
Effects of TP53 Mutations and miRs on Immune Responses in the Tumor Microenvironment Important in Pancreatic Cancer Progression

 , ,
, ,  , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction-Overview of Genes Frequently Mutated in PDAC
2. Interactions of TP53 with the Immune System and Fibroblasts in the PDAC Microenvironment
3. Interactions between Stroma and TP53 and Their Regulation of miRs, LncRNAs and CircRNAs in the PDAC Microenvironment
4. Altered Expression of Growth Factors, Their Receptors and Downstream Signaling Pathways in the PDAC Microenvironment
5. Roles of Hypoxia in the Induction of HIFs, TP53, miRs and LncRNAs in the PDAC Microenvironment
6. TP53 as a Regulator of Metabolism in the PDAC Microenvironment
7. Interactions between TP53 and the Stress-Inducible NUPR1 Oncoprotein
8. Possibility of Treatment of PDAC with Small Molecule Signal Transduction Inhibitors
9. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, H.; Zhang, B.; Li, H. The roles of frequently mutated genes of pancreatic cancer in regulation of tumor microenvironment. Technol. Cancer Res. Treat. 2020, 19, 1533033820920969. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.W.; Jamieson, N.B.; Evans, T.R.; McKay, C.J.; Sansom, O.J.; Morton, J.P.; Carter, C.R. Exploiting inflammation for therapeutic gain in pancreatic cancer. Br. J. Cancer 2013, 108, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Furukawa, S.; Hashimoto, A.; Tsutaho, A.; Fukao, A.; Sakamura, Y.; Parajuli, G.; Onodera, Y.; Otsuka, Y.; Handa, H.; et al. ARF6 and AMAP1 are major targets of KRAS and TP53 mutations to promote invasion, PD-L1 dynamics, and immune evasion of pancreatic cancer. Proc. Nat. Acad. Sci. USA 2019, 116, 17450–17459. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 2017, 66, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Zdanov, S.; Mandapathil, M.; Abu Eid, R.; Adamson-Fadeyi, S.; Wilson, W.; Qian, J.; Carnie, A.; Tarasova, N.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Mutant KRAS conversion of conventional T cells into regulatory T cells. Cancer Immunol. Res. 2016, 4, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef] [Green Version]
- Hafezi, S.; Saber-Ayad, M.; Abdel-Rahman, W.M. Highlights on the role of KRAS mutations in reshaping the microenvironment of pancreatic adenocarcinoma. Int. J. Mol. Sci. 2021, 22, 10219. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Guo, G. Immunomodulatory function of the tumor suppressor p53 in host immune response and the tumor microenvironment. Int. J. Mol. Sci. 2016, 17, 1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparini, C.; Tommasini, A.; Zauli, G. The MDM2 inhibitor Nutlin-3 modulates dendritic cell-induced T cell proliferation. Hum. Immunol. 2012, 73, 342–345. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Tsujii, M.; Kodama, T.; Akasaka, T.; Kondo, J.; Hikita, H.; Inoue, T.; Tsujii, Y.; Maekawa, A.; Yoshii, S.; et al. p53 functional deficiency in human colon cancer cells promotes fibroblast-mediated angiogenesis and tumor growth. Carcinogenesis 2016, 37, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Cremin, C.; Howard, S.; Le, L.; Karsan, A.; Schaeffer, D.F.; Renouf, D.; Schrader, K.A. CDKN2A founder mutation in pancreatic ductal adenocarcinoma patients without cutaneous features of familial atypical multiple mole melanoma (FAMMM) syndrome. Hered. Cancer Clin. Pract. 2018, 16, 7. [Google Scholar] [CrossRef] [Green Version]
- Wartenberg, M.; Cibin, S.; Zlobec, I.; Vassella, E.; Eppenberger-Castori, S.; Terracciano, L.; Eichmann, M.D.; Worni, M.; Gloor, B.; Perren, A.; et al. Integrated genomic and immunophenotypic classification of pancreatic cancer reveals three distinct subtypes with prognostic/predictive significance. Clin. Cancer Res. 2018, 24, 4444–4454. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Bio. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, A.A.; Vimalachandran, D.; Thompson, C.C.; Jenkins, R.E.; Nedjadi, T.; Shekouh, A.; Campbell, F.; Dodson, A.; Prime, W.; Crnogorac-Jurcevic, T.; et al. The expression of S100A8 in pancreatic cancer-associated monocytes is associated with the Smad4 status of pancreatic cancer cells. Proteomics 2007, 7, 1929–1940. [Google Scholar] [CrossRef]
- Nedjadi, T.; Evans, A.; Sheikh, A.; Barerra, L.; Al-Ghamdi, S.; Oldfield, L.; Greenhalf, W.; Neoptolemos, J.P.; Costello, E. S100A8 and S100A9 proteins form part of a paracrine feedback loop between pancreatic cancer cells and monocytes. BMC Cancer 2018, 18, 1255. [Google Scholar] [CrossRef]
- Zheng, S.J.; Lamhamedi-Cherradi, S.E.; Wang, P.; Xu, L.; Chen, Y.H. Tumor suppressor p53 inhibits autoimmune inflammation and macrophage function. Diabetes 2005, 54, 1423–1428. [Google Scholar] [CrossRef] [Green Version]
- Okuda, Y.; Okuda, M.; Bernard, C.C. Regulatory role of p53 in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2003, 135, 29–37. [Google Scholar] [CrossRef]
- Park, J.S.; Lim, M.A.; Cho, M.L.; Ryu, J.G.; Moon, Y.M.; Jhun, J.Y.; Byun, J.K.; Kim, E.K.; Hwang, S.Y.; Ju, J.H.; et al. p53 controls autoimmune arthritis via STAT-mediated regulation of the Th17 cell/Treg cell balance in mice. Arthritis Rheumatol. 2013, 65, 949–959. [Google Scholar] [CrossRef]
- Kawashima, H.; Takatori, H.; Suzuki, K.; Iwata, A.; Yokota, M.; Suto, A.; Minamino, T.; Hirose, K.; Nakajima, H. Tumor suppressor p53 inhibits systemic autoimmune diseases by inducing regulatory T cells. J. Immunol. 2013, 191, 3614–3623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louault, K.; Li, R.R.; DeClerck, Y.A. Cancer-associated fibroblasts: Understanding their heterogeneity. Cancers 2020, 12, 3108. [Google Scholar] [CrossRef]
