
Janus Kinase Signaling Pathway and Its Role in COVID-19 Inflammatory, Vascular, and Thrombotic Manifestations


Abstract
:1. Overview of Vascular, Thrombotic, and Inflammation Manifestations of Coronavirus Disease-19
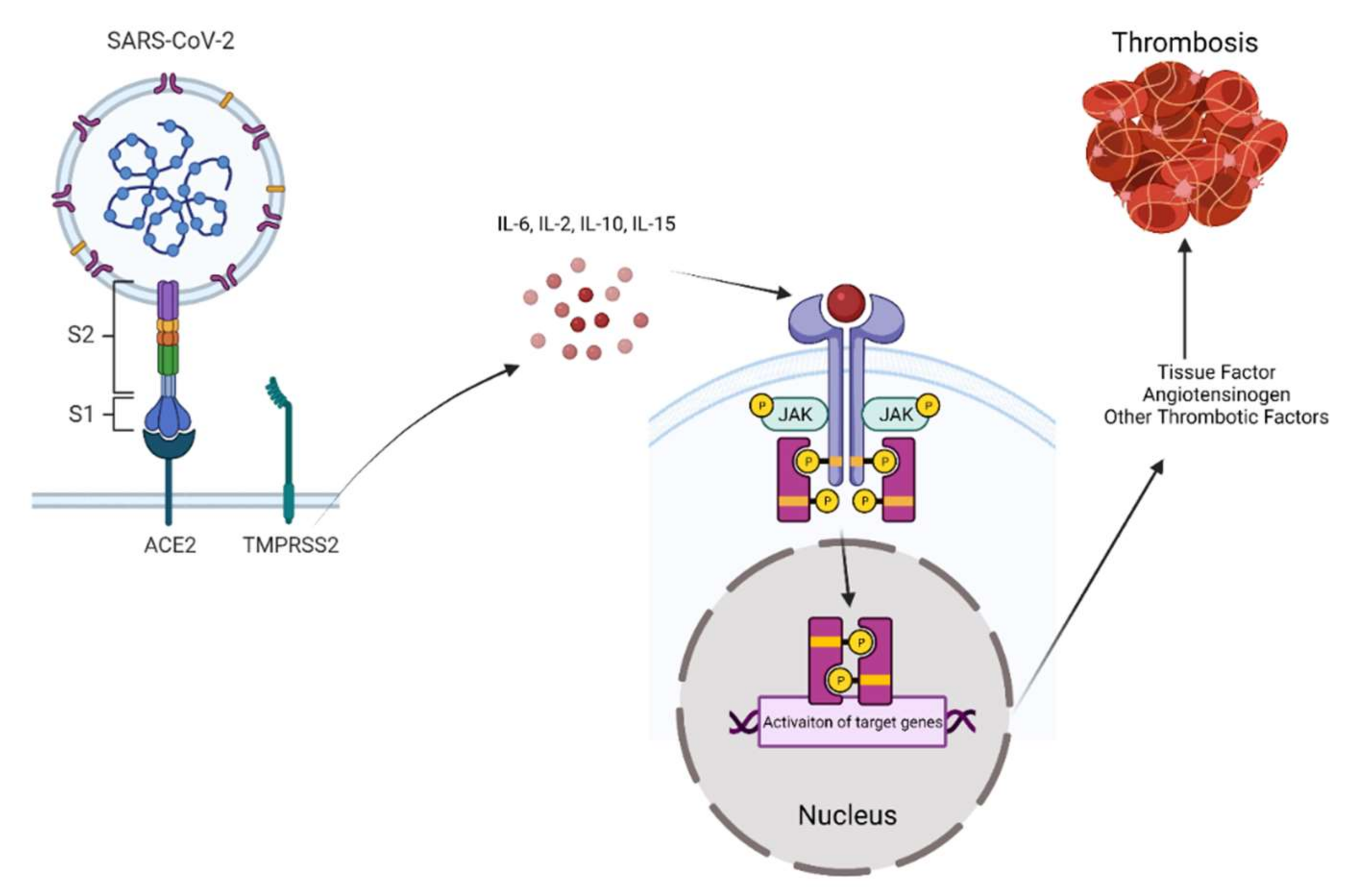
2. SARS-CoV-2 Entry into Target Cells
3. Janus Kinases Signaling towards Inflammation in COVID-19
4. The Role of JAK Signaling in Mediating the Effects of SARS-CoV-2 in Endothelial Cells, Coagulation, and Thrombosis
5. The Role of JAK Signaling in Mediating the Effects of SARS-CoV-2 in Platelets
6. A Controversial Role of JAK Signaling in Mediating the Effects of SARS-CoV-2
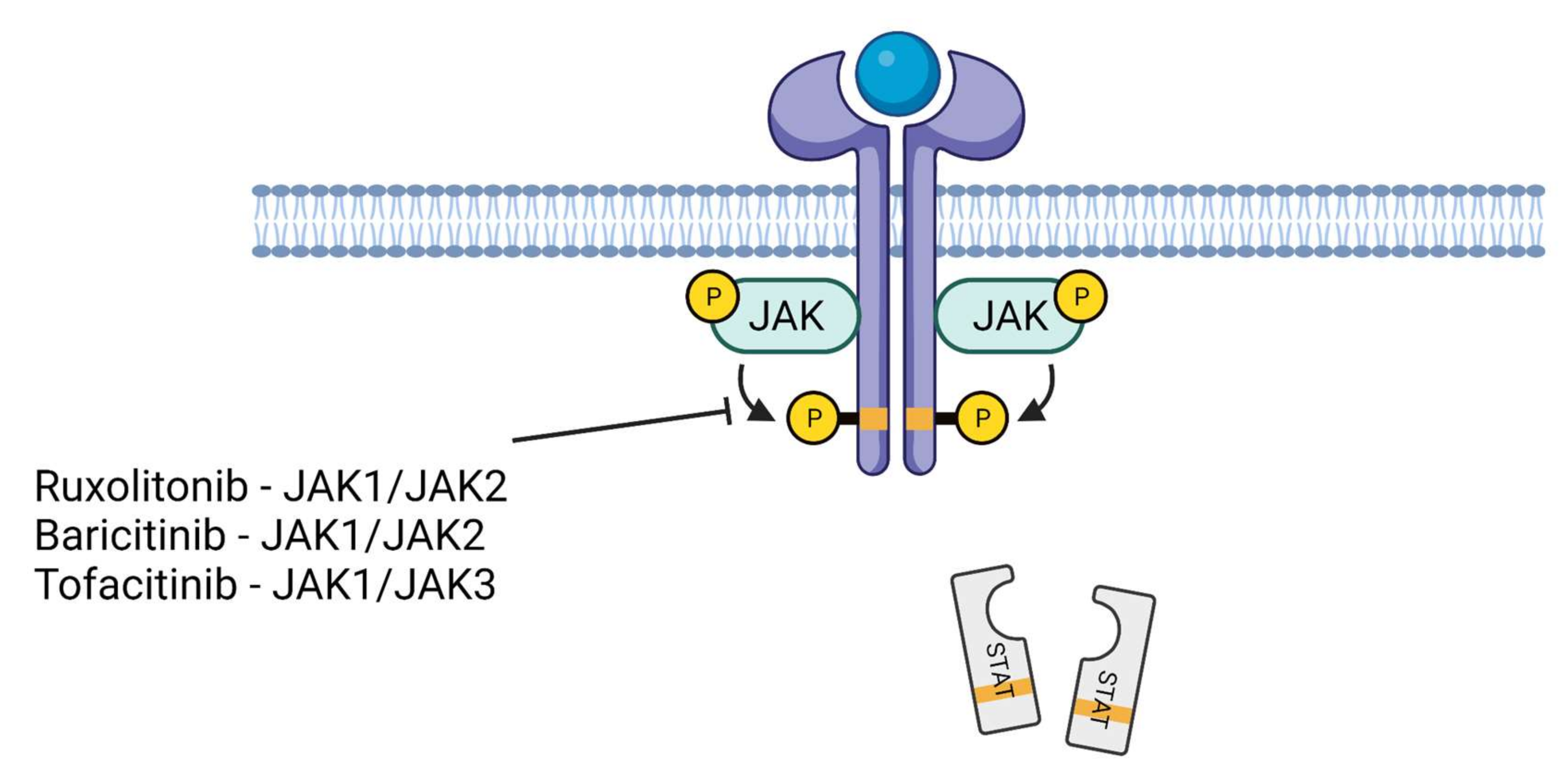
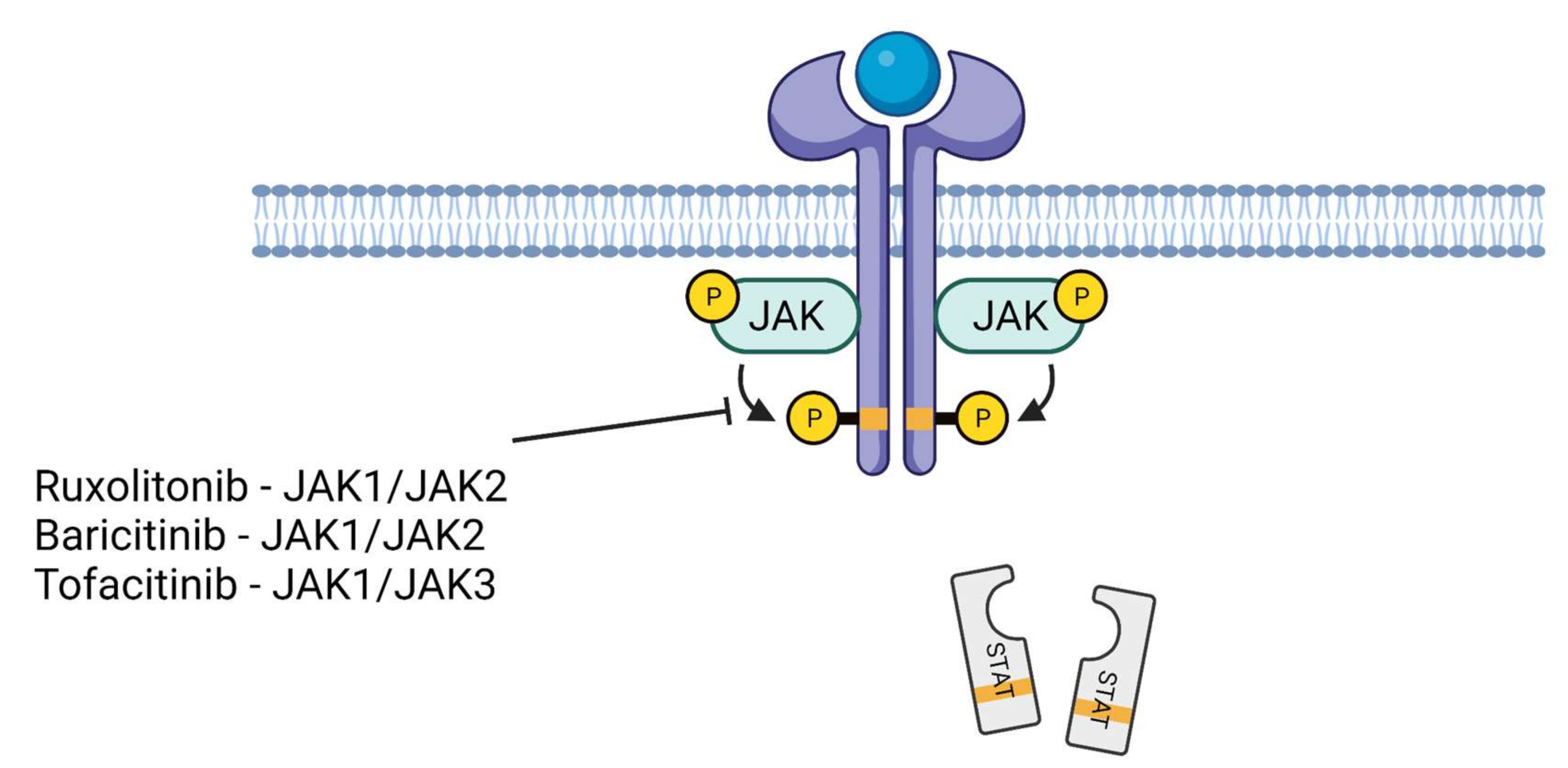
7. JAK Inhibitors in the Treatment of COVID-19
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Piazza, G.; Campia, U.; Hurwitz, S.; Snyder, J.E.; Rizzo, S.M.; Pfeferman, M.B.; Morrison, R.B.; Leiva, O.; Fanikos, J.; Nauffal, V.; et al. Registry of Arterial and Venous Thromboembolic Complications in Patients With COVID-19. J. Am. Coll. Cardiol. 2020, 76, 2060–2072. [Google Scholar] [CrossRef] [PubMed]
- Kamel, M.H.; Yin, W.; Zavaro, C.; Francis, J.M.; Chitalia, V.C. Hyperthrombotic Milieu in COVID-19 Patients. Cells 2020, 9, 2392. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Dolhnikoff, M.; Duarte-Neto, A.N.; de Almeida Monteiro, R.A.; da Silva, L.F.F.; de Oliveira, E.P.; Saldiva, P.H.N.; Mauad, T.; Negri, E.M. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J. Thromb. Haemost. 2020, 18, 1517–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wichmann, D.; Sperhake, J.P.; Lutgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schroder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 1421–1424. [Google Scholar] [CrossRef]
- Llitjos, J.F.; Leclerc, M.; Chochois, C.; Monsallier, J.M.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef] [PubMed]
- Vidale, S. Risk Factors, and Clinical and Etiological Characteristics of Ischemic Strokes in COVID-19-Infected Patients: A Systematic Review of Literature. Cerebrovasc. Dis. 2021, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P.; De Leacy, R.A.; Shigematsu, T.; Ladner, T.R.; Yaeger, K.A.; et al. Large-Vessel Stroke as a Presenting Feature of COVID-19 in the Young. N. Engl. J. Med. 2020, 382, e60. [Google Scholar] [CrossRef] [PubMed]
