
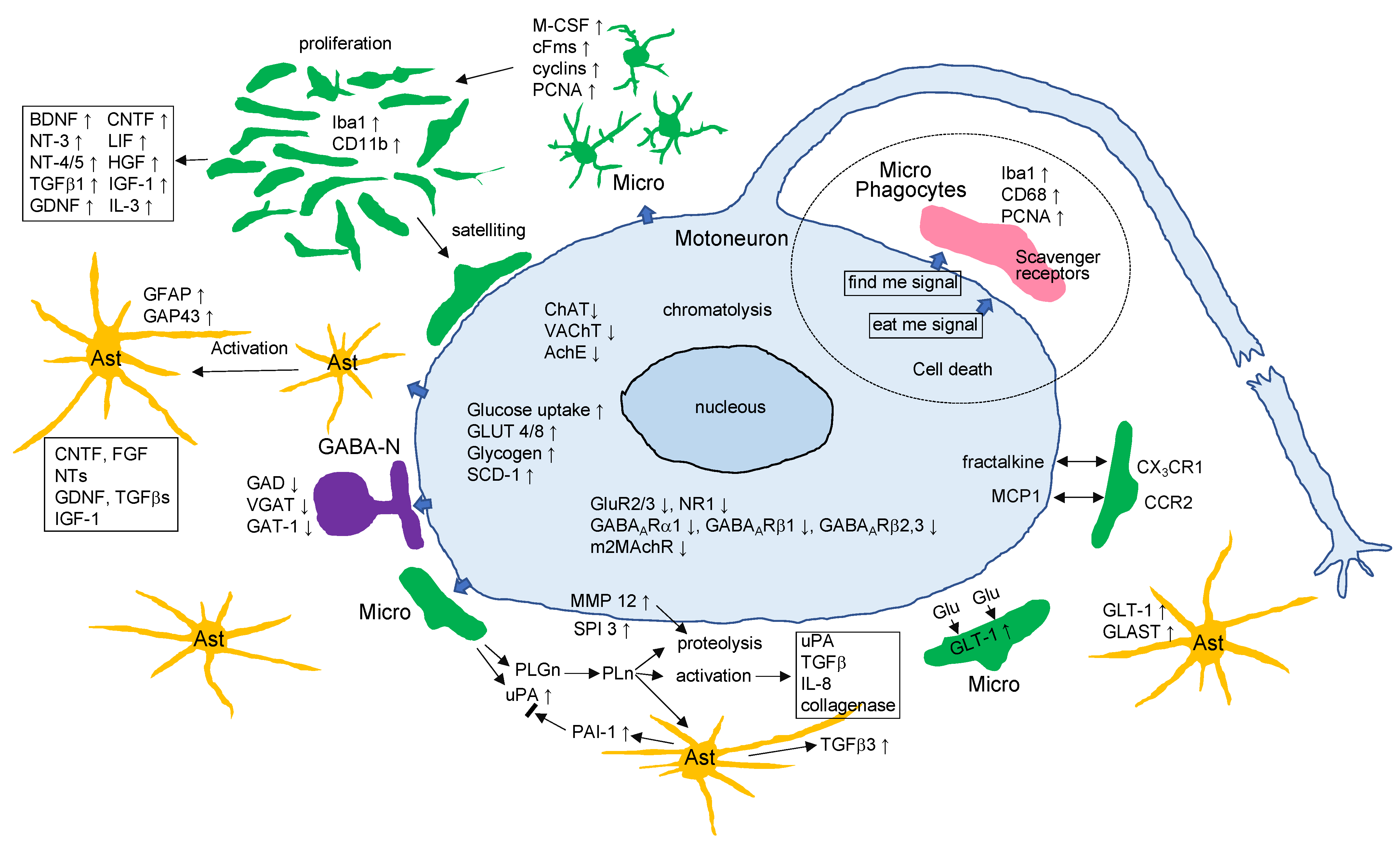
Events Occurring in the Axotomized Facial Nucleus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Damage to Motoneurons
2.1. Chromatolysis
2.2. Metabolism
2.3. Function-Related Molecules
2.4. Survivability and Death

3. Response of Microglia
3.1. Proliferation
3.2. Morphology
3.3. Migratory Property
3.4. Metabolic Features
3.5. Neurotrophic Property
3.6. Protective Ability against Excitotoxicity and Oxidative Stress
3.7. Hazardous Property
3.8. Inflammatory Property
3.9. Phagocytic Ability
4. Response of Astrocytes
4.1. Activation
4.2. Neurosupportive Features
4.3. Energy Metabolism
5. Response of Inhibitory Neurons
6. Tissue Remodeling in the Axotomized Facial Nucleus
7. Mediators Serving between Neurons and around Cells
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kreutzberg, G.W. Microglia: A Sensor for Pathological Events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Moran, L.B.; Graeber, M.B. The Facial Nerve Axotomy Model. Brain Res. Rev. 2004, 44, 154–178. [Google Scholar] [CrossRef] [PubMed]
- Cova, J.L.; Aldskogius, H. Effect of Axotomy on Perineuronal Glial Cells in the Hypoglossal and Dorsal Motor Vagal Nuclei of the Cat. Exp. Neurol. 1986, 93, 662–667. [Google Scholar] [CrossRef]
- Svensson, M.; Eriksson, N.P.; Aldskogius, H. Evidence for Activation of Astrocytes via Reactive Microglial Cells Following Hypoglossal Nerve Transection. J. Neurosci. Res. 1993, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.R.; Mason, C.A. The Seventh Cranial Nerve of the Rat. Visualization of Efferent and Afferent Pathways by Cobalt Precipitation. Brain Res. 1977, 121, 21–41. [Google Scholar] [CrossRef]
- Donoghue, J.P.; Sanes, J.N. Motor Areas of the Cerebral Cortex. J. Clin. Neurophysiol. Off. Publ. Am. Electroencephalogr. Soc. 1994, 11, 382–396. [Google Scholar]
- Cattaneo, L.; Pavesi, G. The Facial Motor System. Neurosci. Biobehav. Rev. 2014, 38, 135–159. [Google Scholar] [CrossRef]
- Petroff, O.A.C. GABA and Glutamate in the Human Brain. Neuroscientist 2002, 8, 562–573. [Google Scholar] [CrossRef]
- Roth, F.C.; Draguhn, A. GABA Metabolism and Transport: Effects on Synaptic Efficacy. Neural Plast. 2012, 2012, 805830. [Google Scholar] [CrossRef]
- Streit, W.J.; Kreutzberg, G.W. Response of Endogenous Glial Cells to Motor Neuron Degeneration Induced by Toxic Ricin. J. Comp. Neurol. 1988, 268, 248–263. [Google Scholar] [CrossRef]
- Sendtner, M.; Arakawa, Y.; Stöckli, K.A.; Kreutzberg, G.W.; Thoenen, H. Effect of Ciliary Neurotrophic Factor (CNTF) on Motoneuron Survival. J. Cell Sci. Suppl. 1991, 15, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Matheson, C.; Lopez, O.T. In Vivo Neurotrophic Effects of GDNF on Neonatal and Adult Facial Motor Neurons. Nature 1995, 373, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Rink, S.; Bendella, H.; Akkin, S.M.; Manthou, M.; Grosheva, M.; Angelov, D.N. Experimental Studies on Facial Nerve Regeneration. Anat. Rec. 2019, 302, 1287–1303. [Google Scholar] [CrossRef] [PubMed]
- Raivich, G.; Jones, L.L.; Kloss, C.U.; Werner, A.; Neumann, H.; Kreutzberg, G.W. Immune Surveillance in the Injured Nervous System: T-Lymphocytes Invade the Axotomized Mouse Facial Motor Nucleus and Aggregate around Sites of Neuronal Degeneration. J. Neurosci. 1998, 18, 5804–5816. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.J.; Lovett-Racke, A.E.; Walker, C.L.; Sanders, V.M. CD4+TCells and Neuroprotection: Relevance to Motoneuron Injury and Disease. J. Neuroimmune Pharmacol. 2015, 10, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W.; Engel, A.K.; Graeber, M.B.; Tetzlaff, W.; Tóth, L. Structural Plasticity in Lesioned Motoneurons. In Post-Lesion Neural Plasticity; Flohr, H., Ed.; Springer: Berlin/Heidelberg, Germany, 1988; pp. 57–64. [Google Scholar]
- Moon, L.D.F. Chromatolysis: Do Injured Axons Regenerate Poorly When Ribonucleases Attack Rough Endoplasmic Reticulum, Ribosomes and RNA? Dev. Neurobiol. 2018, 78, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Guntinas-Lichius, O.; Neiss, W.F.; Schulte, E.; Stennert, E. Quantitative Image Analysis of the Chromatolysis in Rat Facial and Hypoglossal Motoneurons Following Axotomy with and without Reinnervation. Cell Tissue Res. 1996, 286, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, W.; Emmert, H. Glucose Utilization during Chromatolysis: A 14C Deoxyglucose Study. Acta Neuropathol. Suppl. 1981, 7, 29–30. [Google Scholar] [CrossRef]
- Ito, D.; Tanaka, K.; Nagata, E.; Suzuki, S.; Dembo, T.; Fukuuchi, Y. Uncoupling of Cerebral Blood Flow and Glucose Utilization in the Regenerating Facial Nucleus after Axotomy. Neurosci. Res. 1999, 35, 207–215. [Google Scholar] [CrossRef]
- Gómez, O.; Ballester-Lurbe, B.; Mesonero, J.E.; Terrado, J. Glucose Transporters GLUT4 and GLUT8 Are Upregulated after Facial Nerve Axotomy in Adult Mice. J. Anat. 2011, 219, 525–530. [Google Scholar] [CrossRef]
- Angelov, D.N.; Neiss, W.F.; Gunkel, A.; Guntinas-Lichius, O.; Stennert, E. Axotomy Induces Intranuclear Immunolocalization of Neuron-Specific Enolase in Facial and Hypoglossal Neurons of the Rat. J. Neurocytol. 1994, 23, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Tetzlaff, W.; Kreutzberg, G.W. Enzyme Changes in the Rat Facial Nucleus Following a Conditioning Lesion. Exp. Neurol. 1984, 85, 547–564. [Google Scholar] [CrossRef]
- Schmitt, A.B.; Breuer, S.; Liman, J.; Buss, A.; Schlangen, C.; Pech, K.; Hol, E.M.; Brook, G.A.; Noth, J.; Schwaiger, F.-W. Identification of Regeneration-Associated Genes after Central and Peripheral Nerve Injury in the Adult Rat. BMC Neurosci. 2003, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, Y.; Kohsaka, S.; Nakajima, K. Transient Down-Regulation and Restoration of Glycogen Synthase Levels in Axotomized Rat Facial Motoneurons. Brain Res. 2014, 1586, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Broadwell, R.D. Cytochemical Identification of Cerebral Glycogen and Glucose-6-Phosphatase Activity under Normal and Experimental Conditions. II. Choroid Plexus and Ependymal Epithelia, Endothelia and Pericytes. J. Neurocytol. 1986, 15, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, C.; Palay, S.L. The Fine Structure of the Lateral Vestibular Nucleus in the Rat. I. Neurons and Neuroglial Cells. J. Cell Biol. 1968, 36, 151–179. [Google Scholar] [CrossRef]
- Takezawa, Y.; Baba, O.; Kohsaka, S.; Nakajima, K. Accumulation of Glycogen in Axotomized Adult Rat Facial Motoneurons. J. Neurosci. Res. 2015, 93, 913–921. [Google Scholar] [CrossRef]
- Jirmanová, I. Glycogen Deposits in Motorneurones of Young Chickens Following Peripheral Nerve Section. Acta Neuropathol. 1971, 19, 110–120. [Google Scholar] [CrossRef]
- Borke, R.C.; Nau, M.E. Glycogen, Its Transient Occurrence in Neurons of the Rat CNS during Normal Postnatal Development. Brain Res. 1984, 318, 277–284. [Google Scholar] [CrossRef]
- Gilmor, M.L.; Nash, N.R.; Roghani, A.; Edwards, R.H.; Yi, H.; Hersch, S.M.; Levey, A.I. Expression of the Putative Vesicular Acetylcholine Transporter in Rat Brain and Localization in Cholinergic Synaptic Vesicles. J. Neurosci. 1996, 16, 2179–2190. [Google Scholar] [CrossRef]
- Yew, D.T.; Webb, S.E.; Lam, E.T. Neurotransmitters and Peptides in the Developing Human Facial Nucleus. Neurosci. Lett. 1996, 206, 65–68. [Google Scholar] [CrossRef]