- Addadi, Y.; Moskovits, N.; Granot, D.; Lozano, G.; Carmi, Y.; Apte, R.N.; Neeman, M.; Oren, M. p53 status in stromal fibroblasts modulates tumor growth in an SDF1-dependent manner. Cancer Res. 2010, 70, 9650–9658. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, Z.; Li, N.; Li, Y.; Chang, A.; Zhao, T.; Wang, X.; Wang, H.; Gao, S.; Yang, S.; et al. Interleukin 35 expression correlates with microvessel density in pancreatic ductal adenocarcinoma, recruits monocytes, and promotes growth and angiogenesis of xenograft tumors in mice. Gastroenterology 2018, 154, 675–688. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bindea, G.; Kirilovsky, A.; Angell, H.K.; Obenauf, A.C.; Tosolini, M.; Church, S.E.; Maby, P.; Vasaturo, A.; Angelova, M.; et al. The tumor microenvironment and immunoscore are critical determinants of dissemination to distant metastasis. Sci. Transl. Med. 2016, 8, 327ra26. [Google Scholar] [CrossRef]
- Jiang, B.; Zhou, L.; Lu, J.; Wang, Y.; Liu, C.; You, L.; Guo, J. Stroma-targeting therapy in pancreatic cancer: One coin with two sides? Front. Oncol. 2020, 10, 576399. [Google Scholar] [CrossRef]
- Whittle, M.C.; Hingorani, S.R. Fibroblasts in pancreatic ductal adenocarcinoma: Biological mechanisms and therapeutic targets. Gastroenterology 2019, 156, 2085–2096. [Google Scholar] [CrossRef]
- Srivastava, A.; Doppalapudi, S.K.; Patel, H.V.; Srinivasan, R.; Singer, E.A. The roaring 2020s: A new decade of systemic therapy for renal cell carcinoma. Cur. Opin. Oncol. 2022, 34, 234–242. [Google Scholar] [CrossRef]
- Garcia, C.J.; Huang, Y.; Fuentes, N.R.; Turner, M.C.; Monberg, M.E.; Lin, D.; Nguyen, N.D.; Fujimoto, T.N.; Zhao, J.; Lee, J.J.; et al. Stromal HIF2 regulates immune suppression in the pancreatic cancer microenvironment. Gastroenterology 2022, 162, 2018–2031. [Google Scholar] [CrossRef]
- Kwon, J.J.; Nabinger, S.C.; Vega, Z.; Sahu, S.S.; Alluri, R.K.; Abdul-Sater, Z.; Yu, Z.; Gore, J.; Nalepa, G.; Saxena, R.; et al. Pathophysiological role of microRNA-29 in pancreatic cancer stroma. Sci. Rep. 2015, 5, 11450. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, T.; Di Molfetta, D.; Greco, M.R.; Koltai, T.; Alfarouk, K.O.; Reshkin, S.J.; Cardone, R.A. Tumor microenvironment features and chemoresistance in pancreatic ductal adenocarcinoma: Insights into targeting physicochemical barriers and metabolism as therapeutic approaches. Cancers 2021, 13, 6135. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, W.; Chen, S.; Lin, Y. miR-29a sensitizes the response of glioma cells to temozolomide by modulating the P53/MDM2 feedback loop. Cell. Mol. Biol. Lett. 2021, 26, 21. [Google Scholar] [CrossRef]
- Sempere, L.F.; Powell, K.; Rana, J.; Brock, A.A.; Schmittgen, T.D. Role of non-coding RNAs in tumor progression and metastasis in pancreatic cancer. Cancer Metast. Rev. 2021, 40, 761–776. [Google Scholar] [CrossRef]
- Zheng, S.; Hu, C.; Lin, H.; Li, G.; Xia, R.; Zhang, X.; Su, D.; Li, Z.; Zhou, Q.; Chen, R. circCUL2 induces an inflammatory CAF phenotype in pancreatic ductal adenocarcinoma via the activation of the MyD88-dependent NF-κB signaling pathway. J. Exp. Clin. Cancer Res. CR 2022, 41, 71. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Dey, S.; Liu, S.; Factora, T.D.; Taleb, S.; Riverahernandez, P.; Udari, L.; Zhong, X.; Wan, J.; Kota, J. Global targetome analysis reveals critical role of miR-29a in pancreatic cell mediated regulation of PDAC tumor microenvironment. BMC Cancer 2020, 20, 651. [Google Scholar] [CrossRef]
- Jason, J.; Kwon, J.J.; Factora, T.D.; Dey, S.; Kota, J. A systematic review of miR-29 in cancer. Mol. Ther.–Oncolytics 2019, 12, 173–194. [Google Scholar]
- Awaji, M.; Singh, R.K. Cancer-associated fibroblasts’ functional heterogeneity in pancreatic ductal adenocarcinoma. Cancers 2019, 11, 290. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; He, C.; Hua, X.; Kan, A.; Mao, Y.; Sun, S.; Duan, F.; Wang, J.; Huang, P.; Li, S. Oxidative stress induces monocyte-to-myofibroblast transdifferentiation through p38 in pancreatic ductal adenocarcinoma. Clin. Trans. Med. 2020, 10, e41. [Google Scholar] [CrossRef]
- Han, S.; Gonzalo, D.H.; Feely, M.; Rinaldi, C.; Belsare, S.; Zhai, H.; Kalra, K.; Gerber, M.H.; Forsmark, C.E.; Hughes, S.J. Stroma-derived extracellular vesicles deliver tumor-suppressive miRNAs to pancreatic cancer cells. Oncotarget 2017, 9, 5764–5777. [Google Scholar] [CrossRef] [Green Version]
- Rokavec, M.; Li, H.; Jiang, L.; Hermeking, H. The p53/microRNA connection in gastrointestinal cancer. Clin. Exp. Gastroenterol. 2014, 7, 395–413. [Google Scholar]
- Khan, S.; Ebeling, M.C.; Zaman, M.S.; Sikander, M.; Yallapu, M.M.; Chauhan, N.; Yacoubian, A.M.; Behrman, S.W.; Zafar, N.; Kumar, D.; et al. MicroRNA-145 targets MUC13 and suppresses growth and invasion of pancreatic cancer. Oncotarget 2014, 5, 7599–7609. [Google Scholar] [CrossRef]
- Kumari, S.; Khan, S.; Gupta, S.C.; Kashyap, V.K.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. MUC13 contributes to rewiring of glucose metabolism in pancreatic cancer. Oncogenesis 2018, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Schipper, J.; Westerhuis, J.J.; Beddows, I.; Madaj, Z.; Monsma, D.; Hostetter, G.; Kiupel, M.; Conejo-Garcia, J.R.; Sempere, L.F. Loss of microRNA-21 leads to profound stromal remodeling and short survival in K-Ras-driven mouse models of pancreatic cancer. Int. J. Cancer 2020, 147, 2265–2278. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, J.; Li, W.; Zhang, C. Micro-RNA-21 regulates cancer-associated fibroblast-mediated drug resistance in pancreatic cancer. Oncol. Res. 2018, 26, 827–835. [Google Scholar] [CrossRef]
- Wartenberg, M.; Centeno, I.; Haemmig, S.; Vassella, E.; Zlobec, I.; Galván, J.A.; Neuenschwander, M.; Schlup, C.; Gloor, B.; Lugli, A.; et al. PTEN alterations of the stromal cells characterise an aggressive subpopulation of pancreatic cancer with enhanced metastatic potential. Eur. J. Cancer 1990, 65, 80–90. [Google Scholar] [CrossRef]
- Yang, J.; Qiu, B.; Li, X.; Zhang, H.; Liu, W. p53-p66shc/miR-21-Sod2 signaling is critical for the inhibitory effect of betulinic acid on hepatocellular carcinoma. Toxicol. Lett. 2015, 238, 1–10. [Google Scholar] [CrossRef]
- Wang, C.; Li, X.; Zhang, L.; Chen, Y.; Dong, R.; Zhang, J.; Zhao, J.; Guo, X.; Yang, G.; Li, Y.; et al. miR-194-5p down-regulates tumor cell PD-L1 expression and promotes anti-tumor immunity in pancreatic cancer. Int. Immunopharmacol. 2021, 97, 107822. [Google Scholar] [CrossRef]
