; Murat, F.; Confolent, C.; Balzergue, S.; Salse, J. RNA-Seq in Grain Unveils Fate of Neo- and Paleopolyploidization Events in Bread Wheat (Triticum Aestivum L.). Genome Biol. 2011, 12, R119. [Google Scholar] [CrossRef]
- Zhong, H.; Kong, W.; Gong, Z.; Fang, X.; Deng, X.; Liu, C.; Li, Y. Evolutionary Analyses Reveal Diverged Patterns of Squamosa Promoter Binding Protein-like (Spl) Gene Family in Oryza Genus. Front. Plant Sci. 2019, 10, 565. [Google Scholar] [CrossRef]
- Goicoechea, J.L.; Ammiraju, J.S.S.; Marri, P.R.; Chen, M.; Jackson, S.; Yu, Y.; Rounsley, S.; Wing, R.A. The Future of Rice Genomics: Sequencing the Collective Oryza Genome. Rice 2010, 3, 89–97. [Google Scholar] [CrossRef][Green Version]
- Ammiraju, J.S.S.; Lu, F.; Sanyal, A.; Yu, Y.; Song, X.; Jiang, N.; Pontaroli, A.C.; Rambo, T.; Currie, J.; Collura, K.; et al. Dynamic Evolution of Oryza Genomes Is Revealed by Comparative Genomic Analysis of a Genus-Wide Vertical Data Set. Plant Cell 2008, 20, 3191–3209. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | miRNA | Chromosome Location | Precursor Coordinates | |
|---|---|---|---|---|
| Start Position | End Position | |||
| Oryza sativa | miR397A | Chr6 | 30381036 | 30381149 |
| miR397B | Chr2 | 3872846 | 3872974 | |
| miR408 | Chr1 | 13396971 | 13397183 | |
| Oryza barthii | miR397A | Chr6 | 23511907 | 23512020 |
| miR397B | Chr2 | 2992700 | 2992817 | |
| miR408 | Chr1 | 10689652 | 10689860 | |
| Oryza glaberrima | miR397A | Chr6 | 21257361 | 21257474 |
| miR397B | Chr2 | 2962620 | 2962737 | |
| miR408 | Chr1 | 8988098 | 8988197 | |
| Oryza glumaepetula | miR397A | Chr6 | 28979607 | 28979671 |
| miR397B | Chr2 | 3229081 | 3229198 | |
| miR408 | Chr1 | 13778204 | 13778256 | |
| Oryza punctata | miR397A | Chr6 | 32123390 | 32123437 |
| miR397B | Chr2 | 2728396 | 2728465 | |
| miR408 | Chr1 | 12504365 | 12504434 | |
| Oryza rufipogon | miR397A | Chr6 | 25548449 | 25548513 |
| miR397B | Chr2 | 3005958 | 3006075 | |
| miR408 | Chr1 | 11386797 | 11387005 | |
| Oryza brachyantha | miR397A | Chr6 | 19738368 | 19738441 |
| miR397B | Chr2 | 2322369 | 2322422 | |
| miR408 | Chr1 | 9605796 | 9605842 | |
| Zea mays | miR397A | Chr3 | 183569171 | 183569313 |
| miR397B | Chr5 | 84610591 | 84610674 | |
| miR408A | Chr3 | 58127890 | 58128080 | |
| miR408B | Chr8 | 39583938 | 39584084 | |
| Triticum aestivum | miR397 | Chr6A | 106878788 | 106878880 |
| Chr6B | 169172592 | 169172643 | ||
| Chr6D | 88832335 | 88832406 | ||
| miR408 | Chr7B | 632998409 | 632998595 | |
| Sorghum bicolor | miR397 | Chr4 | 4003721 | 4003811 |
| miR408 | Chr3 | 15908453 | 15908657 | |
| Arabidopsis thaliana | miR397A | Chr4 | 2625950 | 2626056 |
| miR397B | Chr4 | 7878652 | 7878760 | |
| miR408 | Chr2 | 19319814 | 19320031 | |
| MIRNA | Plant Species | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O. sativa | O. barthii | O. glaberrima | O. glumaepatula | O. rufipogon | O. brachyantha | O. punctata | S. bicolor | T. aestivum | Z. mays | A. thaliana | ||
| MIR397 | 1 | 3 | ||||||||||
| MIR397A | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||
| MIR397B | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||
| MIR408 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| MIR408A | 1 | |||||||||||
| MIR408B | 1 | |||||||||||
| MIRNA | Plant Species | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O. sativa | O. barthii | O. glaberrima | O. glumaepatula | O. rufipogon | O. brachyantha | O. punctata | S. bicolor | T. aestivum | Z. mays | A. thaliana | ||
| MIR397 | 17 | 6A-34 6B-24 6D-25 | ||||||||||
| MIR397A | 32 | 30 | 25 | 26 | 31 | 18 | 31 | 18 | 28 | |||
| MIR397B | 18 | 18 | 21 | 18 | 16 | 14 | 19 | 21 | 25 | |||
| MIR408 | 21 | 17 | 18 | 17 | 18 | 19 | 22 | 17 | 31 | 30 | ||
| MIR408A | 16 | |||||||||||
| MIR408B | 13 | |||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dash, P.K.; Gupta, P.; Pradhan, S.K.; Shasany, A.K.; Rai, R. Analysis of Homologous Regions of Small RNAs MIR397 and MIR408 Reveals the Conservation of Microsynteny among Rice Crop-Wild Relatives. Cells 2022, 11, 3461. https://doi.org/10.3390/cells11213461
Dash PK, Gupta P, Pradhan SK, Shasany AK, Rai R. Analysis of Homologous Regions of Small RNAs MIR397 and MIR408 Reveals the Conservation of Microsynteny among Rice Crop-Wild Relatives. Cells. 2022; 11(21):3461. https://doi.org/10.3390/cells11213461
Chicago/Turabian StyleDash, Prasanta K., Payal Gupta, Sharat Kumar Pradhan, Ajit Kumar Shasany, and Rhitu Rai. 2022. "Analysis of Homologous Regions of Small RNAs MIR397 and MIR408 Reveals the Conservation of Microsynteny among Rice Crop-Wild Relatives" Cells 11, no. 21: 3461. https://doi.org/10.3390/cells11213461
APA StyleDash, P. K., Gupta, P., Pradhan, S. K., Shasany, A. K., & Rai, R. (2022). Analysis of Homologous Regions of Small RNAs MIR397 and MIR408 Reveals the Conservation of Microsynteny among Rice Crop-Wild Relatives. Cells, 11(21), 3461. https://doi.org/10.3390/cells11213461