- Ichikawa, T.; Ajiki, K.; Matsuura, J.; Misawa, H. Localization of Two Cholinergic Markers, Choline Acetyltransferase and Vesicular Acetylcholine Transporter in the Central Nervous System of the Rat: In Situ Hybridization Histochemistry and Immunohistochemistry. J. Chem. Neuroanat. 1997, 13, 23–39. [Google Scholar] [CrossRef]
- Ichimiya, T.; Yamamoto, S.; Honda, Y.; Kikuchi, R.; Kohsaka, S.; Nakajima, K. Functional Down-Regulation of Axotomized Rat Facial Motoneurons. Brain Res. 2013, 1507, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.M.; Brady, R.; Hersh, L.B.; Hayes, R.C.; Wiley, R.G. Expression of Choline Acetyltransferase and Nerve Growth Factor Receptor within Hypoglossal Motoneurons Following Nerve Injury. J. Comp. Neurol. 1991, 304, 596–607. [Google Scholar] [CrossRef]
- Rende, M.; Giambanco, I.; Buratta, M.; Tonali, P. Axotomy Induces a Different Modulation of Both Low-Affinity Nerve Growth Factor Receptor and Choline Acetyltransferase between Adult Rat Spinal and Brainstem Motoneurons. J. Comp. Neurol. 1995, 363, 249–263. [Google Scholar] [CrossRef]
- Xing, G.-G.; Wang, R.; Yang, B.; Zhang, D. Postnatal Switching of NMDA Receptor Subunits from NR2B to NR2A in Rat Facial Motor Neurons. Eur. J. Neurosci. 2006, 24, 2987–2992. [Google Scholar] [CrossRef]
- Chen, P.; Song, J.; Luo, L.; Cheng, Q.; Xiao, H.; Gong, S. Gene Expression of NMDA and AMPA Receptors in Different Facial Motor Neurons. Laryngoscope 2016, 126, E6–E11. [Google Scholar] [CrossRef]
- Eleore, L.; Vassias, I.; Vidal, P.-P.; De Waele, C. Modulation of the Glutamatergic Receptors (AMPA and NMDA) and of Glutamate Vesicular Transporter 2 in the Rat Facial Nucleus after Axotomy. Neuroscience 2005, 136, 147–160. [Google Scholar] [CrossRef]
- Tang, F.R.; Sim, M.K. Expression of Glutamate Receptor Subunits 2/3 and 4 in the Hypoglossal Nucleus of the Rat after Neurectomy. Exp. Brain Res. 1997, 117, 453–456. [Google Scholar] [CrossRef]
- García Del Caño, G.; Gerrikagoitia, I.; Sarasa, M.; Matute, C.; Martínez-Millán, L. Ionotropic Glutamate Receptor Subunits Are Differentially Regulated in the Motoneuronal Pools of the Rat Hypoglossal Nucleus in Response to Axotomy. J. Neurocytol. 2000, 29, 509–523. [Google Scholar] [CrossRef]
- Alvarez, F.J.; Dewey, D.E.; Carr, P.A.; Cope, T.C.; Fyffe, R.E. Downregulation of Metabotropic Glutamate Receptor 1a in Motoneurons after Axotomy. Neuroreport 1997, 8, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- DeFeudis, F.V. Vertebrate GABA Receptors. Neurochem. Res. 1978, 3, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.; Kerr, D.I. GABA-Receptors in Peripheral Tissues. Life Sci. 1990, 46, 1489–1501. [Google Scholar] [CrossRef]
- Kudo, Y.; Abe, N.; Goto, S.; Fukuda, H. The Chloride-Dependent Depression by GABA in the Frog Spinal Cord. Eur. J. Pharmacol. 1975, 32, 251–259. [Google Scholar] [CrossRef]
- Bowery, N.G.; Brown, D.A. Depolarizing Actions of Gamma-Aminobutyric Acid and Related Compounds on Rat Superior Cervical Ganglia In Vitro. Br. J. Pharmacol. 1974, 50, 205–218. [Google Scholar] [CrossRef]
- Matsumoto, R.R. GABA Receptors: Are Cellular Differences Reflected in Function? Brain Res. Rev. 1989, 14, 203–225. [Google Scholar] [CrossRef]
- Chebib, M.; Johnston, G.A. The “ABC” of GABA Receptors: A Brief Review. Clin. Exp. Pharmacol. Physiol. 1999, 26, 937–940. [Google Scholar] [CrossRef]
- Kikuchi, R.; Hamanoue, M.; Koshimoto, M.; Kohsaka, S.; Nakajima, K. Response of the GABAergic System to Axotomy of the Rat Facial Nerve. Neurochem. Res. 2018, 43, 324–339. [Google Scholar] [CrossRef]
- Nakajima, K. Responses of Several GABAA Receptors to Facial Nerve Injury in Rat. J. Clin. Neurol. Neurosurg. Spine 2020, 3, 121–126. [Google Scholar]
- Vassias, I.; Lecolle, S.; Vidal, P.-P.; De Waele, C. Modulation of GABA Receptor Subunits in Rat Facial Motoneurons after Axotomy. Mol. Brain Res. 2005, 135, 260–275. [Google Scholar] [CrossRef]
- Hoover, D.B.; Baisden, R.H.; Lewis, J.V. Axotomy-Induced Loss of M2 Muscarinic Receptor MRNA in the Rat Facial Motor Nucleus Precedes a Decrease in Concentration of Muscarinic Receptors. Histochem. J. 1996, 28, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Miles, G.B.; Hartley, R.; Todd, A.J.; Brownstone, R.M. Spinal Cholinergic Interneurons Regulate the Excitability of Motoneurons during Locomotion. Proc. Natl. Acad. Sci. USA 2007, 104, 2448–2453. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.B.; Hancock, J.C. Effect of Facial Nerve Transection on Acetylcholinesterase, Choline Acetyltransferase and [3H]Quinuclidinyl Benzilate Binding in Rat Facial Nuclei. Neuroscience 1985, 15, 481–487. [Google Scholar] [CrossRef]
- Olsson, T.; Kristensson, K. Uptake and Retrograde Axonal Transport of Horseradish Peroxidase in Normal and Axotomized Motor Neurons during Postnatal Development. Neuropathol. Appl. Neurobiol. 1979, 5, 377–387. [Google Scholar] [CrossRef]
- Rossiter, J.P.; Riopelle, R.J.; Bisby, M.A. Axotomy-Induced Apoptotic Cell Death of Neonatal Rat Facial Motoneurons: Time Course Analysis and Relation to NADPH-Diaphorase Activity. Exp. Neurol. 1996, 138, 33–44. [Google Scholar] [CrossRef]
- Tao, R.; Aldskogius, H. Influence of FK506, Cyclosporin A, Testosterone and Nimodipine on Motoneuron Survival Following Axotomy. Restor. Neurol. Neurosci. 1998, 12, 239–246. [Google Scholar]
- Søreide, A.J. Variations in the Axon Reaction in Animals of Different Ages. A Light Microscopic Study on the Facial Nucleus of the Rat. Acta Anat. 1981, 110, 40–47. [Google Scholar] [CrossRef]
- Koshimoto, M.; Ishijima, T.; Nakajima, K. Response of Microglia to Motoneuron Cell Death in Axotomized Infant Rat Facial Nucleus. In Advances in Medicine and Biology; Berhardt, L.V., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2019; Volume 152, pp. 133–157. ISBN 978-1-53616-403-9. [Google Scholar]
- Aldskogius, H.; Thomander, L. Selective Reinnervation of Somatotopically Appropriate Muscles after Facial Nerve Transection and Regeneration in the Neonatal Rat. Brain Res. 1986, 375, 126–134. [Google Scholar] [CrossRef]
- Thompson, W.J.; Soileau, L.C.; Balice-Gordon, R.J.; Sutton, L.A. Selective Innervation of Types of Fibres in Developing Rat Muscle. J. Exp. Biol. 1987, 132, 249–263. [Google Scholar] [CrossRef]
- Vanderluit, J.L.; McPhail, L.T.; Fernandes, K.J.L.; Kobayashi, N.R.; Tetzlaff, W. In Vivo Application of Mitochondrial Pore Inhibitors Blocks the Induction of Apoptosis in Axotomized Neonatal Facial Motoneurons. Cell Death Differ. 2003, 10, 969–976. [Google Scholar] [CrossRef]
- De Bilbao, F.; Dubois-Dauphin, M. Time Course of Axotomy-Induced Apoptotic Cell Death in Facial Motoneurons of Neonatal Wild Type and Bcl-2 Transgenic Mice. Neuroscience 1996, 71, 1111–1119. [Google Scholar] [CrossRef]
- Vanderluit, J.L.; McPhail, L.T.; Fernandes, K.J.; McBride, C.B.; Huguenot, C.; Roy, S.; Robertson, G.S.; Nicholson, D.W.; Tetzlaff, W. Caspase-3 Is Activated Following Axotomy of Neonatal Facial Motoneurons and Caspase-3 Gene Deletion Delays Axotomy-Induced Cell Death in Rodents. Eur. J. Neurosci. 2000, 12, 3469–3480. [Google Scholar] [CrossRef] [PubMed]
- Keramaris, E.; Vanderluit, J.L.; Bahadori, M.; Mousavi, K.; Davis, R.J.; Flavell, R.; Slack, R.S.; Park, D.S. C-Jun N-Terminal Kinase 3 Deficiency Protects Neurons from Axotomy-Induced Death in Vivo through Mechanisms Independent of c-Jun Phosphorylation. J. Biol. Chem. 2005, 280, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Horiuchi, M.; Ikeda, R.H.J.; Xu, J.; Bannerman, P.; Pleasure, D.; Penninger, J.M.; Tournier, C.; Itoh, A. ZPK/DLK and MKK4 Form the Critical Gateway to Axotomy-Induced Motoneuron Death in Neonates. J. Neurosci. 2014, 34, 10729–10742. [Google Scholar] [CrossRef]