- Xi, Q.; Chen, Y.; Yang, G.Z.; Zhang, J.Y.; Zhang, L.J.; Guo, X.D.; Zhao, J.Y.; Xue, Z.Y.; Li, Y.; Zhang, R. miR-128 regulates tumor cell CD47 expression and promotes anti-tumor immunity in pancreatic cancer. Front. Immunol. 2020, 11, 890. [Google Scholar] [CrossRef]
- Yin, Z.; Ma, T.; Huang, B.; Lin, L.; Zhou, Y.; Yan, J.; Zou, Y.; Chen, S. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-β signaling pathway. J. Exp. Clin. Cancer Res. CR 2019, 38, 310. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ishak Gabra, M.B.; Hanse, E.A.; Lowman, X.H.; Tran, T.Q.; Li, H.; Milman, N.; Liu, J.; Reid, M.A.; Locasale, J.W.; et al. MiR-135 suppresses glycolysis and promotes pancreatic cancer cell adaptation to metabolic stress by targeting phosphofructokinase-1. Nat. Commun. 2019, 10, 809. [Google Scholar] [CrossRef] [Green Version]
- Binenbaum, Y.; Fridman, E.; Yaari, Z.; Milman, N.; Schroeder, A.; Ben David, G.; Shlomi, T.; Gil, Z. Transfer of miRNA in macrophage-derived exosomes induces drug resistance in pancreatic adenocarcinoma. Cancer Res. 2018, 78, 5287–5299. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Shen, W.W.; Cao, X.M.; Ding, W.Y.; Yan, L.P.; Gao, L.J.; Li, X.L.; Zhong, T.Y. Novel mechanism of miRNA-365-regulated trophoblast apoptosis in recurrent miscarriage. J. Cell. Mol. Med. 2017, 21, 2412–2425. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Gonzalo, D.H.; Feely, M.; Delitto, D.; Behrns, K.E.; Beveridge, M.; Zhang, D.; Thomas, R.; Trevino, J.G.; Schmittgen, T.D.; et al. The pancreatic tumor microenvironment drives changes in miRNA expression that promote cytokine production and inhibit migration by the tumor associated stroma. Oncotarget 2016, 8, 54054–54067. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, E.; Gandellini, P. Unveiling the ups and downs of miR-205 in physiology and cancer: Transcriptional and post-transcriptional mechanisms. Cell Death Dis. 2020, 11, 980. [Google Scholar] [CrossRef]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.S.; et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef]
- Singh, S.K.; Chen, N.M.; Hessmann, E.; Siveke, J.; Lahmann, M.; Singh, G.; Voelker, N.; Vogt, S.; Esposito, I.; Schmidt, A.; et al. Antithetical NFATc1-Sox2 and p53-miR200 signaling networks govern pancreatic cancer cell plasticity. EMBO J. 2015, 34, 517–530. [Google Scholar] [CrossRef]
- Long, L.M.; Zhan, J.K.; Wang, H.Q.; Li, S.; Chen, Y.Y.; Liu, Y.S. The clinical significance of miR-34a in pancreatic ductal carcinoma and associated molecular and cellular mechanisms. Pathobiol. J. Immunopatho. Mol. Cellul. Biol. 2017, 84, 38–48. [Google Scholar] [CrossRef]
- Si, L.; Xu, L.; Yin, L.; Qi, Y.; Han, X.; Xu, Y.; Zhao, Y.; Liu, K.; Peng, J. Potent effects of dioscin against pancreatic cancer via miR-149-3P-mediated inhibition of the Akt1 signalling pathway. Br. J. Pharmacol. 2017, 174, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Prives, C. Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene 2007, 26, 2220–2225. [Google Scholar] [CrossRef] [Green Version]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P., 4th; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [Green Version]
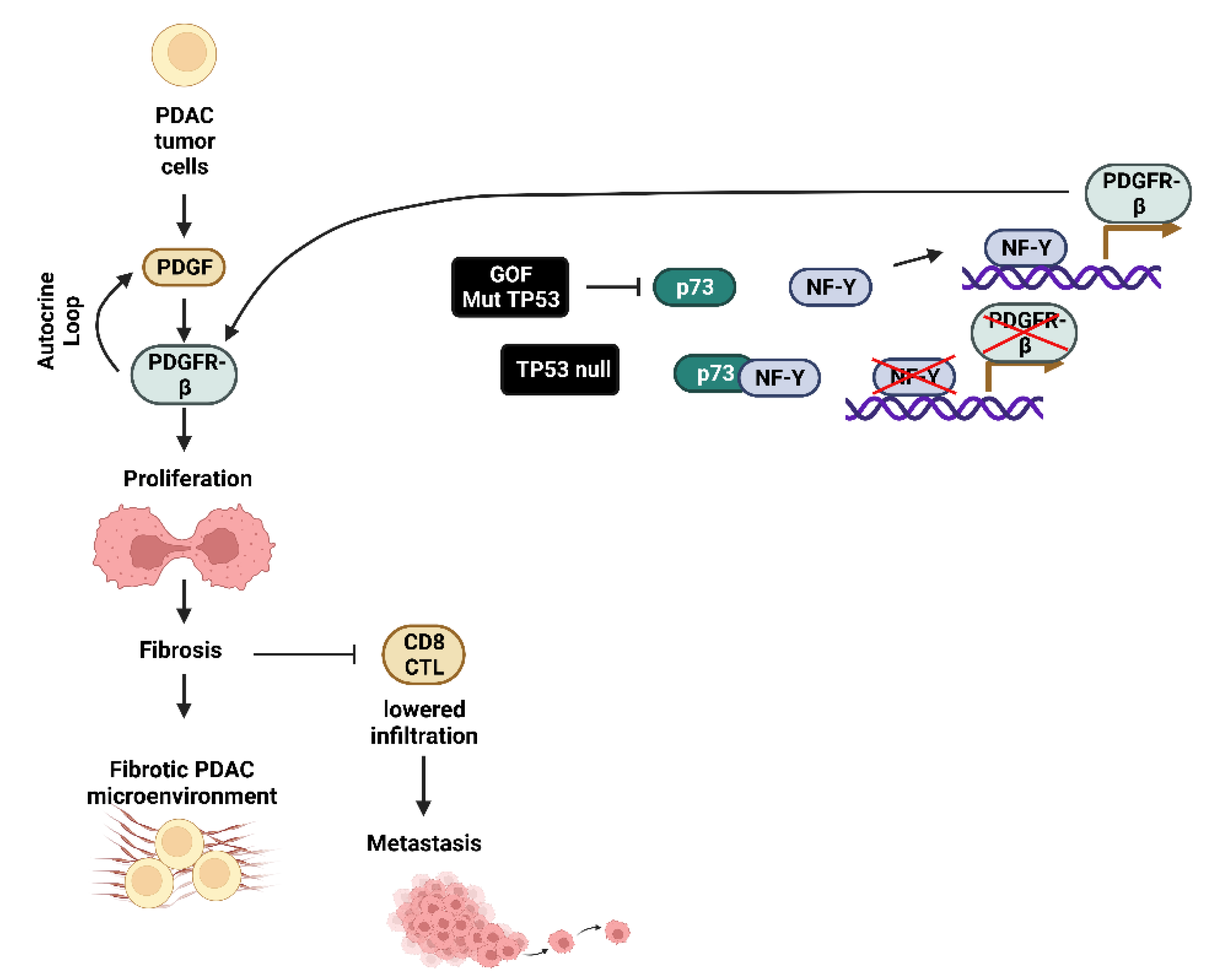
- Maddalena, M.; Mallel, G.; Nataraj, N.B.; Shreberk-Shaked, M.; Hassin, O.; Mukherjee, S.; Arandkar, S.; Rotkopf, R.; Kapsack, A.; Lambiase, G.; et al. TP53 missense mutations in PDAC are associated with enhanced fibrosis and an immunosuppressive microenvironment. Proc. Nat. Acad. Sci. USA 2021, 118, e2025631118. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic ductal adenocarcinoma: Current and evolving therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Prabhu, L.; Mundade, R.; Korc, M.; Loehrer, P.J.; Lu, T. Critical role of NF-κB in pancreatic cancer. Oncotarget 2014, 5, 10969–10975. [Google Scholar] [CrossRef] [Green Version]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar]
- Wang, S.; Zheng, Y.; Yang, F.; Zhu, L.; Zhu, X.Q.; Wang, Z.F.; Wu, X.L.; Zhou, C.H.; Yan, J.Y.; Hu, B.Y. The molecular biology of pancreatic adenocarcinoma: Translational challenges and clinical perspectives. Signal. Transduct. Target. Ther. 2021, 6, 249m. [Google Scholar] [CrossRef]
- Renaldi, K.; Simadibrata, M.; Rahadiani, N.; Handjari, D.R.; William, A.; Sinuraya, F.; Makmun, D. Prognostic value of COX-2, NF-κB, and Sp1 tissue expressions in pancreatic ductal adenocarcinoma: A systematic review and meta-analysis. Turkish J. Gastroenterol. 2021, 32, 956–970. [Google Scholar] [CrossRef]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nature Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Sun, Q.; Fan, G.; Zhuo, Q.; Dai, W.; Ye, Z.; Ji, S.; Xu, W.; Liu, W.; Hu, Q.; Zhang, Z.; et al. Pin1 promotes pancreatic cancer progression and metastasis by activation of NF-κB-IL-18 feedback loop. Cell Prolif. 2020, 53, e12816. [Google Scholar] [CrossRef] [PubMed]
- Garg, B.; Giri, B.; Modi, S.; Sethi, V.; Castro, I.; Umland, O.; Ban, Y.; Lavania, S.; Dawra, R.; Banerjee, S.; et al. NFκB in pancreatic stellate cells reduces infiltration of tumors by cytotoxic T cells and killing of cancer cells, via up-regulation of CXCL12. Gastroenterology 2018, 155, 880–891.e8. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Activated p53 induces NF-κB DNA binding but suppresses its transcriptional activation. Biochem. Biophys. Res. Commun. 2008, 372, 137–141. [Google Scholar] [CrossRef]
- Ak, P.; Levine, A.J. p53 and NF-κB: Different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010, 24, 3643–3652. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef]
- Schwitalla, S.; Ziegler, P.K.; Horst, D.; Becker, V.; Kerle, I.; Begus-Nahrmann, Y.; Lechel, A.; Rudolph, K.L.; Langer, R.; Slotta-Huspenina, J.; et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 2013, 23, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Denley, S.M.; Jamieson, N.B.; McCall, P.; Oien, K.A.; Morton, J.P.; Carter, C.R.; Edwards, J.; McKay, C.J. Activation of the IL-6R/Jak/stat pathway is associated with a poor outcome in resected pancreatic ductal adenocarcinoma. J. Gastrointest. Surg. 2013, 17, 887–898. [Google Scholar] [CrossRef]