- Lodigiani, C.; Iapichino, G.; Carenzo, L.; Cecconi, M.; Ferrazzi, P.; Sebastian, T.; Kucher, N.; Studt, J.D.; Sacco, C.; Bertuzzi, A.; et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb. Res. 2020, 191, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Umapathi, T.; Kor, A.C.; Venketasubramanian, N.; Lim, C.C.; Pang, B.C.; Yeo, T.T.; Lee, C.C.; Lim, P.L.; Ponnudurai, K.; Chuah, K.L.; et al. Large artery ischaemic stroke in severe acute respiratory syndrome (SARS). J. Neurol. 2004, 251, 1227–1231. [Google Scholar] [CrossRef] [Green Version]
- Bozzani, A.; Arici, V.; Tavazzi, G.; Mojoli, F.; Bruno, R.; Sterpetti, A.V.; Ragni, F. Acute Thrombosis of Lower Limbs Arteries in the Acute Phase and After Recovery From COVID19. Ann. Surg. 2021, 273, e159–e160. [Google Scholar] [CrossRef]
- Lax, S.F.; Skok, K.; Zechner, P.; Kessler, H.H.; Kaufmann, N.; Koelblinger, C.; Vander, K.; Bargfrieder, U.; Trauner, M. Pulmonary Arterial Thrombosis in COVID-19 With Fatal Outcome: Results From a Prospective, Single-Center, Clinicopathologic Case Series. Ann. Intern. Med. 2020, 173, 350–361. [Google Scholar] [CrossRef]
- Stefan, N.; Birkenfeld, A.L.; Schulze, M.B.; Ludwig, D.S. Obesity and impaired metabolic health in patients with COVID-19. Nat. Rev. Endocrinol. 2020, 16, 341–342. [Google Scholar] [CrossRef] [Green Version]
- Cevik, M.; Tate, M.; Lloyd, O.; Maraolo, A.E.; Schafers, J.; Ho, A. SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: A systematic review and meta-analysis. Lancet Microbe. 2021, 2, e13–e22. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe. 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef]
- Sims, J.T.; Krishnan, V.; Chang, C.Y.; Engle, S.M.; Casalini, G.; Rodgers, G.H.; Bivi, N.; Nickoloff, B.J.; Konrad, R.J.; de Bono, S.; et al. Characterization of the cytokine storm reflects hyperinflammatory endothelial dysfunction in COVID-19. J. Allergy Clin. Immunol. 2021, 147, 107–111. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Smilowitz, N.R.; Kunichoff, D.; Garshick, M.; Shah, B.; Pillinger, M.; Hochman, J.S.; Berger, J.S. C-reactive protein and clinical outcomes in patients with COVID-19. Eur. Heart J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Pang, J.; Ji, P.; Zhong, Z.; Li, H.; Li, B.; Zhang, J. Elevated interleukin-6 is associated with severity of COVID-19: A meta-analysis. J. Med. Virol. 