- Ferri, C.C.; Moore, F.A.; Bisby, M.A. Effects of Facial Nerve Injury on Mouse Motoneurons Lacking the p75 Low-Affinity Neurotrophin Receptor. J. Neurobiol. 1998, 34, 1–9. [Google Scholar] [CrossRef]
- Raivich, G.; Liu, Z.Q.; Kloss, C.U.; Labow, M.; Bluethmann, H.; Bohatschek, M. Cytotoxic Potential of Proinflammatory Cytokines: Combined Deletion of TNF Receptors TNFR1 and TNFR2 Prevents Motoneuron Cell Death after Facial Axotomy in Adult Mouse. Exp. Neurol. 2002, 178, 186–193. [Google Scholar] [CrossRef]
- Sendtner, M.; Kreutzberg, G.W.; Thoenen, H. Ciliary Neurotrophic Factor Prevents the Degeneration of Motor Neurons after Axotomy. Nature 1990, 345, 440–441. [Google Scholar] [CrossRef]
- Li, M.; Sendtner, M.; Smith, A. Essential Function of LIF Receptor in Motor Neurons. Nature 1995, 378, 724–727. [Google Scholar] [CrossRef]
- Sendtner, M.; Holtmann, B.; Kolbeck, R.; Thoenen, H.; Barde, Y.A. Brain-Derived Neurotrophic Factor Prevents the Death of Motoneurons in Newborn Rats after Nerve Section. Nature 1992, 360, 757–759. [Google Scholar] [CrossRef]
- Koliatsos, V.E.; Cayouette, M.H.; Berkemeier, L.R.; Clatterbuck, R.E.; Price, D.L.; Rosenthal, A. Neurotrophin 4/5 Is a Trophic Factor for Mammalian Facial Motor Neurons. Proc. Natl. Acad. Sci. USA 1994, 91, 3304–3308. [Google Scholar] [CrossRef]
- Henderson, C.E.; Phillips, H.S.; Pollock, R.A.; Davies, A.M.; Lemeulle, C.; Armanini, M.; Simmons, L.; Moffet, B.; Vandlen, R.A. GDNF: A Potent Survival Factor for Motoneurons Present in Peripheral Nerve and Muscle. Science 1994, 266, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, P.; Carceller, F.; Giménez-Gallego, G. Acidic Fibroblast Growth Factor Prevents Post-Axotomy Neuronal Death of the Newborn Rat Facial Nerve. Neurosci. Lett. 1995, 197, 183–186. [Google Scholar] [CrossRef]
- Koyama, J.; Yokouchi, K.; Fukushima, N.; Kawagishi, K.; Higashiyama, F.; Moriizumi, T. Neurotrophic Effect of Hepatocyte Growth Factor on Neonatal Facial Motor Neurons. Neurol. Res. 2003, 25, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Svensson, M.; Eriksson, P.; Persson, J.K.; Molander, C.; Arvidsson, J.; Aldskogius, H. The Response of Central Glia to Peripheral Nerve Injury. Brain Res. Bull. 1993, 30, 499–506. [Google Scholar] [CrossRef]
- Søreide, A.J. Variations in the Perineuronal Glial Changes after Different Types of Nerve Lesion: Light and Electron Microscopic Investigations on the Facial Nucleus of the Rat. Neuropathol. Appl. Neurobiol. 1981, 7, 195–204. [Google Scholar] [CrossRef]
- Streit, W.J.; Kreutzberg, G.W. Lectin Binding by Resting and Reactive Microglia. J. Neurocytol. 1987, 16, 249–260. [Google Scholar] [CrossRef]
- Graeber, M.B.; López-Redondo, F.; Ikoma, E.; Ishikawa, M.; Imai, Y.; Nakajima, K.; Kreutzberg, G.W.; Kohsaka, S. The Microglia/Macrophage Response in the Neonatal Rat Facial Nucleus Following Axotomy. Brain Res. 1998, 813, 241–253. [Google Scholar] [CrossRef]
- Tao, R.; Aldskogius, H. Glial Cell Responses, Complement and Apolipoprotein J Expression Following Axon Injury in the Neonatal Rat. J. Neurocytol. 1999, 28, 559–570. [Google Scholar] [CrossRef]
- Conde, J.R.; Streit, W.J. Effect of Aging on the Microglial Response to Peripheral Nerve Injury. Neurobiol. Aging 2006, 27, 1451–1461. [Google Scholar] [CrossRef]
- Rieske, E.; Graeber, M.B.; Tetzlaff, W.; Czlonkowska, A.; Streit, W.J.; Kreutzberg, G.W. Microglia and Microglia-Derived Brain Macrophages in Culture: Generation from Axotomized Rat Facial Nuclei, Identification and Characterization In Vitro. Brain Res. 1989, 492, 1–14. [Google Scholar] [CrossRef]
- Nakajima, K.; Graeber, M.B.; Sonoda, M.; Tohyama, Y.; Kohsaka, S.; Kurihara, T. In Vitro Proliferation of Axotomized Rat Facial Nucleus-Derived Activated Microglia in an Autocrine Fashion. J. Neurosci. Res. 2006, 84, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Flügel, A.; Bradl, M.; Kreutzberg, G.W.; Graeber, M.B. Transformation of Donor-Derived Bone Marrow Precursors into Host Microglia during Autoimmune CNS Inflammation and during the Retrograde Response to Axotomy. J. Neurosci. Res. 2001, 66, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Giulian, D.; Ingeman, J.E. Colony-Stimulating Factors as Promoters of Ameboid Microglia. J. Neurosci. 1988, 8, 4707–4717. [Google Scholar] [CrossRef] [PubMed]
- Wiktor-Jedrzejczak, W.; Bartocci, A.; Ferrante, A.W.J.; Ahmed-Ansari, A.; Sell, K.W.; Pollard, J.W.; Stanley, E.R. Total Absence of Colony-Stimulating Factor 1 in the Macrophage-Deficient Osteopetrotic (Op/Op) Mouse. Proc. Natl. Acad. Sci. USA 1990, 87, 4828–4832. [Google Scholar] [CrossRef] [PubMed]
- Raivich, G.; Moreno-Flores, M.T.; Möller, J.C.; Kreutzberg, G.W. Inhibition of Posttraumatic Microglial Proliferation in a Genetic Model of Macrophage Colony-Stimulating Factor Deficiency in the Mouse. Eur. J. Neurosci. 1994, 6, 1615–1618. [Google Scholar] [CrossRef]
- Yamamoto, S.; Nakajima, K.; Kohsaka, S. Macrophage-Colony Stimulating Factor as an Inducer of Microglial Proliferation in Axotomized Rat Facial Nucleus. J. Neurochem. 2010, 115, 1057–1067. [Google Scholar] [CrossRef]
- Guan, Z.; Kuhn, J.A.; Wang, X.; Colquitt, B.; Solorzano, C.; Vaman, S.; Guan, A.K.; Evans-Reinsch, Z.; Braz, J.; Devor, M.; et al. Injured Sensory Neuron-Derived CSF1 Induces Microglial Proliferation and DAP12-Dependent Pain. Nat. Neurosci. 2016, 19, 94–101. [Google Scholar] [CrossRef]
- Gushchina, S.; Pryce, G.; Yip, P.K.; Wu, D.; Pallier, P.; Giovannoni, G.; Baker, D.; Bo, X. Increased Expression of Colony-Stimulating Factor-1 in Mouse Spinal Cord with Experimental Autoimmune Encephalomyelitis Correlates with Microglial Activation and Neuronal Loss. Glia 2018, 66, 2108–2125. [Google Scholar] [CrossRef]
- Raivich, G.; Gehrmann, J.; Kreutzberg, G.W. Increase of Macrophage Colony-Stimulating Factor and Granulocyte-Macrophage Colony-Stimulating Factor Receptors in the Regenerating Rat Facial Nucleus. J. Neurosci. Res. 1991, 30, 682–686. [Google Scholar] [CrossRef]
- Raivich, G.; Haas, S.; Werner, A.; Klein, M.A.; Kloss, C.; Kreutzberg, G.W. Regulation of MCSF Receptors on Microglia in the Normal and Injured Mouse Central Nervous System: A Quantitative Immunofluorescence Study Using Confocal Laser Microscopy. J. Comp. Neurol. 1998, 395, 342–358. [Google Scholar] [CrossRef]
- Roussel, M.F.; Dull, T.J.; Rettenmier, C.W.; Ralph, P.; Ullrich, A.; Sherr, C.J. Transforming Potential of the C-Fms Proto-Oncogene (CSF-1 Receptor). Nature 1987, 325, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.P. M-CSF, c-Fms, and Signaling in Osteoclasts and Their Precursors. Ann. N. Y. Acad. Sci. 2006, 1068, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Liu, W.; Brosnan, C.F.; Dickson, D.W. GM-CSF Promotes Proliferation of Human Fetal and Adult Microglia in Primary Cultures. Glia 1994, 12, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, A.; Vigliani, M.-C.; Magrassi, L.; Vercelli, A.; Rossi, F. G-CSF Administration to Adult Mice Stimulates the Proliferation of Microglia but Does Not Modify the Outcome of Ischemic Injury. Neurobiol. Dis. 2011, 41, 640–649. [Google Scholar] [CrossRef]