- Li, N.; Grivennikov, S.I.; Karin, M. The unholy trinity: Inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell 2011, 19, 429–431. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.; Wright, K.L.; Ma, Y.; Wright, G.M.; Huang, M.; Irby, R.; Briggs, J.; Karras, J.; Cress, W.D.; Pardoll, D.; et al. Role of Stat3 in regulating p53 expression and function. Mol. Cell. Biol. 2005, 25, 7432–7440. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Tang, M.Y.; Chen, W.; Wang, Z.; Wang, S.L. High JAK2 protein expression predicts a poor prognosis in patients with resectable pancreatic ductal adenocarcinoma. Dis. Markers 2020, 2020, 7656031. [Google Scholar] [CrossRef]
- Doi, T.; Ishikawa, T.; Okayama, T.; Oka, K.; Mizushima, K.; Yasuda, T.; Sakamoto, N.; Katada, K.; Kamada, K.; Uchiyama, K.; et al. The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines. Oncol. Rep. 2017, 37, 1545–1554. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Talukder, A.; Savage, N.M.; Singh, N.; Liu, K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. Oncoimmunology 2017, 6, e1291106. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Nemazanyy, I.; White, S.M.; Chen, H.; Nguyen, C.; Graham, G.T.; Saur, D.; Pende, M.; Yi, C. A Yap-Myc-Sox2-p53 regulatory network dictates metabolic homeostasis and differentiation in Kras-driven pancreatic ductal adenocarcinomas. Dev. Cell 2019, 51, 113–128.e9. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhou, C.; Cheng, L.; Yan, B.; Chen, K.; Chen, X.; Zong, L.; Lei, J.; Duan, W.; Xu, Q.; et al. Inhibiting YAP expression suppresses pancreatic cancer progression by disrupting tumor-stromal interactions. J. Exp. Clin. Cancer Res. CR 2018, 37, 69. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Gao, M.; Nipper, M.; Deng, J.; Sharkey, F.E.; Johnson, R.L.; Crawford, H.C.; Chen, Y.; Wang, P. Activation of the intrinsic fibroinflammatory program in adult pancreatic acinar cells triggered by Hippo signaling disruption. PLoS Biol. 2019, 17, e3000418. [Google Scholar] [CrossRef]
- Raj, N.; Bam, R. Reciprocal crosstalk between YAP1/Hippo pathway and the p53 family proteins: Mechanisms and outcomes in cancer. Front. Cell Dev. Biol. 2019, 7, 159. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [Green Version]
- Ram Makena, M.; Gatla, H.; Verlekar, D.; Sukhavasi, S.; Pandey, M.K.; Pramanik, K.C. Wnt/β-catenin signaling: The culprit in pancreatic carcinogenesis and therapeutic resistance. Int. J. Mol. Sci. 2019, 20, 4242. [Google Scholar] [CrossRef] [Green Version]
- Palamaris, K.; Felekouras, E.; Sakellariou, S. Epithelial to mesenchymal transition: Key regulator of pancreatic ductal adenocarcinoma progression and chemoresistance. Cancers 2021, 13, 5532. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, H.S.; Kim, N.G.; Lee, I.; Choi, H.S.; Li, X.Y.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Na, J.; et al. p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci. Signal. 2011, 4, ra71. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.P.; Li, X.; Deng, J.; Zhang, Y.; Dai, B.; Allton, K.L.; Hughes, T.G.; Siangco, C.; Augustine, J.J.; Kang, Y.; et al. Oncogenic KRAS recruits an expansive transcriptional network through mutant p53 to drive pancreatic cancer metastasis. Cancer Discov. 2021, 11, 2094–2111. [Google Scholar] [CrossRef]
- Zhang, Q.; Lou, Y.; Zhang, J.; Fu, Q.; Wei, T.; Sun, X.; Chen, Q.; Yang, J.; Bai, X.; Liang, T. Hypoxia-inducible factor-2α promotes tumor progression and has crosstalk with Wnt/β-catenin signaling in pancreatic cancer. Mol. Cancer 2017, 16, 119. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tang, T.; Yang, X.; Qin, P.; Wang, P.; Zhang, H.; Bai, M.; Wu, R.; Li, F. Tumor-derived exosomal long noncoding RNA LINC01133, regulated by Periostin, contributes to pancreatic ductal adenocarcinoma epithelial-mesenchymal transition through the Wnt/β-catenin pathway by silencing AXIN2. Oncogene 2021, 40, 3164–3179. [Google Scholar] [CrossRef]
- Fu, Z.; Chen, C.; Zhou, Q.; Wang, Y.; Zhao, Y.; Zhao, X.; Li, W.; Zheng, S.; Ye, H.; Wang, L.; et al. LncRNA HOTTIP modulates cancer stem cell properties in human pancreatic cancer by regulating HOXA9. Cancer Lett. 2017, 410, 68–81. [Google Scholar] [CrossRef]
- Yue, H.; Liu, L.; Song, Z. miR-212 regulated by HIF-1α promotes the progression of pancreatic cancer. Exp. Ther. Med. 2019, 17, 2359–2365. [Google Scholar] [CrossRef]
- Tiwari, A.; Tashiro, K.; Dixit, A.; Soni, A.; Vogel, K.; Hall, B.; Shafqat, I.; Slaughter, J.; Param, N.; Le, A.; et al. Loss of HIF1A from pancreatic cancer cells increases expression of PPP1R1B and degradation of p53 to promote invasion and metastasis. Gastroenterology 2020, 159, 1882–1897.e5. [Google Scholar] [CrossRef]
- Li, H.; Wang, X.; Wen, C.; Huo, Z.; Wang, W.; Zhan, Q.; Cheng, D.; Chen, H.; Deng, X.; Peng, C.; et al. Long noncoding RNA NORAD, a novel competing endogenous RNA, enhances the hypoxia-induced epithelial-mesenchymal transition to promote metastasis in pancreatic cancer. Mol. Cancer 2017, 16, 169. [Google Scholar] [CrossRef]
- Li, S.; Li, X.; Zhao, H.; Gao, M.; Wang, F.; Li, W. Overexpression of microRNA-125a-3p effectively inhibits the cell growth and invasion of lung cancer cells by regulating the mouse double minute 2 homolog/p53 signaling pathway. Mol. Med. Rep. 2015, 12, 5482–5486. [Google Scholar] [CrossRef]
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.; Le Quesne, J.; Acevedo, J.D.B.; et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E10869–E10878. [Google Scholar] [CrossRef] [Green Version]
- Zampieri, C.; Panatta, E.; Corbo, V.; Mauriello, A.; Melino, G.; Amelio, I. p53 mutations define the chromatin landscape to confer drug tolerance in pancreatic cancer. Mol. Oncol. 2022, 16, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2alpha regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Lv, X.; Yan, Y.; Zhao, Y.; Ma, R.; He, M.; Wei, M. Hypoxia-mediated cancer stem cell resistance and targeted therapy. Biomed. Pharmacother. 2020, 130, 110623. [Google Scholar] [CrossRef] [PubMed]