2021, 93, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Rosas, I.O.; Brau, N.; Waters, M.; Go, R.C.; Hunter, B.D.; Bhagani, S.; Skiest, D.; Aziz, M.S.; Cooper, N.; Douglas, I.S.; et al. Tocilizumab in Hospitalized Patients with Severe COVID-19 Pneumonia. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Investigators, R.-C.; Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; van Bentum-Puijk, W.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with COVID-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Amraei, R.; Yin, W.; Napoleon, M.A.; Suder, E.L.; Berrigan, J.; Zhao, Q.; Olejnik, J.; Chandler, K.B.; Xia, C.; Feldman, J.; et al. CD209L/L-SIGN and CD209/DC-SIGN Act as Receptors for SARS-CoV-2. ACS Cent. Sci 2021, 7, 1156–1165. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Anton-Plagaro, C.; Shoemark, D.K.; Simon-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef] [PubMed]
- Beyerstedt, S.; Casaro, E.B.; Rangel, E.B. COVID-19: Angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 905–919. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Jafarzadeh, A.; Chauhan, P.; Saha, B.; Jafarzadeh, S.; Nemati, M. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: Lessons from SARS and MERS, and potential therapeutic interventions. Life Sci. 2020, 257, 118102. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Satarker, S.; Nampoothiri, M. Structural Proteins in Severe Acute Respiratory Syndrome Coronavirus-2. Arch. Med. Res. 2020, 51, 482–491. [Google Scholar] [CrossRef]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martinez-Colon, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response to severe COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Stebbing, J.; Sanchez Nievas, G.; Falcone, M.; Youhanna, S.; Richardson, P.; Ottaviani, S.; Shen, J.X.; Sommerauer, C.; Tiseo, G.; Ghiadoni, L.; et al. JAK inhibition reduces SARS-CoV-2 liver infectivity and modulates inflammatory responses to reduce morbidity and mortality. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Croce, K.; Libby, P. Intertwining of thrombosis and inflammation in atherosclerosis. Curr. Opin. Hematol. 2007, 14, 55–61. [Google Scholar] [CrossRef]