- Ganter, S.; Northoff, H.; Männel, D.; Gebicke-Härter, P.J. Growth Control of Cultured Microglia. J. Neurosci. Res. 1992, 33, 218–230. [Google Scholar] [CrossRef]
- Suzumura, A.; Sawada, M.; Itoh, Y.; Marunouchi, T. Interleukin-4 Induces Proliferation and Activation of Microglia but Suppresses Their Induction of Class II Major Histocompatibility Complex Antigen Expression. J. Neuroimmunol. 1994, 53, 209–218. [Google Scholar] [CrossRef]
- Ringheim, G.E. Mitogenic Effects of Interleukin-5 on Microglia. Neurosci. Lett. 1995, 201, 131–134. [Google Scholar] [CrossRef]
- Sherr, C.J. Mammalian G1 Cyclins. Cell 1993, 73, 1059–1065. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kohsaka, S.; Nakajima, K. Role of Cell Cycle-Associated Proteins in Microglial Proliferation in the Axotomized Rat Facial Nucleus. Glia 2012, 60, 570–581. [Google Scholar] [CrossRef]
- Fischer, P.M.; Endicott, J.; Meijer, L. Cyclin-Dependent Kinase Inhibitors. Prog. Cell Cycle Res. 2003, 5, 235–248. [Google Scholar]
- Flanary, B.E.; Streit, W.J. Effects of Axotomy on Telomere Length, Telomerase Activity, and Protein in Activated Microglia. J. Neurosci. Res. 2005, 82, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Liva, S.M.; Kahn, M.A.; Dopp, J.M.; De Vellis, J. Signal Transduction Pathways Induced by GM-CSF in Microglia: Significance in the Control of Proliferation. Glia 1999, 26, 344–352. [Google Scholar] [CrossRef]
- Suh, H.-S.; Kim, M.-O.; Lee, S.C. Inhibition of Granulocyte-Macrophage Colony-Stimulating Factor Signaling and Microglial Proliferation by Anti-CD45RO: Role of Hck Tyrosine Kinase and Phosphatidylinositol 3-Kinase/Akt. J. Immunol. 2005, 174, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- Mander, P.K.; Jekabsone, A.; Brown, G.C. Microglia Proliferation Is Regulated by Hydrogen Peroxide from NADPH Oxidase. J. Immunol. 2006, 176, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. The Role of Microglia in Brain Injury. Neurotoxicology 1996, 17, 671–678. [Google Scholar]
- Von Bernhardi, R.; Heredia, F.; Salgado, N.; Muñoz, P. Microglia Function in the Normal Brain. Adv. Exp. Med. Biol. 2016, 949, 67–92. [Google Scholar] [CrossRef]
- Savage, J.C.; Carrier, M.; Tremblay, M.-È. Morphology of Microglia Across Contexts of Health and Disease. Methods Mol. Biol. 2019, 2034, 13–26. [Google Scholar] [CrossRef]
- Del Rio-Hortega, P. Microglia. In Cytology and Cellular Pathology of the Nervous System; Penfield, W., Ed.; Hocker: New York, NY, USA, 1932; Volume 2, pp. 483–534. [Google Scholar]
- Kalla, R.; Bohatschek, M.; Kloss, C.U.A.; Krol, J.; Von Maltzan, X.; Raivich, G. Loss of Microglial Ramification in Microglia-Astrocyte Cocultures: Involvement of Adenylate Cyclase, Calcium, Phosphatase, and Gi-Protein Systems. Glia 2003, 41, 50–63. [Google Scholar] [CrossRef]
- Wilms, H.; Hartmann, D.; Sievers, J. Ramification of Microglia, Monocytes and Macrophages In Vitro: Influences of Various Epithelial and Mesenchymal Cells and Their Conditioned Media. Cell Tissue Res. 1997, 287, 447–458. [Google Scholar] [CrossRef]
- Schilling, T.; Nitsch, R.; Heinemann, U.; Haas, D.; Eder, C. Astrocyte-Released Cytokines Induce Ramification and Outward K+ Channel Expression in Microglia via Distinct Signalling Pathways. Eur. J. Neurosci. 2001, 14, 463–473. [Google Scholar] [CrossRef]
- Tanaka, J.; Toku, K.; Sakanaka, M.; Maeda, N. Morphological Differentiation of Microglial Cells in Culture: Involvement of Insoluble Factors Derived from Astrocytes. Neurosci. Res. 1999, 34, 207–215. [Google Scholar] [CrossRef]
- Schiefer, J.; Kampe, K.; Dodt, H.U.; Zieglgänsberger, W.; Kreutzberg, G.W. Microglial Motility in the Rat Facial Nucleus Following Peripheral Axotomy. J. Neurocytol. 1999, 28, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, C.N.; Gan, W.-B. Microglia Dynamics and Function in the CNS. Curr. Opin. Neurobiol. 2010, 20, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Glabinski, A.R.; Ransohoff, R.M. Chemokines and Chemokine Receptors in CNS Pathology. J. Neurovirol. 1999, 5, 3–12. [Google Scholar] [CrossRef]
- Skuljec, J.; Sun, H.; Pul, R.; Bénardais, K.; Ragancokova, D.; Moharregh-Khiabani, D.; Kotsiari, A.; Trebst, C.; Stangel, M. CCL5 Induces a Pro-Inflammatory Profile in Microglia in Vitro. Cell. Immunol. 2011, 270, 164–171. [Google Scholar] [CrossRef]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for Neuronally Derived Fractalkine in Mediating Interactions between Neurons and CX3CR1-Expressing Microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef]
- Jung, S.; Aliberti, J.; Graemmel, P.; Sunshine, M.J.; Kreutzberg, G.W.; Sher, A.; Littman, D.R. Analysis of Fractalkine Receptor CX(3)CR1 Function by Targeted Deletion and Green Fluorescent Protein Reporter Gene Insertion. Mol. Cell. Biol. 2000, 20, 4106–4114. [Google Scholar] [CrossRef]
- Flügel, A.; Hager, G.; Horvat, A.; Spitzer, C.; Singer, G.M.; Graeber, M.B.; Kreutzberg, G.W.; Schwaiger, F.W. Neuronal MCP-1 Expression in Response to Remote Nerve Injury. J. Cereb. Blood Flow Metab. 2001, 21, 69–76. [Google Scholar] [CrossRef]
- Kalsbeek, M.J.T.; Mulder, L.; Yi, C.-X. Microglia Energy Metabolism in Metabolic Disorder. Mol. Cell. Endocrinol. 2016, 438, 27–35. [Google Scholar] [CrossRef]
- Maher, F.; Vannucci, S.J.; Simpson, I.A. Glucose Transporter Proteins in Brain. FASEB J. 1994, 8, 1003–1011. [Google Scholar] [CrossRef]
- Vannucci, S.J.; Maher, F.; Simpson, I.A. Glucose Transporter Proteins in Brain: Delivery of Glucose to Neurons and Glia. Glia 1997, 21, 2–21. [Google Scholar] [CrossRef]
- Nijland, P.G.; Michailidou, I.; Witte, M.E.; Mizee, M.R.; Van der Pol, S.M.A.; Van Het Hof, B.; Reijerkerk, A.; Pellerin, L.; Van der Valk, P.; De Vries, H.E.; et al. Cellular Distribution of Glucose and Monocarboxylate Transporters in Human Brain White Matter and Multiple Sclerosis Lesions. Glia 2014, 62, 1125–1141. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Streit, W.J.; Kreutzberg, G.W. Axotomy of the Rat Facial Nerve Leads to Increased CR3 Complement Receptor Expression by Activated Microglial Cells. J. Neurosci. Res. 1988, 21, 18–24. [Google Scholar] [CrossRef]
- Nakajima, K.; Kohsaka, S. Microglia: Neuroprotective and Neurotrophic Cells in the Central Nervous System. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2004, 4, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Elkabes, S.; DiCicco-Bloom, E.M.; Black, I.B. Brain Microglia/Macrophages Express Neurotrophins That Selectively Regulate Microglial Proliferation and Function. J. Neurosci. 1996, 16, 2508–2521. [Google Scholar] [CrossRef] [PubMed]
- Miwa, T.; Furukawa, S.; Nakajima, K.; Furukawa, Y.; Kohsaka, S. Lipopolysaccharide Enhances Synthesis of Brain-Derived Neurotrophic Factor in Cultured Rat Microglia. J. Neurosci. Res. 1997, 50, 1023–1029. [Google Scholar] [CrossRef]
- Nakajima, K.; Honda, S.; Tohyama, Y.; Imai, Y.; Kohsaka, S.; Kurihara, T. Neurotrophin Secretion from Cultured Microglia. J. Neurosci. Res. 2001, 65, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Welser-Alves, J.V.; Milner, R. Microglia Are the Major Source of TNF-α and TGF-b1 in Postnatal Glial Cultures; Regulation by Cytokines, Lipopolysaccharide, and Vitronectin. Neurochem. Int. 2013, 63, 47–53. [Google Scholar] [CrossRef]