- Koifman, G.; Aloni-Grinstein, R.; Rotter, V. p53 balances between tissue hierarchy and anarchy. J. Mol. Cell Biol. 2019, 11, 553–563. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Kamat, C.D.; Green, D.E.; Warnke, L.; Thorpe, J.E.; Ceriello, A.; Ihnat, M.A. Mut-p53 facilitates pro-angiogenic, hyperproliferative phenotype in response to chronic relative hypoxia. Cancer Lett. 2007, 249, 209–219. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Wang, J.; Zhang, T.; Xu, D.; Hu, W.; Feng, Z. The interplay between tumor suppressor p53 and hypoxia signaling pathways in cancer. Front. Cell Dev. Biol. 2021, 9, 648808. [Google Scholar] [CrossRef]
- Yu, L.; Wu, M.; Zhu, G.; Xu, Y. Emerging roles of the tumor suppressor p53 in metabolism. Front. Cell Dev. Biol. 2022, 9, 762742. [Google Scholar] [CrossRef]
- Yan, L.; Raj, P.; Yao, W.; Ying, H. Glucose metabolism in pancreatic cancer. Cancers 2019, 11, 1460. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mRNA by hypoxia-inducible factor-1 a. Interaction between H-ras and hypoxia. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Shibaji, T.; Nagao, M.; Ikeda, N.; Kanehiro, H.; Hisanaga, M.; Ko, S.; Fukumoto, A.; Nakajima, Y. Prognostic significance of HIF-1 alpha overexpression in human pancreatic cancer. Anticancer Res. 2003, 23, 4721–4727. [Google Scholar]
- Zhang, L.; Hill, R.P. Hypoxia enhances metastatic efficiency by upregulating Mdm2 in KHT cells and increasing resistance to apoptosis. Cancer Res. 2004, 64, 4180–4189. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Longtine, M.S.; Sadovsky, Y.; Nelson, D.M. Hypoxia downregulates p53 but induces apoptosis and enhances expression of BAD in cultures of human syncytiotrophoblasts. Am. J. Physiol. Cell Physiol. 2010, 299, C968–C976. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, X.; Sejas, D.P.; Bagby, G.C.; Pang, Q. Hypoxia-induced nucleophosmin protects cell death through inhibition of p53. J. Biol. Chem. 2004, 279, 41275–41279. [Google Scholar] [CrossRef] [Green Version]
- Leszczynska, K.B.; Foskolou, I.P.; Abraham, A.G.; Anbalagan, S.; Tellier, C.; Haider, S.; Span, P.N.; O’Neill, E.E.; Buffa, F.M.; Hammond, E.M. Hypoxia-induced p53 modulates both apoptosis and radiosensitivity via AKT. J. Clin. Investig. 2015, 125, 2385–2398. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Nat. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [Green Version]
- Viotti, J.; Duplan, E.; Caillava, C.; Condat, J.; Goiran, T.; Giordano, C.; Marie, Y.; Idbaih, A.; Delattre, J.Y.; Honnorat, J.; et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene 2014, 33, 1764–1775. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A.; et al. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 2017, 8, 1823. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Lotterman, C.D.; Bao, C.; Hruban, R.H.; Karim, B.; Mendell, J.T.; Huso, D.; Lowenstein, C.J. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc. Nat. Acad. Sci. USA 2010, 107, 6334–6339. [Google Scholar] [CrossRef] [Green Version]
- Schito, L.; Semenza, G.L. Hypoxia-inducible factors: Master regulators of cancer progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choueiri, T.K.; Kaelin, W.G. Targeting the HIF2–VEGF axis in renal cell carcinoma. Nat. Med. 2020, 26, 1519–1530. [Google Scholar] [CrossRef]
- de Heer, E.C.; Jalving, M.; Harris, A.L. HIFs, angiogenesis, and metabolism: Elusive enemies in breast cancer. J. Clin. Investig. 2020, 130, 5074–5087. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Xie, C.; Jiang, C. The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 2019, 15, 21–32. [Google Scholar] [CrossRef]
- Chen, X.; Zeh, H.J.; Kang, R.; Kroemer, G.; Tang, D. Cell death in pancreatic cancer: From pathogenesis to therapy. Nature reviews. Gastroenterol. Hepatol. 2021, 18, 804–823. [Google Scholar] [CrossRef] [PubMed]
- Daniel, S.K.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin. Trans. Med. 2019, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [Green Version]
- Chaika, N.V.; Gebregiworgis, T.; Lewallen, M.E.; Purohit, V.; Radhakrishnan, P.; Liu, X.; Zhang, B.; Mehla, K.; Brown, R.B.; Caffrey, T.; et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc. Nat. Acad. Sci. USA 2012, 109, 13787–13792. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Cui, J.; Du, J.; Wei, D.; Jia, Z.; Zhang, J.; Zhu, Z.; Gao, Y.; Xie, K. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin. Cancer Res. 2014, 20, 4370–4380. [Google Scholar] [CrossRef] [Green Version]
- Parmakhtiar, B.; Burger, R.A.; Kim, J.H.; Fruehauf, J.P. HIF inactivation of p53 in ovarian cancer can be reversed by topotecan, restoring cisplatin and paclitaxel sensitivity. Mol. Cancer Res. 2019, 17, 1675–1686. [Google Scholar] [CrossRef]
- Xiao, X.; Huang, X.; Ye, F.; Chen, B.; Song, C.; Wen, J.; Zhang, Z.; Zheng, G.; Tang, H.; Xie, X. The miR-34a-LDHA axis regulates glucose metabolism and tumor growth in breast cancer. Sci. Rep. 2016, 6, 21735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.R.; Roe, J.S.; Lee, J.E.; Cho, E.J.; Youn, H.D. p53 regulates glucose metabolism by miR-34a. Biochem. Biophys. Res. Commun. 2013, 437, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, N.B.; Morran, D.C.; Morton, J.P.; Ali, A.; Dickson, E.J.; Carter, C.R.; Sansom, O.J.; Evans, T.R.; McKay, C.J.; Oien, K.A. MicroRNA molecular profiles associated with diagnosis, clinicopathologic criteria, and overall survival in patients with resectable pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2012, 18, 534–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Zhang, L.F.; Zhang, H.W.; Hu, S.; Lu, M.H.; Liang, S.; Li, B.; Li, Y.; Li, D.; Wang, E.D.; et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 2012, 31, 1985–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, O.A.; Chivukula, R.R.; Mullendore, M.; Wentzel, E.A.; Feldmann, G.; Lee, K.H.; Liu, S.; Leach, S.D.; Maitra, A.; Mendell, J.T. Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev. 2010, 24, 2754–2759. [Google Scholar] [CrossRef] [Green Version]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the MVA pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P., 4th; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsmotti, A.M.; Culp-Hill, R.; et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell 2019, 176, 564–580.e19. [Google Scholar] [CrossRef] [Green Version]