- O'Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein. Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, R.; Lahiri, D.; Deb, S.; Bandyopadhyay, D.; Shome, G.; Sarkar, S.; Paria, S.R.; Thakurta, T.G.; Singla, P.; Biswas, S.C. COVID-19: Are we dealing with a multisystem vasculopathy in disguise of a viral infection? J. Thromb. Thrombolysis 2020, 50, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Prinz, D.; Klein, K.; List, J.; Knab, V.M.; Menzl, I.; Leidenfrost, N.; Heller, G.; Polic, B.; Putz, E.M.; Witalisz-Siepracka, A.; et al. Loss of NKG2D in murine NK cells leads to increased perforin production upon long-term stimulation with IL-2. Eur. J. Immunol. 2020, 50, 880–890. [Google Scholar] [CrossRef] [Green Version]
- Guillon, A.; Hiemstra, P.S.; Si-Tahar, M. Pulmonary immune responses against SARS-CoV-2 infection: Harmful or not? Intensive Care Med. 2020, 46, 1897–1900. [Google Scholar] [CrossRef]
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Siddiqi, H.K.; Libby, P.; Ridker, P.M. COVID-19—A vascular disease. Trends Cardiovasc. Med. 2021, 31, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Domingo, P.; Mur, I.; Pomar, V.; Corominas, H.; Casademont, J.; de Benito, N. The four horsemen of a viral Apocalypse: The pathogenesis of SARS-CoV-2 infection (COVID-19). EBioMedicine 2020, 58, 102887. [Google Scholar] [CrossRef]
- Gao, Y.M.; Xu, G.; Wang, B.; Liu, B.C. Cytokine storm syndrome in coronavirus disease 2019: A narrative review. J. Intern. Med. 2021, 289, 147–161. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Canas, C.A.; Canas, F.; Bautista-Vargas, M.; Bonilla-Abadia, F. Role of Tissue Factor in the Pathogenesis of COVID-19 and the Possible Ways to Inhibit It. Clin. Appl. Thromb. Hemost. 2021, 27, 10760296211003983. [Google Scholar] [CrossRef]
- Satou, R.; Gonzalez-Villalobos, R.A. JAK-STAT and the renin-angiotensin system: The role of the JAK-STAT pathway in blood pressure and intrarenal renin-angiotensin system regulation. JAKSTAT 2012, 1, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Park, D.W.; Lyu, J.H.; Kim, J.S.; Chin, H.; Bae, Y.S.; Baek, S.H. Role of JAK2-STAT3 in TLR2-mediated tissue factor expression. J. Cell Biochem. 2013, 114, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pendurthi, U.R.; Yi, G.; Rao, L.V.M. SARS-CoV-2 infection induces the activation of tissue factor-mediated coagulation via activation of acid sphingomyelinase. Blood 2021, 138, 344–349. [Google Scholar] [CrossRef]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Escher, R.; Breakey, N.; Lammle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef]