- Matsushita, Y.; Nakajima, K.; Tohyama, Y.; Kurihara, T.; Kohsaka, S. Activation of Microglia by Endotoxin Suppresses the Secretion of Glial Cell Line-Derived Neurotrophic Factor (GDNF) through the Action of Protein Kinase C Alpha (PKCalpha) and Mitogen-Activated Protein Kinases (MAPKS). J. Neurosci. Res. 2008, 86, 1959–1971. [Google Scholar] [CrossRef]
- Harada, T.; Harada, C.; Kohsaka, S.; Wada, E.; Yoshida, K.; Ohno, S.; Mamada, H.; Tanaka, K.; Parada, L.F.; Wada, K. Microglia-Müller Glia Cell Interactions Control Neurotrophic Factor Production during Light-Induced Retinal Degeneration. J. Neurosci. 2002, 22, 9228–9236. [Google Scholar] [CrossRef]
- Nakanishi, M.; Niidome, T.; Matsuda, S.; Akaike, A.; Kihara, T.; Sugimoto, H. Microglia-Derived Interleukin-6 and Leukaemia Inhibitory Factor Promote Astrocytic Differentiation of Neural Stem/Progenitor Cells. Eur. J. Neurosci. 2007, 25, 649–658. [Google Scholar] [CrossRef]
- Hamanoue, M.; Takemoto, N.; Matsumoto, K.; Nakamura, T.; Nakajima, K.; Kohsaka, S. Neurotrophic Effect of Hepatocyte Growth Factor on Central Nervous System Neurons in Vitro. J. Neurosci. Res. 1996, 43, 554–564. [Google Scholar] [CrossRef]
- Beilharz, E.J.; Russo, V.C.; Butler, G.; Baker, N.L.; Connor, B.; Sirimanne, E.S.; Dragunow, M.; Werther, G.A.; Gluckman, P.D.; Williams, C.E.; et al. Co-Ordinated and Cellular Specific Induction of the Components of the IGF/IGFBP Axis in the Rat Brain Following Hypoxic-Ischemic Injury. Mol. Brain Res. 1998, 59, 119–134. [Google Scholar] [CrossRef]
- Gebicke-Haerter, P.J.; Appel, K.; Taylor, G.D.; Schobert, A.; Rich, I.N.; Northoff, H.; Berger, M. Rat Microglial Interleukin-3. J. Neuroimmunol. 1994, 50, 203–214. [Google Scholar] [CrossRef]
- Nakajima, K.; Tohyama, Y.; Maeda, S.; Kohsaka, S.; Kurihara, T. Neuronal Regulation by Which Microglia Enhance the Production of Neurotrophic Factors for GABAergic, Catecholaminergic, and Cholinergic Neurons. Neurochem. Int. 2007, 50, 807–820. [Google Scholar] [CrossRef]
- Kiefer, R.; Lindholm, D.; Kreutzberg, G.W. Interleukin-6 and Transforming Growth Factor-Beta 1 MRNAs Are Induced in Rat Facial Nucleus Following Motoneuron Axotomy. Eur. J. Neurosci. 1993, 5, 775–781. [Google Scholar] [CrossRef]
- Makwana, M.; Jones, L.L.; Cuthill, D.; Heuer, H.; Bohatschek, M.; Hristova, M.; Friedrichsen, S.; Ormsby, I.; Bueringer, D.; Koppius, A.; et al. Endogenous Transforming Growth Factor Beta 1 Suppresses Inflammation and Promotes Survival in Adult CNS. J. Neurosci. 2007, 27, 11201–11213. [Google Scholar] [CrossRef]
- Kobayashi, N.R.; Bedard, A.M.; Hincke, M.T.; Tetzlaff, W. Increased Expression of BDNF and TrkB MRNA in Rat Facial Motoneurons after Axotomy. Eur. J. Neurosci. 1996, 8, 1018–1029. [Google Scholar] [CrossRef]
- Burazin, T.C.; Gundlach, A.L. Up-Regulation of GDNFR-Alpha and c-Ret MRNA in Facial Motor Neurons Following Facial Nerve Injury in the Rat. Mol. Brain Res. 1998, 55, 331–336. [Google Scholar] [CrossRef]
- Haas, C.A.; Hofmann, H.D.; Kirsch, M. Expression of CNTF/LIF-Receptor Components and Activation of STAT3 Signaling in Axotomized Facial Motoneurons: Evidence for a Sequential Postlesional Function of the Cytokines. J. Neurobiol. 1999, 41, 559–571. [Google Scholar] [CrossRef]
- Streit, W.J. Microglia as Neuroprotective, Immunocompetent Cells of the CNS. Glia 2002, 40, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Lalancette-Hébert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective Ablation of Proliferating Microglial Cells Exacerbates Ischemic Injury in the Brain. J. Neurosci. 2007, 27, 2596–2605. [Google Scholar] [CrossRef] [PubMed]
- Runge, E.M.; Iyer, A.K.; Setter, D.O.; Kennedy, F.M.; Sanders, V.M.; Jones, K.J. CD4+ T cell Expression of the IL-10 Receptor Is Necessary for Facial Motoneuron Survival after Axotomy. J. Neuroinflamm. 2020, 17, 121. [Google Scholar] [CrossRef]
- Kalla, R.; Liu, Z.; Xu, S.; Koppius, A.; Imai, Y.; Kloss, C.U.; Kohsaka, S.; Gschwendtner, A.; Möller, J.C.; Werner, A.; et al. Microglia and the Early Phase of Immune Surveillance in the Axotomized Facial Motor Nucleus: Impaired Microglial Activation and Lymphocyte Recruitment but No Effect on Neuronal Survival or Axonal Regeneration in Macrophage-Colony Stimulating Factor-Defici. J. Comp. Neurol. 2001, 436, 182–201. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S. Synaptic Release of Excitatory Amino Acid Neurotransmitter Mediates Anoxic Neuronal Death. J. Neurosci. 1984, 4, 1884–1891. [Google Scholar] [CrossRef] [PubMed]
- Récasens, M.; Guiramand, J.; Aimar, R.; Abdulkarim, A.; Barbanel, G. Metabotropic Glutamate Receptors as Drug Targets. Curr. Drug Targets 2007, 8, 651–681. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of Glutamate Transporters Reveals a Major Role for Astroglial Transport in Excitotoxicity and Clearance of Glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef]
- Hansson, E.; Muyderman, H.; Leonova, J.; Allansson, L.; Sinclair, J.; Blomstrand, F.; Thorlin, T.; Nilsson, M.; Rönnbäck, L. Astroglia and Glutamate in Physiology and Pathology: Aspects on Glutamate Transport, Glutamate-Induced Cell Swelling and Gap-Junction Communication. Neurochem. Int. 2000, 37, 317–329. [Google Scholar] [CrossRef]
- López-Redondo, F.; Nakajima, K.; Honda, S.; Kohsaka, S. Glutamate Transporter GLT-1 Is Highly Expressed in Activated Microglia Following Facial Nerve Axotomy. Mol. Brain Res. 2000, 76, 429–435. [Google Scholar] [CrossRef]
- Nakajima, K.; Tohyama, Y.; Kohsaka, S.; Kurihara, T. Ability of Rat Microglia to Uptake Extracellular Glutamate. Neurosci. Lett. 2001, 307, 171–174. [Google Scholar] [CrossRef]
- Nakajima, K.; Yamamoto, S.; Kohsaka, S.; Kurihara, T. Neuronal Stimulation Leading to Upregulation of Glutamate Transporter-1 (GLT-1) in Rat Microglia in Vitro. Neurosci. Lett. 2008, 436, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.A.; Liu, J.; Miller, J.W.; Rothstein, J.D.; Farrell, K.; Stein, B.A.; Longuemare, M.C. Neuronal Regulation of Glutamate Transporter Subtype Expression in Astrocytes. J. Neurosci. 1997, 17, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kanamatsu, T.; Koshimoto, M.; Kohsaka, S. Microglia Derived from the Axotomized Adult Rat Facial Nucleus Uptake Glutamate and Metabolize It to Glutamine in Vitro. Neurochem. Int. 2017, 102, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Ling, E.-A. Antioxidants and Neuroprotection in the Adult and Developing Central Nervous System. Curr. Med. Chem. 2008, 15, 3068–3080. [Google Scholar] [CrossRef]
- Fan, J.; Dawson, T.M.; Dawson, V.L. Cell Death Mechanisms of Neurodegeneration. Adv. Neurobiol. 2017, 15, 403–425. [Google Scholar] [CrossRef] [PubMed]
- Nogradi, B.; Meszlenyi, V.; Patai, R.; Polgar, T.F.; Spisak, K.; Kristof, R.; Siklos, L. Diazoxide Blocks or Reduces Microgliosis When Applied Prior or Subsequent to Motor Neuron Injury in Mice. Brain Res. 2020, 1741, 146875. [Google Scholar] [CrossRef]
- Lee, K.H.; Cha, M.; Lee, B.H. Crosstalk between Neuron and Glial Cells in Oxidative Injury and Neuroprotection. Int. J. Mol. Sci. 2021, 22, 13315. [Google Scholar] [CrossRef]
- Oshiro, S.; Kawamura, K.I.; Zhang, C.; Sone, T.; Morioka, M.S.; Kobayashi, S.; Nakajima, K. Microglia and Astroglia Prevent Oxidative Stress-Induced Neuronal Cell Death: Implications for Aceruloplasminemia. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 109–117. [Google Scholar] [CrossRef][Green Version]