- Guerra, B.; Recio, C.; Aranda-Tavío, H.; Guerra-Rodríguez, M.; Gatrcía-Castellano, J.M.; Fernández-Pérez, L. The mevalonate pathway, a metabolic target in cancer therapy. Front. Oncol. 2021, 11, 626971. [Google Scholar] [CrossRef]
- Kaymak, I.; Maier, C.R.; Schmitz, W.; Campbell, A.D.; Dankworth, B.; Ade, C.P.; Walz, S.; Paauwe, M.; Kalogirou, C.; Marouf, H.; et al. Mevalonate pathway provides ubiquinone to maintain pyrimidine synthesis and survival in p53-deficient cancer cells exposed to metabolic stress. Cancer Res. 2020, 80, 189–203. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, C.; Wang, J.; Hu, W.; Feng, Z. The regulation of ferroptosis by tumor suppressor p53 and its pathway. Int. J. Mol. Sci. 2020, 21, 8387. [Google Scholar] [CrossRef]
- Li, J.; Lama, R.; Galster, S.L.; Inigo, J.R.; Wu, J.; Chandra, D.; Chemler, S.R.; Wang, X. Small molecule MMRi62 induces ferroptosis and inhibits metastasis in pancreatic cancer via degradation of ferritin heavy chain and mutant p53. Mol. Cancer Ther. 2022, 21, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Birsen, R.; Larrue, C.; Decroocq, J.; Johnson, N.; Guiraud, N.; Gotanegre, M.; Cantero-Aguilar, L.; Grignano, E.; Huynh, T.; Fontenay, M.; et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 2022, 107, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Sun, L.; Hu, L.; Cogdell, D.; Lu, L.; Gao, C.; Tian, W.; Zhang, Z.; Kang, Y.; Fleming, J.B.; Zhang, W. MIR506 induces autophagy-related cell death in pancreatic cancer cells by targeting the STAT3 pathway. Autophagy 2017, 13, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhang, Z.; Lu, Y.; Song, K.; Liu, X.; Xia, F.; Sun, W. Downregulation of ULK1 by microRNA-372 inhibits the survival of human pancreatic adenocarcinoma cells. Cancer Sci. 2017, 108, 1811–1819. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Chen, W.; Peng, J.; Li, Y.; Zhuang, Y.; Zhu, Z.; Shao, C.; Yang, W.; Yao, H.; Zhang, S. LncRNA PVT1 triggers Cyto-protective autophagy and promotes pancreatic ductal adenocarcinoma development via the miR-20a-5p/ULK1 Axis. Mol. Cancer 2018, 17, 98. [Google Scholar] [CrossRef]
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Kroemer, G.; Klionsky, D.J.; Zeh, H.J.; Kang, R.; Wang, J.; Tang, D. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 2020, 16, 2069–2083. [Google Scholar] [CrossRef]
- Huang, C.; Santofimia-Castaño, P.; Iovanna, J. NUPR1: A critical regulator of the antioxidant system. Cancers 2021, 13, 3670. [Google Scholar] [CrossRef]
- Peuget, S.; Bonacci, T.; Soubeyran, P.; Iovanna, J.; Dusetti, N.J. Oxidative stress-induced p53 activity is enhanced by a redox-sensitive TP53INP1 SUMOylation. Cell Death Differ. 2014, 21, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Labuschagne, C.F.; Zani, F.; Vousden, K.H. Control of metabolism by p53—Cancer and beyond. Biochim. Biophys. Rev. Cancer 2018, 1870, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Li, A.X.; Sanders, A.J.; Ye, L.; Frewer, K.; Hargest, R.; Jiang, W.G. NUPR1 and its potential role in cancer and pathological conditions. Int. J. Oncol. 2021, 58, 21. [Google Scholar] [CrossRef] [PubMed]
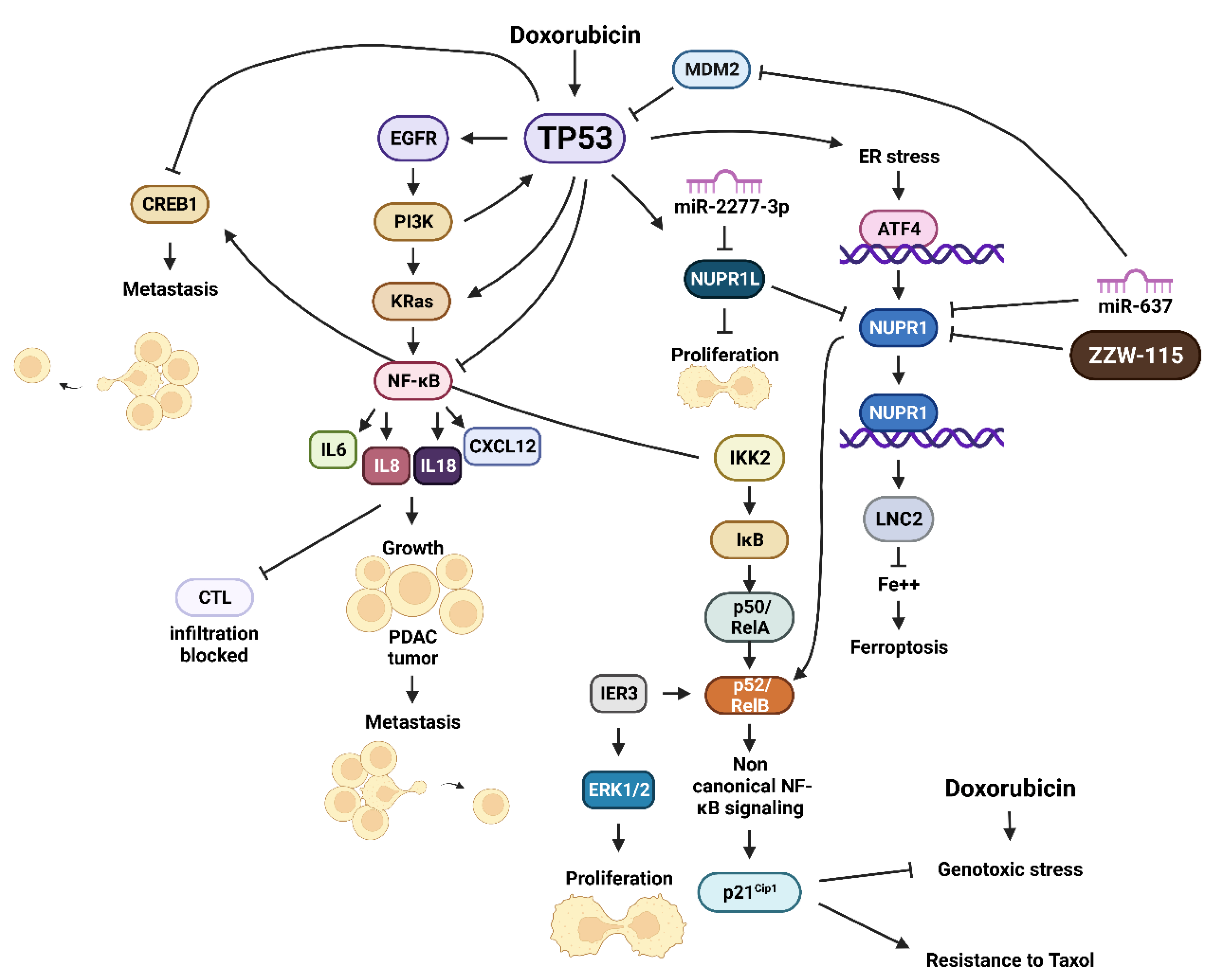
- Clark, D.W.; Mitra, A.; Fillmore, R.A.; Jiang, W.G.; Samant, R.S.; Fodstad, O.; Shevde, L.A. NUPR1 interacts with p53, transcriptionally regulates p21 and rescues breast epithelial cells from doxorubicin-induced genotoxic stress. Cur. Cancer Drug Targets 2008, 8, 421–430. [Google Scholar] [CrossRef]
- Hamidi, T.; Algul, H.; Cano, C.E.; Sandi, M.J.; Molejon, M.I.; Riemann, M.; Calvo, E.L.; Lomberk, G.; Dagorn, J.C.; Weih, F.; et al. Nuclear protein 1 promotes pancreatic cancer development and protects cells from stress by inhibiting apoptosis. J. Clin. Investig. 2012, 122, 2092–2103. [Google Scholar] [CrossRef] [Green Version]
- Cáceres, M.; Quesada, R.; Iglesias, M.; Real, F.X.; Villamonte, M.; de Villarreal, J.M.; Pérez, M.; Andaluz, A.; Moll, X.; Berjano, E.; et al. Pancreatic duct ligation reduces premalignant pancreatic lesions in a Kras model of pancreatic adenocarcinoma in mice. Sci. Rep. 2020, 10, 18344. [Google Scholar] [CrossRef]
- Grasso, D.; Garcia, M.N.; Hamidi, T.; Cano, C.; Calvo, E.; Lomberk, G.; Urrutia, R.; Iovanna, J.L. Genetic inactivation of the pancreatic-inducible gene NUPR1 impairs PanIN formation by modulating KRasG12D-induced senescence. Cell Death Differ. 2014, 21, 1633–1641. [Google Scholar] [CrossRef] [Green Version]
- Grasso, D.; Bintz, J.; Lomberk, G.; Molejon, M.I.; Loncle, C.; Garcia, M.N.; Lopez, M.B.; Urrutia, R.; Iovanna, J.L. Pivotal role of the chromatin protein Nupr1 in Kras-induced senescence and transformation. Sci. Rep. 2015, 5, 17549. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.N.; Grasso, D.; Lopez-Millan, M.B.; Hamidi, T.; Loncle, C.; Tomasini, R.; Lomberk, G.; Porteu, F.; Urrutia, R.; Iovanna, J.L. IER3 supports KRASG12D-dependent pancreatic cancer development by sustaining ERK1/2 phosphorylation. J. Clin. Investig. 2014, 124, 4709–4722. [Google Scholar] [CrossRef] [Green Version]
- Cano, C.E.; Hamidi, T.; Garcia, M.N.; Grasso, D.; Loncle, C.; Garcia, S.; Calvo, E.; Lomberk, G.; Dusetti, N.; Bartholin, L.; et al. Genetic inactivation of NUPR1 acts as a dominant suppressor event in two-hit model of pancreatic carcinogenesis. Gut 2014, 63, 984–995. [Google Scholar] [CrossRef]
- Huang, C.; Iovanna, J.; Santofimia-Castaño, P. Targeting fibrosis: The bridge that connects pancreatitis and pancreatic cancer. Int. J. Mol. Sci. 2021, 22, 4970. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Song, X.; Kuang, F.; Zhang, Q.; Xie, Y.; Kang, R.; Kroemer, G.; Tang, D. NUPR1 is a critical repressor of ferroptosis. Nat. Commun. 2021, 12, 647. [Google Scholar] [CrossRef] [PubMed]
- Santofimia-Castaño, P.; Xia, Y.; Lan, W.; Zhou, Z.; Huang, C.; Peng, L.; Soubeyran, P.; Velázquez-Campoy, A.; Abián, O.; Rizzuti, B.; et al. Ligand-based design identifies a potent NUPR1 inhibitor exerting anticancer activity via necroptosis. J. Clin. Investig. 2019, 129, 2500–2513. [Google Scholar] [CrossRef] [PubMed]