- Matsuishi, Y.; Mathis, B.J.; Shimojo, N.; Subrina, J.; Okubo, N.; Inoue, Y. Severe COVID-19 Infection Associated with Endothelial Dysfunction Induces Multiple Organ Dysfunction: A Review of Therapeutic Interventions. Biomedicines 2021, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Freedman, J.E. Platelets and Immunity: Going Viral. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1605–1607. [Google Scholar] [CrossRef]
- Barrett, T.J.; Lee, A.H.; Xia, Y.; Lin, L.H.; Black, M.; Cotzia, P.; Hochman, J.; Berger, J.S. Platelet and Vascular Biomarkers Associate With Thrombosis and Death in Coronavirus Disease. Circ. Res. 2020, 127, 945–947. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.J.; Bilaloglu, S.; Cornwell, M.; Burgess, H.M.; Virginio, V.W.; Drenkova, K.; Ibrahim, H.; Yuriditsky, E.; Aphinyanaphongs, Y.; Lifshitz, M.; et al. Platelets Contribute to Disease Severity in COVID-19. J. Thromb. Haemost. 2021. [Google Scholar] [CrossRef] [PubMed]
- Fard, M.B.; Fard, S.B.; Ramazi, S.; Atashi, A.; Eslamifar, Z. Thrombosis in COVID-19 infection: Role of platelet activation-mediated immunity. Thromb. J. 2021, 19, 59. [Google Scholar] [CrossRef]
- Tibbles, H.E.; Vassilev, A.; Wendorf, H.; Schonhoff, D.; Zhu, D.; Lorenz, D.; Waurzyniak, B.; Liu, X.P.; Uckun, F.M. Role of a JAK3-dependent biochemical signaling pathway in platelet activation and aggregation. J. Biol. Chem. 2001, 276, 17815–17822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets Can Associate with SARS-CoV-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Campbell, R.A.; Boilard, E.; Rondina, M.T. Is there a role for the ACE2 receptor in SARS-CoV-2 interactions with platelets? J. Thromb. Haemost. 2021, 19, 46–50. [Google Scholar] [CrossRef]
- Lee, H.K.; Jung, O.; Hennighausen, L. Activation of Interferon-Stimulated Transcriptomes and ACE2 Isoforms in Human Airway Epithelium Is Curbed by Janus Kinase Inhibitors. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Luo, J.; Lu, S.; Yu, M.; Zhu, L.; Zhu, C.; Li, C.; Fang, J.; Zhu, X.; Wang, X. The potential involvement of JAK-STAT signaling pathway in the COVID-19 infection assisted by ACE2. Gene 2021, 768, 145325. [Google Scholar] [CrossRef]