- Evans, C.A.; Harbuz, M.S.; Ostenfeld, T.; Norrish, A.; Blackwell, J.M. Nramp1 Is Expressed in Neurons and Is Associated with Behavioural and Immune Responses to Stress. Neurogenetics 2001, 3, 69–78. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Heme Oxygenase 1 Is Required for Mammalian Iron Reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef]
- Dringen, R. Oxidative and Antioxidative Potential of Brain Microglial Cells. Antioxid. Redox Signal. 2005, 7, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Vilhardt, F.; Haslund-Vinding, J.; Jaquet, V.; McBean, G. Microglia Antioxidant Systems and Redox Signalling. Br. J. Pharmacol. 2017, 174, 1719–1732. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A.; Gilbert, D.L. Production of Superoxide Anions by a CNS Macrophage, the Microglia. FEBS Lett. 1987, 223, 284–288. [Google Scholar] [CrossRef]
- Chao, C.C.; Hu, S.; Molitor, T.W.; Shaskan, E.G.; Peterson, P.K. Activated Microglia Mediate Neuronal Cell Injury via a Nitric Oxide Mechanism. J. Immunol. 1992, 149, 2736–2741. [Google Scholar] [PubMed]
- Banati, R.B.; Gehrmann, J.; Schubert, P.; Kreutzberg, G.W. Cytotoxicity of Microglia. Glia 1993, 7, 111–118. [Google Scholar] [CrossRef]
- Alleva, C.; Machtens, J.P.; Kortzak, D.; Weyand, I.; Fahlke, C. Molecular Basis of Coupled Transport and Anion Conduction in Excitatory Amino Acid Transporters. Neurochem Res. 2022, 47, 9–22. [Google Scholar] [CrossRef]
- Takeuchi, H.; Jin, S.; Suzuki, H.; Doi, Y.; Liang, J.; Kawanokuchi, J.; Mizuno, T.; Sawada, M.; Suzumura, A. Blockade of Microglial Glutamate Release Protects against Ischemic Brain Injury. Exp. Neurol. 2008, 214, 144–146. [Google Scholar] [CrossRef]
- Noda, M.; Nakanishi, H.; Akaike, N. Glutamate Release from Microglia via Glutamate Transporter Is Enhanced by Amyloid-Beta Peptide. Neuroscience 1999, 92, 1465–1474. [Google Scholar] [CrossRef]
- Lindhout, I.A.; Murray, T.E.; Richards, C.M.; Klegeris, A. Potential Neurotoxic Activity of Diverse Molecules Released by Microglia. Neurochem. Int. 2021, 148, 105117. [Google Scholar] [CrossRef]
- Hogg, N. Free Radicals in Disease. Semin. Reprod. Endocrinol. 1998, 16, 241–248. [Google Scholar] [CrossRef]
- Mead, E.L.; Mosley, A.; Eaton, S.; Dobson, L.; Heales, S.J.; Pocock, J.M. Microglial Neurotransmitter Receptors Trigger Superoxide Production in Microglia; Consequences for Microglial-Neuronal Interactions. J. Neurochem. 2012, 121, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Paakkari, I.; Lindsberg, P. Nitric Oxide in the Central Nervous System. Ann. Med. 1995, 27, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Ramdial, K.; Franco, M.C.; Estevez, A.G. Cellular Mechanisms of Peroxynitrite-Induced Neuronal Death. Brain Res. Bull. 2017, 133, 4–11. [Google Scholar] [CrossRef]
- Streit, W.J.; Graeber, M.B.; Kreutzberg, G.W. Functional Plasticity of Microglia: A Review. Glia 1988, 1, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, J.; Matsumoto, Y.; Kreutzberg, G.W. Microglia: Intrinsic Immuneffector Cell of the Brain. Brain Res. Rev. 1995, 20, 269–287. [Google Scholar] [CrossRef]
- Kaur, G.; Han, S.J.; Yang, I.; Crane, C. Microglia and Central Nervous System Immunity. Neurosurg. Clin. N. Am. 2010, 21, 43–51. [Google Scholar] [CrossRef]
- Suzumura, A.; Sawada, M. Microglia as immunoregulatory cells in the central nervous system. In Topical Issues in Microglia Research; Ling, E., Tan, C., Tan, C., Eds.; Singapore Neuroscience Association: Singapore, 1996; pp. 189–202. [Google Scholar]
- Aloisi, F. Cytokine production. In Neuroglia, 2nd ed.; Kettenmann, H., Ransom, B., Eds.; Oxford University Press: Oxford, UK, 2005; pp. 285–301. [Google Scholar]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active Sensor and Versatile Effector Cells in the Normal and Pathologic Brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Dickson, D.W.; Lee, S.C.; Mattiace, L.A.; Yen, S.H.; Brosnan, C. Microglia and Cytokines in Neurological Disease, with Special Reference to AIDS and Alzheimer’s Disease. Glia 1993, 7, 75–83. [Google Scholar] [CrossRef]
- Stojkovska, I.; Wagner, B.M.; Morrison, B.E. Parkinson’s Disease and Enhanced Inflammatory Response. Exp. Biol. Med. 2015, 240, 1387–1395. [Google Scholar] [CrossRef]
- Evans, M.C.; Couch, Y.; Sibson, N.; Turner, M.R. Inflammation and Neurovascular Changes in Amyotrophic Lateral Sclerosis. Mol. Cell. Neurosci. 2013, 53, 34–41. [Google Scholar] [CrossRef]
- Patterson, P.H. Cytokines in Alzheimer’s Disease and Multiple Sclerosis. Curr. Opin. Neurobiol. 1995, 5, 642–646. [Google Scholar] [CrossRef]
- Streit, W.J.; Semple-Rowland, S.L.; Hurley, S.D.; Miller, R.C.; Popovich, P.G.; Stokes, B.T. Cytokine MRNA Profiles in Contused Spinal Cord and Axotomized Facial Nucleus Suggest a Beneficial Role for Inflammation and Gliosis. Exp. Neurol. 1998, 152, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Aldskogius, H.; Kozlova, E.N. Central Neuron-Glial and Glial-Glial Interactions Following Axon Injury. Prog. Neurobiol. 1998, 55, 1–26. [Google Scholar] [CrossRef]
- Ishijima, T.; Nakajima, K. Inflammatory Cytokines TNFα, IL-1β, and IL-6 Are Induced in Endotoxin- Stimulated Microglia through Different Signaling Cascades. Sci. Prog. 2021, 104, 003685042110549. [Google Scholar] [CrossRef]
- Yoshino, Y.; Yamamoto, S.; Kohsaka, S.; Oshiro, S.; Nakajima, K. Superoxide Anion Contributes to the Induction of Tumor Necrosis Factor Alpha (TNFα) through Activation of the MKK3/6-P38 MAPK Cascade in Rat Microglia. Brain Res. 2011, 1422, 1–12. [Google Scholar] [CrossRef]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef]
- Hopkins, S.J.; Rothwell, N.J. Cytokines and the Nervous System. I: Expression and Recognition. Trends Neurosci. 1995, 18, 83–88. [Google Scholar] [CrossRef]
- Klein, M.A.; Möller, J.C.; Jones, L.L.; Bluethmann, H.; Kreutzberg, G.W.; Raivich, G. Impaired Neuroglial Activation in Interleukin-6 Deficient Mice. Glia 1997, 19, 227–233. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia, the First Line of Defence in Brain Pathologies. Arzneimittelforschung 1995, 45, 357–360. [Google Scholar]
- Napoli, I.; Neumann, H. Microglial Clearance Function in Health and Disease. Neuroscience 2009, 158, 1030–1038. [Google Scholar] [CrossRef]
- Sokolowski, J.D.; Mandell, J.W. Phagocytic Clearance in Neurodegeneration. Am. J. Pathol. 2011, 178, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial Phagocytosis of Neurons in Neurodegeneration, and Its Regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Snider, W.D.; Thanedar, S. Target Dependence of Hypoglossal Motor Neurons during Development in Maturity. J. Comp. Neurol. 1989, 279, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Ikeda, K.; Shiojima, T.; Kinoshita, M. CNQX Prevents Spinal Motor Neuron Death Following Sciatic Nerve Transection in Newborn Rats. J. Neurol. Sci. 1995, 134, 21–25. [Google Scholar] [CrossRef]
- Damoiseaux, J.G.; Döpp, E.A.; Calame, W.; Chao, D.; MacPherson, G.G.; Dijkstra, C.D. Rat Macrophage Lysosomal Membrane Antigen Recognized by Monoclonal Antibody ED1. Immunology 1994, 83, 140–147. [Google Scholar]
- Mattsson, P.; Morgan, B.P.; Svensson, M. Complement activation and CD59 expression in the motor facial nucleus following intracranial transection of the facial nerve in the adult rat. J. Neuroimmunol. 1998, 91, 180–189. [Google Scholar] [CrossRef]
- Llewellyn-Smith, I.J.; Martin, C.L.; Arnolda, L.F.; Minson, J.B. Tracer-Toxins: Cholera Toxin B-Saporin as a Model. J. Neurosci. Methods 2000, 103, 83–90. [Google Scholar] [CrossRef]