- Santofimia-Castaño, P.; Xia, Y.; Peng, L.; Velázquez-Campoy, A.; Abián, O.; Lan, W.; Lomberk, G.; Urrutia, R.; Rizzuti, B.; Soubeyran, P.; et al. Targeting the stress-induced protein NUPR1 to treat pancreatic adenocarcinoma. Cells 2019, 8, 1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, W.; Santofimia-Castaño, P.; Swayden, M.; Xia, Y.; Zhou, Z.; Audebert, S.; Camoin, L.; Huang, C.; Peng, L.; Jiménez-Alesanco, A.; et al. ZZW-115-dependent inhibition of NUPR1 nuclear translocation sensitizes cancer cells to genotoxic agents. JCI Insight 2020, 5, e138117. [Google Scholar] [CrossRef]
- Lopez, M.B.; Garcia, M.N.; Grasso, D.; Bintz, J.; Molejon, M.I.; Velez, G.; Lomberk, G.; Neira, J.L.; Urrutia, R.; Iovanna, J. Functional Characterization of Nupr1L, A novel p53-regulated isoform of the high-mobility group (HMG)-related protumoral protein Nupr1. J. Cell. Physiol. 2015, 230, 2936–2950. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Lei, F.; Zeng, Q.; Gao, Z.; Niu, P.; Ning, J.; Li, J.; Zhang, J. Functional passenger-strand miRNAs in exosomes derived from human colon cancer cells and their heterogeneous paracrine effects. Int. J. Biol. Sci. 2020, 16, 1044–1058. [Google Scholar] [CrossRef]
- Emma, M.R.; Iovanna, J.L.; Bachvarov, D.; Puleio, R.; Loria, G.R.; Augello, G.; Candido, S.; Libra, M.; Gulino, A.; Cancila, V.; et al. NUPR1, a new target in liver cancer: Implication in controlling cell growth, migration, invasion and sorafenib resistance. Cell Death Dis. 2016, 7, e2269. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, Y.; Wang, X.; Zhou, X.; Li, S.; Wang, M.; Ren, J. Nuclear protein 1 knockdown inhibits proliferation and migration of HepG2 cell line. Chin. J. Cell. Mol. Immunol. 2015, 31, 782–786. [Google Scholar]
- Lan, W.; Santofimia-Castaño, P.; Xia, Y.; Zhou, Z.; Huang, C.; Fraunhoffer, N.; Barea, D.; Cervello, M.; Giannitrapani, L.; Montalto, G.; et al. Targeting NUPR1 with the small compound ZZW-115 is an efficient strategy to treat hepatocellular carcinoma. Cancer Lett. 2020, 486, 8–17. [Google Scholar] [CrossRef]
- Augello, G.; Emma, M.R.; Azzolina, A.; Puleio, R.; Condorelli, L.; Cusimano, A.; Giannitrapani, L.; McCubrey, J.A.; Iovanna, J.L.; Cervello, M. The NUPR1/p73 axis contributes to sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2021, 519, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Dis. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in advanced solid tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Cervello, M.; Emma, M.R.; Augello, G.; Cusimano, A.; Giannitrapani, L.; Soresi, M.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Gulino, A.; et al. New landscapes and horizons in hepatocellular carcinoma therapy. Aging 2020, 12, 3053–3094. [Google Scholar] [CrossRef]
- Genovese, G.; Carugo, A.; Tepper, J.; Robinson, F.S.; Li, L.; Svelto, M.; Nezi, L.; Corti, D.; Minelli, R.; Pettazzoni, P.; et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 2017, 542, 362–366. [Google Scholar] [CrossRef]
- Falcomatà, C.; Bärthel, S.; Widholz, S.A.; Schneeweis, C.; Montero, J.J.; Toska, A.; Mir, J.; Kaltenbacher, T.; Heetmeyer, J.; Swietlik, J.J.; et al. Selective multi-kinase inhibition sensitizes mesenchymal pancreatic cancer to immune checkpoint blockade by remodeling the tumor microenvironment. Nat. Cancer 2022, 3, 318–336. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Stalnecker, C.A.; Grover, K.R.; Edwards, A.C.; Coleman, M.F.; Yang, R.; DeLiberty, J.M.; Papke, B.; Goodwin, C.M.; Pierobon, M.; Petricoin, E.F.; et al. Concurrent inhibition of IGF1R and ERK increases pancreatic cancer sensitivity to autophagy inhibitors. Cancer Res. 2022, 82, 586–598. [Google Scholar] [CrossRef]
- Woodfield, S.E.; Shi, Y.; Patel, R.H.; Chen, Z.; Shah, A.P.; Srivastava, R.K.; Whitlock, R.S.; Ibarra, A.M.; Larson, S.R.; Sarabia, S.F.; et al. MDM4 inhibition: A novel therapeutic strategy to reactivate p53 in hepatoblastoma. Sci. Rep. 2021, 11, 2967. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, X. Mdm2 and MdmX partner to regulate p53. FEBS Lett. 2012, 586, 1390–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical overview of MDM2/X-targeted therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Conradt, L.; Henrich, A.; Wirth, M.; Reichert, M.; Lesina, M.; Algül, H.; Schmid, R.M.; Krämer, O.H.; Saur, D.; Schneider, G. Mdm2 inhibitors synergize with topoisomerase II inhibitors to induce p53-independent pancreatic cancer cell death. Int. J. Cancer 2013, 132, 2248–2257. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.L.; Lertpiriyapong, K.; Yang, L.V.; Martelli, A.M.; Cocco, L.; Ratti, S.; Falasca, M.; Murata, R.M.; Rosalen, P.L.; Lombardi, P.; et al. Introduction of WT-TP53 into pancreatic cancer cells alters sensitivity to chemotherapeutic drugs, targeted therapeutics and nutraceuticals. Adv. Biol. Regul. 2018, 69, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.L.; Akula, S.M.; Martelli, A.M.; Cocco, L.; Ratti, S.; Libra, M.; Candido, S.; Montalto, G.; Cervello, M.; Gizak, A.; et al. Sensitivity of pancreatic cancer cells to chemotherapeutic drugs, signal transduction inhibitors and nutraceuticals can be regulated by WT-TP53. Adv. Biol. Regul. 2021, 79, 100780. [Google Scholar] [CrossRef]
- Candido, S.; Abrams, S.L.; Steelman, L.S.; Lertpiriyapong, K.; Martelli, A.M.; Cocco, L.; Ratti, S.; Follo, M.Y.; Murata, R.M.; Rosalen, P.L.; et al. Effects of the MDM-2 inhibitor Nutlin-3a on PDAC cells containing and lacking WT-TP53 on sensitivity to chemotherapy, signal transduction inhibitors and nutraceuticals. Adv. Biol. Regul. 2019, 72, 22–40. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, Y.; Gao, H.; Xu, W.; Zhang, C.; Lai, J.; Liu, X.; Sun, Y.; Huang, H. Berberine inhibits cell proliferation by interfering with wild-type and mutant P53 in human glioma cells. OncoTargets Ther. 2020, 13, 12151–12162. [Google Scholar] [CrossRef]