- Chen, D.Y.; Khan, N.; Close, B.J.; Goel, R.K.; Blum, B.; Tavares, A.H.; Kenney, D.; Conway, H.L.; Ewoldt, J.K.; Chitalia, V.C.; et al. SARS-CoV-2 Disrupts Proximal Elements in the JAK-STAT Pathway. J. Virol. 2021, 95, e0086221. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef] [PubMed]
- Astuti, I.; Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Ciurtin, C.; Scully, M.; Levi, M.; Chambers, R.C. JAK inhibitors in COVID-19: The need for vigilance regarding increased inherent thrombotic risk. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef] [PubMed]
- Cantini, F.; Niccoli, L.; Matarrese, D.; Nicastri, E.; Stobbione, P.; Goletti, D. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J. Infect. 2020, 81, 318–356. [Google Scholar] [CrossRef]
- La Rosee, F.; Bremer, H.C.; Gehrke, I.; Kehr, A.; Hochhaus, A.; Birndt, S.; Fellhauer, M.; Henkes, M.; Kumle, B.; Russo, S.G.; et al. The Janus kinase 1/2 inhibitor ruxolitinib in COVID-19 with severe systemic hyperinflammation. Leukemia 2020, 34, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wei, J.; Zou, L.; Jiang, T.; Wang, G.; Chen, L.; Huang, L.; Meng, F.; Huang, L.; Wang, N.; et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): A multicenter, single-blind, randomized controlled trial. J. Allergy Clin. Immunol. 2020, 146, 137–146.e3. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, P.O.; Quirk, D.; Furtado, R.H.; Maia, L.N.; Saraiva, J.F.; Antunes, M.O.; Kalil Filho, R.; Junior, V.M.; Soeiro, A.M.; Tognon, A.P.; et al. Tofacitinib in Patients Hospitalized with COVID-19 Pneumonia. N. Engl. J. Med. 2021, 385, 406–415. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Inhibitor Used/JAK Selectivity | Approval for Other Diseases | End Point/Treatment Duration/Site | Clinical Trial or Reference Number | Outcome |
|---|---|---|---|---|
| baricitinib/JAK1 or JAK2 | FDA- and EU-approved for rheumatoid arthritis, EU-approved for atopic dermatitis | COVID-19 recovery time/14 days/USA | clinical trial number: NCT04401579 | baricitinib with remdesivir was superior to remdesivir alone in reducing recovery time |