- Nakajima, K.; Yamamoto, S.; Takezawa, Y.; Kohsaka, S. Transformation of Resident Microglia into Phagocytic Cells in the Transected and Cycloheximide-Treated Rat Facial Nucleus. Curr. Trends Neurol. 2015, 9, 37–43. [Google Scholar]
- Ravichandran, K.S. Find-Me and Eat-Me Signals in Apoptotic Cell Clearance: Progress and Conundrums. J. Exp. Med. 2010, 207, 1807–1817. [Google Scholar] [CrossRef]
- Naeini, M.B.; Bianconi, V.; Pirro, M.; Sahebkar, A. The Role of Phosphatidylserine Recognition Receptors in Multiple Biological Functions. Cell. Mol. Biol. Lett. 2020, 25, 23. [Google Scholar] [CrossRef]
- Pontejo, S.M.; Murphy, P.M. Chemokines Act as Phosphatidylserine-Bound “Find-Me” Signals in Apoptotic Cell Clearance. PLoS Biol. 2021, 19, e3001259. [Google Scholar] [CrossRef]
- Maruoka, M.; Suzuki, J. Regulation of Phospholipid Dynamics in Brain. Neurosci. Res. 2021, 167, 30–37. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; Lerner, A. The Second Phase of Brain Trauma Can Be Controlled by Nutraceuticals That Suppress DAMP-Mediated Microglial Activation. Expert Rev Neurother. 2021, 21, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Aderem, A.; Underhill, D.M. Mechanisms of Phagocytosis in Macrophages. Annu. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Phagocytosis: Phenotypically Simple Yet a Mechanistically Complex Process. Int. Rev. Immunol. 2020, 39, 118–150. [Google Scholar] [CrossRef]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP Acting at P2Y6 Receptors Is a Mediator of Microglial Phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef]
- Gu, B.J.; Wiley, J.S. P2X7 as a Scavenger Receptor for Innate Phagocytosis in the Brain. Br. J. Pharmacol. 2018, 175, 4195–4208. [Google Scholar] [CrossRef]
- Graeber, M.B.; Kreutzberg, G.W. Astrocytes Increase in Glial Fibrillary Acidic Protein during Retrograde Changes of Facial Motor Neurons. J. Neurocytol. 1986, 15, 363–373. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, K.K.W. Glial Fibrillary Acidic Protein: From Intermediate Filament Assembly and Gliosis to Neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef]
- Tetzlaff, W.; Graeber, M.B.; Bisby, M.A.; Kreutzberg, G.W. Increased Glial Fibrillary Acidic Protein Synthesis in Astrocytes during Retrograde Reaction of the Rat Facial Nucleus. Glia 1988, 1, 90–95. [Google Scholar] [CrossRef]
- Laskawi, R.; Wolff, J.R. Changes in Glial Fibrillary Acidic Protein Immunoreactivity in the Rat Facial Nucleus Following Various Types of Nerve Lesions. Eur. Arch. Otorhinolaryngol. 1996, 253, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Kreutzberg, G.W. Delayed Astrocyte Reaction Following Facial Nerve Axotomy. J. Neurocytol. 1988, 17, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Tetzlaff, W.; Streit, W.J.; Kreutzberg, G.W. Microglial Cells but Not Astrocytes Undergo Mitosis Following Rat Facial Nerve Axotomy. Neurosci. Lett. 1988, 85, 317–321. [Google Scholar] [CrossRef]
- Liedtke, W.; Edelmann, W.; Bieri, P.L.; Chiu, F.C.; Cowan, N.J.; Kucherlapati, R.; Raine, C.S. GFAP Is Necessary for the Integrity of CNS White Matter Architecture and Long-Term Maintenance of Myelination. Neuron 1996, 17, 607–615. [Google Scholar] [CrossRef]
- Wilhelmsson, U.; Li, L.; Pekna, M.; Berthold, C.-H.; Blom, S.; Eliasson, C.; Renner, O.; Bushong, E.; Ellisman, M.; Morgan, T.E.; et al. Absence of Glial Fibrillary Acidic Protein and Vimentin Prevents Hypertrophy of Astrocytic Processes and Improves Post-Traumatic Regeneration. J. Neurosci. 2004, 24, 5016–5021. [Google Scholar] [CrossRef]
- Rohlmann, A.; Laskawi, R.; Hofer, A.; Dermietzel, R.; Wolff, J.R. Astrocytes as Rapid Sensors of Peripheral Axotomy in the Facial Nucleus of Rats. Neuroreport 1994, 5, 409–412. [Google Scholar] [CrossRef]
- Graeber, M.; Kreutzberg, G. Astrocytic Reactions Accompanying Motor Neuron Regeneration. In Advances in Neural Regeneration Research; Wiley-Liss: Hoboken, NJ, USA, 1990; pp. 215–224. [Google Scholar]
- Müller, H.W.; Junghans, U.; Kappler, J. Astroglial Neurotrophic and Neurite-Promoting Factors. Pharmacol. Ther. 1995, 65, 1–18. [Google Scholar] [CrossRef]
- Yamagata, K.; Tagami, M.; Torii, Y.; Takenaga, F.; Tsumagari, S.; Itoh, S.; Yamori, Y.; Nara, Y. Sphingosine 1-Phosphate Induces the Production of Glial Cell Line-Derived Neurotrophic Factor and Cellular Proliferation in Astrocytes. Glia 2003, 41, 199–206. [Google Scholar] [CrossRef]
- Corbett, G.T.; Roy, A.; Pahan, K. Sodium Phenylbutyrate Enhances Astrocytic Neurotrophin Synthesis via Protein Kinase C (PKC)-Mediated Activation of CAMP-Response Element-Binding Protein (CREB): Implications for Alzheimer Disease Therapy. J. Biol. Chem. 2013, 288, 8299–8312. [Google Scholar] [CrossRef]
- Gehrmann, J.; Yao, D.L.; Bonetti, B.; Bondy, C.A.; Brenner, M.; Zhou, J.; Kreutzberg, G.W.; Webster, H.D. Expression of Insulin-like Growth Factor-I and Related Peptides during Motoneuron Regeneration. Exp. Neurol. 1994, 128, 202–210. [Google Scholar] [CrossRef]
- Saad, B.; Constam, D.B.; Ortmann, R.; Moos, M.; Fontana, A.; Schachner, M. Astrocyte-Derived TGF-Beta 2 and NGF Differentially Regulate Neural Recognition Molecule Expression by Cultured Astrocytes. J. Cell Biol. 1991, 115, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Constam, D.B.; Philipp, J.; Malipiero, U.V.; Ten Dijke, P.; Schachner, M.; Fontana, A. Differential Expression of Transforming Growth Factor-Beta 1, -Beta 2, and -Beta 3 by Glioblastoma Cells, Astrocytes, and Microglia. J. Immunol. 1992, 148, 1404–1410. [Google Scholar] [PubMed]
- Krieglstein, K.; Strelau, J.; Schober, A.; Sullivan, A.; Unsicker, K. TGF-Beta and the Regulation of Neuron Survival and Death. J. Physiol. Paris 2002, 96, 25–30. [Google Scholar] [CrossRef]
- Li, S.; Gu, X.; Yi, S. The Regulatory Effects of Transforming Growth Factor-β on Nerve Regeneration. Cell Transplant. 2017, 26, 381–394. [Google Scholar] [CrossRef]
- Zhao, Z.; Alam, S.; Oppenheim, R.W.; Prevette, D.M.; Evenson, A.; Parsadanian, A. Overexpression of Glial Cell Line-Derived Neurotrophic Factor in the CNS Rescues Motoneurons from Programmed Cell Death and Promotes Their Long-Term Survival Following Axotomy. Exp. Neurol. 2004, 190, 356–372. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The Role of Astrocytic Glutamate Transporters GLT-1 and GLAST in Neurological Disorders: Potential Targets for Neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.R.; Binder, D.K. Astrocyte Glutamate Uptake and Signaling as Novel Targets for Antiepileptogenic Therapy. Front. Neurol. 2020, 11, 1006. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Norenberg, M.D. Glutamine Synthetase: Role in Neurological Disorders. Adv. Neurobiol. 2016, 13, 327–350. [Google Scholar] [CrossRef]
- Yamashita, T.; Kohmura, E.; Yuguchi, T.; Shimada, S.; Tanaka, K.; Hayakawa, T.; Tohyama, M. Changes in Glutamate/Aspartate Transporter (GLAST/GluT-1) MRNA Expression Following Facial Nerve Transection. Mol. Brain Res. 1996, 38, 294–299. [Google Scholar] [CrossRef]
- Persson, M.; Rönnbäck, L. Microglial Self-Defence Mediated through GLT-1 and Glutathione. Amino Acids 2012, 42, 207–219. [Google Scholar] [CrossRef]
- Drukarch, B.; Schepens, E.; Stoof, J.C.; Langeveld, C.H.; Van Muiswinkel, F.L. Astrocyte-Enhanced Neuronal Survival Is Mediated by Scavenging of Extracellular Reactive Oxygen Species. Free Radic. Biol. Med. 1998, 25, 217–220. [Google Scholar] [CrossRef]