- Hsieh Li, S.M.; Liu, S.T.; Chang, Y.L.; Ho, C.L.; Huang, S.M. Metformin causes cancer cell death through downregulation of p53-dependent differentiated embryo chondrocyte 1. J. Biomed. Sci. 2018, 25, 81. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Jin, S.; Hao, J.; Shi, Y.; Li, W.; Jiang, L. Metformin attenuates ischemia/reperfusion-induced apoptosis of cardiac cells by downregulation of p53/microRNA-34a via activation of SIRT1. Can. J. Physiol. Pharmacol. 2021, 99, 875–884. [Google Scholar] [CrossRef]
- Kubra, K.T.; Uddin, M.A.; Akhter, M.S.; Leo, A.J.; Siejka, A.; Barabutis, N. P53 mediates the protective effects of metformin in inflamed lung endothelial cells. Int. Immunopharmacol. 2021, 101, 108367. [Google Scholar] [CrossRef]
- Gralewska, P.; Gajek, A.; Marczak, A.; Rogalska, A. Metformin affects sensitivity through induction of apoptosis in epithelial ovarian cancer cell lines. Int. J. Mol. Sci. 2021, 22, 10557. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Chen, K.; Jiang, Z.; Chen, X.; Sun, L.; Li, J.; Lei, J.; Xu, Q.; Ma, J.; Li, X.; et al. Desmoplasia suppression by metforminmediated AMPK activation inhibits pancreatic cancer progression. Cancer Lett. 2017, 385, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Li, J.; Chen, K.; Jiang, Z.; Cheng, L.; Zhou, C.; Yan, B.; Cao, J.; Ma, Q.; Duan, W. Metformin suppresses tumor angiogenesis and enhances the chemosensitivity of gemcitabine in a genetically engineered mouse model of pancreatic cancer. Life Sci. 2018, 208, 253–261. [Google Scholar] [CrossRef]
- Duan, W.; Qian, W.; Zhou, C.; Cao, J.; Qin, T.; Xiao, Y.; Cheng, L.; Li, J.; Chen, K.; Li, X.; et al. Metformin suppresses the invasive ability of pancreatic cancer cells by blocking autocrine TGF-β1 signaling. Oncol. Rep. 2018, 40, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Candido, S.; Abrams, S.L.; Steelman, L.; Lertpiriyapong, K.; Martelli, A.M.; Cocco, L.; Ratti, S.; Follo, M.Y.; Murata, R.M.; Rosalen, P.L.; et al. Metformin influences drug sensitivity in pancreatic cancer cells. Adv. Biol. Regul. 2018, 68, 13–30. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, C.; Ying, Y.; Luo, L.; Huang, D.; Luo, Z. Metformin and berberine, two versatile drugs in treatment of common metabolic diseases. Oncotarget 2017, 9, 10135–10146. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Deguchi, Y.; Wei, D.; Liu, F.; Moussalli, M.J.; Deguchi, E.; Li, D.; Wang, H.; Valentin, L.A.; Colby, J.K.; et al. Rapid acceleration of KRAS-mutant pancreatic carcinogenesis via remodeling of tumor immune microenvironment by PPARδ. Nat. Commun. 2022, 13, 2665. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Lertpiriyapong, K.; Steelman, L.S.; Abrams, S.L.; Yang, L.V.; Murata, R.M.; Rosalen, P.L.; Scalisi, A.; Neri, L.M.; Cocco, L.; et al. Effects of resveratrol, curcumin, berberine and other nutraceuticals on aging, cancer development, cancer stem cells and microRNAs. Aging 2017, 9, 1477–1536. [Google Scholar] [CrossRef] [Green Version]
- Akula, S.M.; Candido, S.; Libra, M.; Abrams, S.L.; Steelman, L.S.; Lertpiriyapong, K.; Ramazzotti, G.; Ratti, S.; Follo, M.Y.; Martelli, A.M.; et al. Abilities of berberine and chemically modified berberines to interact with metformin and inhibit proliferation of pancreatic cancer cells. Adv. Biol. Regul. 2019, 73, 100633. [Google Scholar] [CrossRef]
- Akula, S.M.; Candido, S.; Abrams, S.L.; Steelman, L.S.; Lertpiriyapong, K.; Cocco, L.; Ramazzotti, G.; Ratti, S.; Follo, M.Y.; Martelli, A.M.; et al. Abilities of β-Estradiol to interact with chemotherapeutic drugs, signal transduction inhibitors and nutraceuticals and alter the proliferation of pancreatic cancer cells. Adv. Biol. Regul. 2019, 75, 100672. [Google Scholar] [CrossRef]
- Abrams, S.L.; Akula, S.M.; Steelman, L.S.; Follo, M.L.; Cocco, L.; Ratti, S.; Martelli, A.M.; Libra, M.; Falzone, L.; Candido, S.; et al. Effects of the MDM2 inhibitor Nutlin-3a on sensitivity of pancreatic cancer cells to berberine and modified berberines in the presence and absence of WT-TP53. Adv. Biol. Regul. 2022, 83, 100840. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.L.; Duda, P.; Akula, S.M.; Steelman, L.S.; Follo, M.L.; Cocco, L.; Ratti, S.; Martelli, A.M.; Montalto, G.; Emma, M.R.; et al. Effects of the mutant TP53 reactivator APR-246 on therapeutic sensitivity of pancreatic cancer cells in the presence and absence of WT-TP53. Cells 2022, 11, 794. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Abrams, S.L.; Steelman, L.S.; Cocco, L.; Ratti, S.; Martelli, A.M.; Lombardi, P.; Gizak, A.; Duda, P. APR-246-The mutant TP53 reactivator-Increases the effectiveness of berberine and modified berberines to inhibit the proliferation of pancreatic cancer cells. Biomolecules 2022, 12, 276. [Google Scholar] [CrossRef] [PubMed]
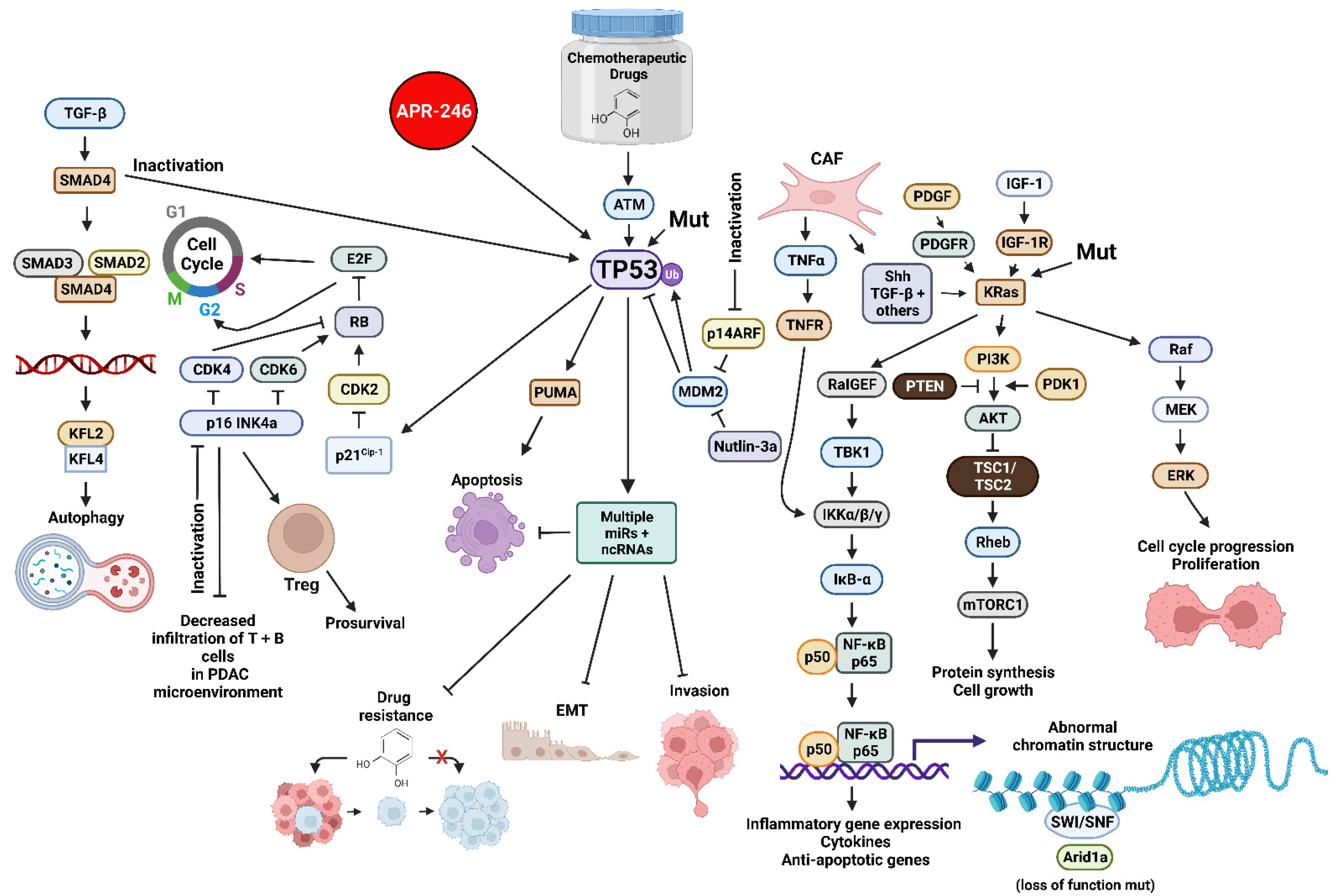
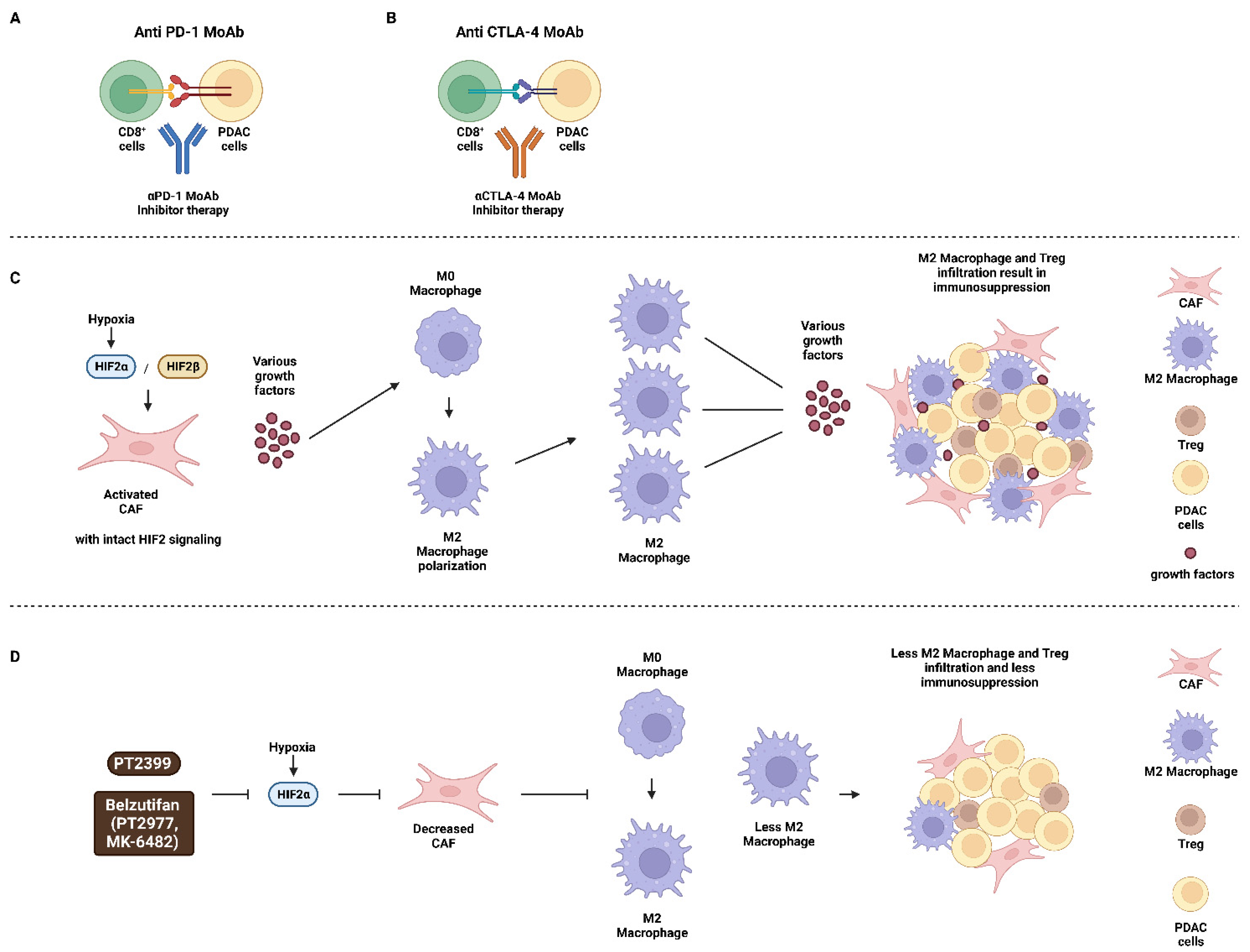
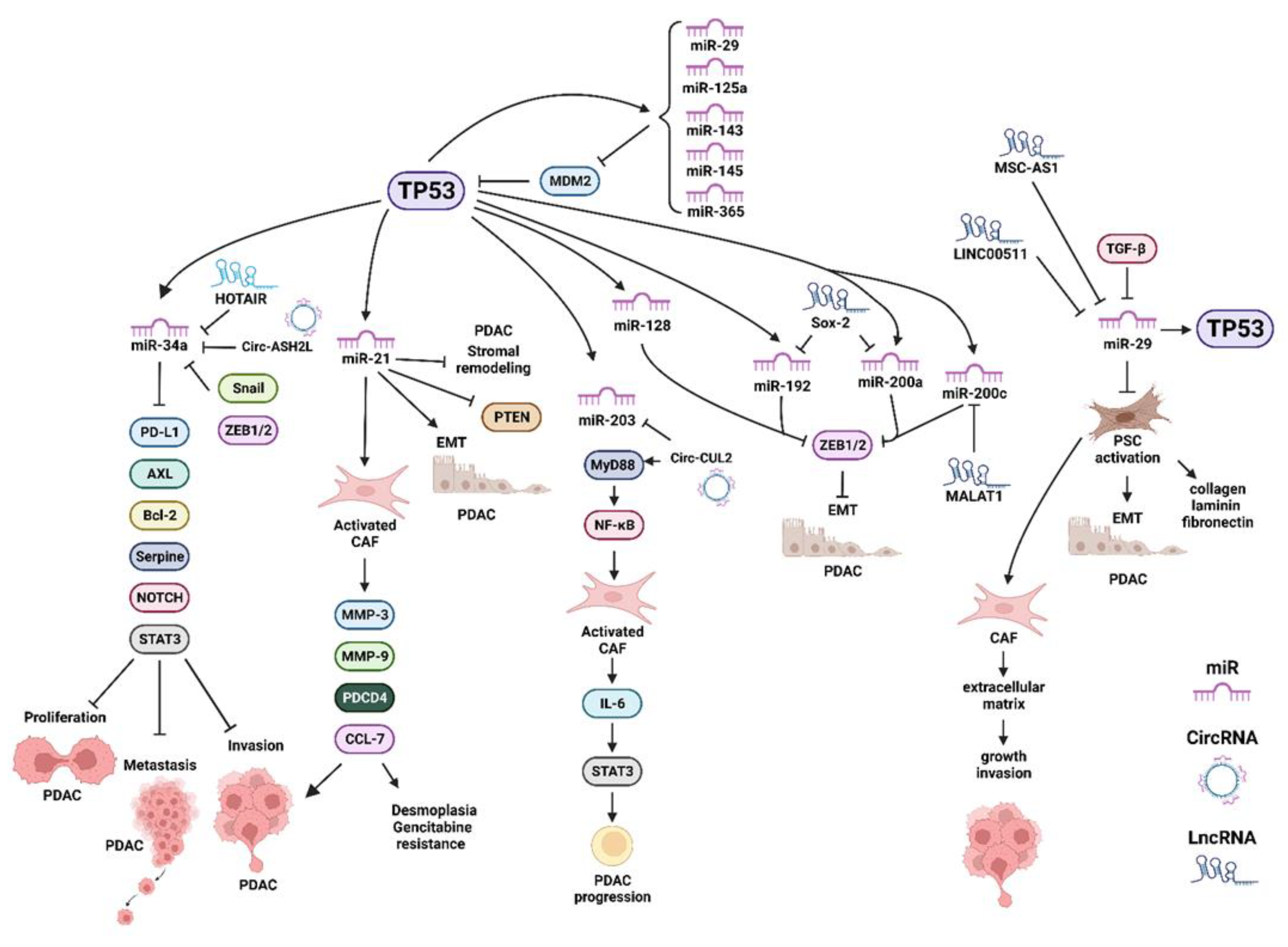
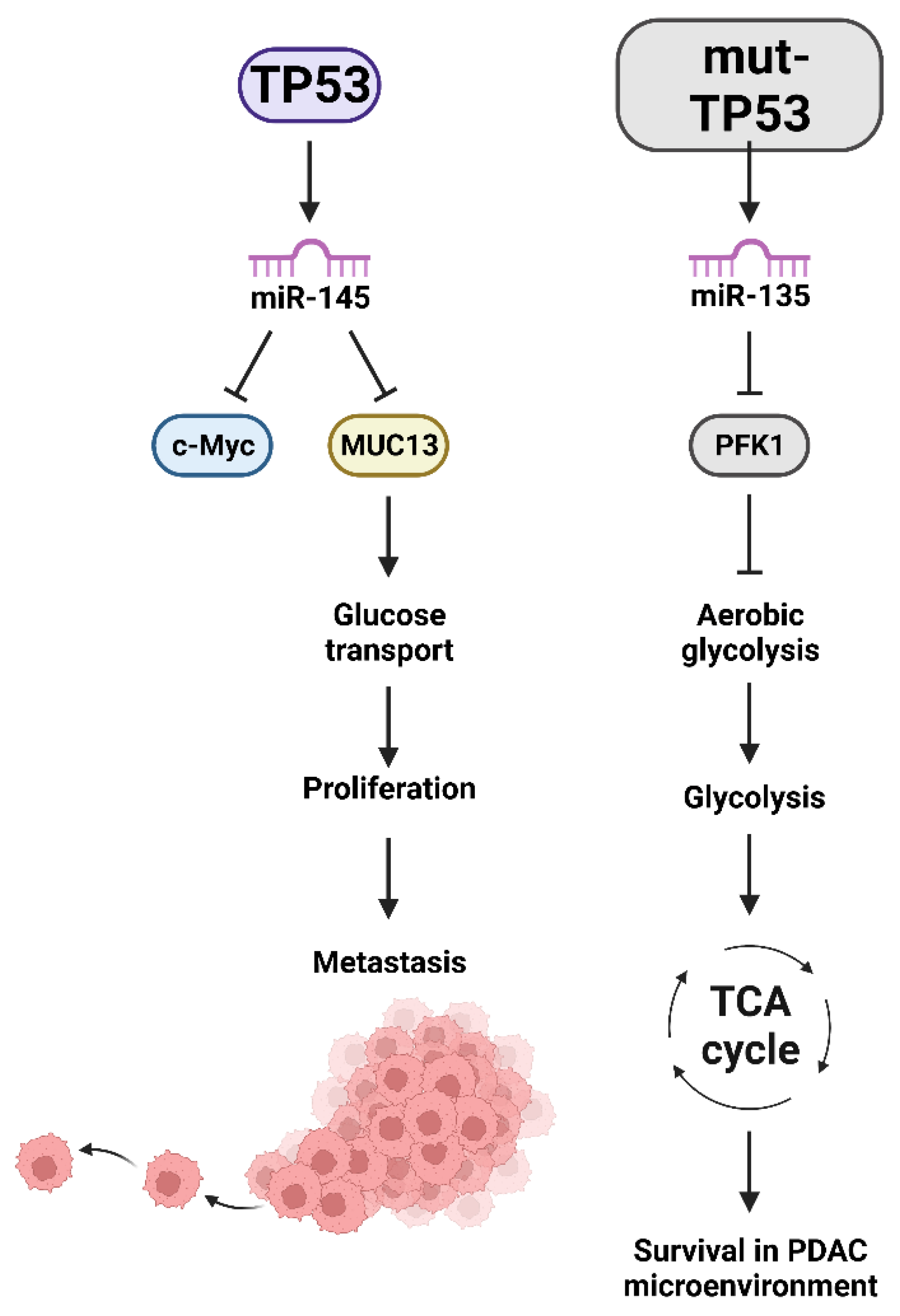
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCubrey, J.A.; Yang, L.V.; Abrams, S.L.; Steelman, L.S.; Follo, M.Y.; Cocco, L.; Ratti, S.; Martelli, A.M.; Augello, G.; Cervello, M. Effects of TP53 Mutations and miRs on Immune Responses in the Tumor Microenvironment Important in Pancreatic Cancer Progression. Cells 2022, 11, 2155. https://doi.org/10.3390/cells11142155
McCubrey JA, Yang LV, Abrams SL, Steelman LS, Follo MY, Cocco L, Ratti S, Martelli AM, Augello G, Cervello M. Effects of TP53 Mutations and miRs on Immune Responses in the Tumor Microenvironment Important in Pancreatic Cancer Progression. Cells. 2022; 11(14):2155. https://doi.org/10.3390/cells11142155
Chicago/Turabian StyleMcCubrey, James A., Li V. Yang, Stephen L. Abrams, Linda S. Steelman, Matilde Y. Follo, Lucio Cocco, Stefano Ratti, Alberto M. Martelli, Giuseppa Augello, and Melchiorre Cervello. 2022. "Effects of TP53 Mutations and miRs on Immune Responses in the Tumor Microenvironment Important in Pancreatic Cancer Progression" Cells 11, no. 14: 2155. https://doi.org/10.3390/cells11142155
APA StyleMcCubrey, J. A., Yang, L. V., Abrams, S. L., Steelman, L. S., Follo, M. Y., Cocco, L., Ratti, S., Martelli, A. M., Augello, G., & Cervello, M. (2022). Effects of TP53 Mutations and miRs on Immune Responses in the Tumor Microenvironment Important in Pancreatic Cancer Progression. Cells, 11(14), 2155. https://doi.org/10.3390/cells11142155