| reduced mortality in moderate to severe COVID-19 pneumonia/14 days/Europe | reference: [39] | baricitinib administration reduced mortality by over 71% | ||
| recovery time in mild COVID-19 (not requiring mechanical ventilation)/28 days/USA | clinical trial number: NCT04421027 (https://investor.lilly.com/news-releases/news-release-details/lilly-and-incyte-announce-results-phase-3-cov-barrier-study (accessed on 30 December 2021)) | 38% reduction in mortality by day 28 in patients treated with baricitinib in addition to corticosteroids and remdesivir | ||
| ruxolitinib/JAK1 or JAK2 | FDA- and EU-approved for myelofibrosis and polycythemia vera, FDA-approved for graft-versus-host disease | chest CT improvement and mortality/14 days/China | reference: [77] | chest CT improvement within 14 days of treatment |
| multi-organ failure/14 days/USA | vlinical trial number: NCT04348071 | less incidence of organ failure upon treatment, leading to an ongoing phase III clinical trial (NCT04362137) | ||
| tofacitinib/JAK1 or JAK3 | FDA- and EU-approved for rheumatoid arthritis, psoriatic arthritis, and ulcerative colitis | respiratory failure and death in patients hospitalized with COVID-19 pneumonia/28 days/USA | vlinical trial number: NCT04469114 | lower risk of death or respiratory failure as compared to placebo-treated patients |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravid, J.D.; Leiva, O.; Chitalia, V.C. Janus Kinase Signaling Pathway and Its Role in COVID-19 Inflammatory, Vascular, and Thrombotic Manifestations. Cells 2022, 11, 306. https://doi.org/10.3390/cells11020306
Ravid JD, Leiva O, Chitalia VC. Janus Kinase Signaling Pathway and Its Role in COVID-19 Inflammatory, Vascular, and Thrombotic Manifestations. Cells. 2022; 11(2):306. https://doi.org/10.3390/cells11020306
Chicago/Turabian StyleRavid, Jonathan D., Orly Leiva, and Vipul C. Chitalia. 2022. "Janus Kinase Signaling Pathway and Its Role in COVID-19 Inflammatory, Vascular, and Thrombotic Manifestations" Cells 11, no. 2: 306. https://doi.org/10.3390/cells11020306
APA StyleRavid, J. D., Leiva, O., & Chitalia, V. C. (2022). Janus Kinase Signaling Pathway and Its Role in COVID-19 Inflammatory, Vascular, and Thrombotic Manifestations. Cells, 11(2), 306. https://doi.org/10.3390/cells11020306