- Schreiner, B.; Romanelli, E.; Liberski, P.; Ingold-Heppner, B.; Sobottka-Brillout, B.; Hartwig, T.; Chandrasekar, V.; Johannssen, H.; Zeilhofer, H.U.; Aguzzi, A.; et al. Astrocyte Depletion Impairs Redox Homeostasis and Triggers Neuronal Loss in the Adult CNS. Cell Rep. 2015, 12, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lachance, B.B.; Mattson, M.P.; Jia, X. Glucose Metabolic Crosstalk and Regulation in Brain Function and Diseases. Prog. Neurobiol. 2021, 204, 102089. [Google Scholar] [CrossRef] [PubMed]
- Selwyn, R.G.; Cooney, S.J.; Khayrullina, G.; Hockenbury, N.; Wilson, C.M.; Jaiswal, S.; Bermudez, S.; Armstrong, R.C.; Byrnes, K.R. Outcome after Repetitive Mild Traumatic Brain Injury Is Temporally Related to Glucose Uptake Profile at Time of Second Injury. J. Neurotrauma 2016, 33, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C. Astrocytes as Key Regulators of Brain Energy Metabolism: New Therapeutic Perspectives. Front. Physiol. 2021, 12, 825816. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Glucose Transporters in Brain in Health and Disease. Pflug. Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- Dienel, G.A. Lactate Shuttling and Lactate Use as Fuel after Traumatic Brain Injury: Metabolic Considerations. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2014, 34, 1736–1748. [Google Scholar] [CrossRef]
- Buddhala, C.; Hsu, C.-C.; Wu, J.-Y. A Novel Mechanism for GABA Synthesis and Packaging into Synaptic Vesicles. Neurochem. Int. 2009, 55, 9–12. [Google Scholar] [CrossRef]
- McIntire, S.L.; Reimer, R.J.; Schuske, K.; Edwards, R.H.; Jorgensen, E.M. Identification and Characterization of the Vesicular GABA Transporter. Nature 1997, 389, 870–876. [Google Scholar] [CrossRef]
- Chaudhry, F.A.; Reimer, R.J.; Bellocchio, E.E.; Danbolt, N.C.; Osen, K.K.; Edwards, R.H.; Storm-Mathisen, J. The Vesicular GABA Transporter, VGAT, Localizes to Synaptic Vesicles in Sets of Glycinergic as Well as GABAergic Neurons. J. Neurosci. 1998, 18, 9733–9750. [Google Scholar] [CrossRef]
- Li, Y.Q.; Takada, M.; Kaneko, T.; Mizuno, N. Distribution of GABAergic and Glycinergic Premotor Neurons Projecting to the Facial and Hypoglossal Nuclei in the Rat. J. Comp. Neurol. 1997, 378, 283–294. [Google Scholar] [CrossRef]
- Aldes, L.D.; Chronister, R.B.; Marco, L.A. Distribution of Glutamic Acid Decarboxylase and Gamma-Aminobutyric Acid in the Hypoglossal Nucleus in the Rat. J. Neurosci. Res. 1988, 19, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Ohno, K.; Yamada, J.; Ikeda, M.; Okabe, A.; Sato, K.; Hashimoto, K.; Fukuda, A. Induction of NMDA and GABAA Receptor-Mediated Ca2+ Oscillations with KCC2 mRNA Downregulation in Injured Facial Motoneurons. J Neurophysiol. 2003, 89, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Möller, J.C.; Klein, M.A.; Haas, S.; Jones, L.L.; Kreutzberg, G.W.; Raivich, G. Regulation of Thrombospondin in the Regenerating Mouse Facial Motor Nucleus. Glia 1996, 17, 121–132. [Google Scholar] [CrossRef]
- Angelov, D.N.; Walther, M.; Streppel, M.; Guntinas-Lichius, O.; Neiss, W.F.; Probstmeier, R.; Pesheva, P. Tenascin-R Is Antiadhesive for Activated Microglia That Induce Downregulation of the Protein after Peripheral Nerve Injury: A New Role in Neuronal Protection. J. Neurosci. 1998, 18, 6218–6229. [Google Scholar] [CrossRef] [PubMed]
- Sajjan, S.; Holsinger, R.M.D.; Fok, S.; Ebrahimkhani, S.; Rollo, J.L.; Banati, R.B.; Graeber, M.B. Up-Regulation of Matrix Metallopeptidase 12 in Motor Neurons Undergoing Synaptic Stripping. Neuroscience 2014, 274, 331–340. [Google Scholar] [CrossRef]
- Gesase, A.P.; Kiyama, H. Peripheral Nerve Injury Induced Expression of MRNA for Serine Protease Inhibitor 3 in the Rat Facial and Hypoglossal Nuclei but Not in the Spinal Cord. Ital. J. Anat. Embryol. 2007, 112, 157–168. [Google Scholar]
- Nakajima, K.; Reddington, M.; Kohsaka, S.; Kreutzberg, G.W. Induction of Urokinase-Type Plasminogen Activator in Rat Facial Nucleus by Axotomy of the Facial Nerve. J. Neurochem. 1996, 66, 2500–2505. [Google Scholar] [CrossRef]
- Nakajima, K.; Tohyama, Y.; Kurihara, T.; Kohsaka, S. Axotomy-Dependent Urokinase Induction in the Rat Facial Nucleus: Possible Stimulation of Microglia by Neurons. Neurochem. Int. 2005, 46, 107–116. [Google Scholar] [CrossRef]
- Nakajima, K.; Tsuzaki, N.; Nagata, K.; Takemoto, N.; Kohsaka, S. Production and Secretion of Plasminogen in Cultured Rat Brain Microglia. FEBS Lett. 1992, 308, 179–182. [Google Scholar] [CrossRef]
- Inoue, K.; Nakajima, K.; Morimoto, T.; Kikuchi, Y.; Koizumi, S.; Illes, P.; Kohsaka, S. ATP Stimulation of Ca2+-Dependent Plasminogen Release from Cultured Microglia. Br. J. Pharmacol. 1998, 123, 1304–1310. [Google Scholar] [CrossRef]
- Nagata, K.; Nakajima, K.; Kohsaka, S. Plasminogen Promotes the Development of Rat Mesencephalic Dopaminergic Neurons in Vitro. Dev. Brain Res. 1993, 75, 31–37. [Google Scholar] [CrossRef]
- Nagata, K.; Nakajima, K.; Takemoto, N.; Saito, H.; Kohsaka, S. Microglia-Derived Plasminogen Enhances Neurite Outgrowth from Explant Cultures of Rat Brain. Int. J. Dev. Neurosci. 1993, 11, 227–237. [Google Scholar] [CrossRef]
- Blasi, F.; Vassalli, J.D.; Danø, K. Urokinase-Type Plasminogen Activator: Proenzyme, Receptor, and Inhibitors. J. Cell Biol. 1987, 104, 801–804. [Google Scholar] [CrossRef] [PubMed]
- HE, C.S.; Wilhelm, S.M.; Pentland, A.P.; Marmer, B.L.; Grant, G.A.; Eisen, A.Z.; Goldberg, G.I. Tissue Cooperation in a Proteolytic Cascade Activating Human Interstitial Collagenase. Proc. Natl. Acad. Sci. USA 1989, 86, 2632–2636. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hatakeyama, S.; Ikesue, A.; Miyai, H. Generation of Interleukin-8 by Plasmin from AVLPR-Interleukin-8, the Human Fibroblast-Derived Neutrophil Chemotactic Factor. FEBS Lett. 1991, 282, 412–414. [Google Scholar] [CrossRef]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making Sense of Latent TGFbeta Activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef]
- Reddington, M.; Priller, J.; Treichel, J.; Haas, C.; Kreutzberg, G.W. Astrocytes and Microglia as Potential Targets for Calcitonin Gene Related Peptide in the Central Nervous System. Can. J. Physiol. Pharmacol. 1995, 73, 1047–1049. [Google Scholar] [CrossRef]
- Liu, Y.; Honda, S.; Kohsaka, S.; Nakajima, K. Plasminogen Enhances the Secretion of Plasminogen Activator Inhibitor-1 from Cultured Rat Astrocytes. Neurosci. Lett. 2000, 282, 137–140. [Google Scholar] [CrossRef]
- Nakajima, K.; Yamamoto, S.; Tohyama, Y.; Kohsaka, S. Close Association of P38 and JNK with Plasminogen-Dependent Upregulation of PAI-1 in Rat Astrocytes in Vitro. Neurosci. Lett. 2010, 471, 66–69. [Google Scholar] [CrossRef]
- Maeda, S.; Nakajima, K.; Tohyama, Y.; Kohsaka, S. Characteristic Response of Astrocytes to Plasminogen/Plasmin to Upregulate Transforming Growth Factor Beta 3 (TGFβ3) Production/Secretion through Proteinase-Activated Receptor-1 (PAR-1) and the Downstream Phosphatidylinositol 3-Kinase (PI3K)-Akt/PKB Signa. Brain Res. 2009, 1305, 1–13. [Google Scholar] [CrossRef]
- Flanders, K.C.; Ren, R.F.; Lippa, C.F. Transforming Growth Factor-Betas in Neurodegenerative Disease. Prog. Neurobiol. 1998, 54, 71–85. [Google Scholar] [CrossRef]
- Blinzinger, K.; Kreutzberg, G. Displacement of Synaptic Terminals from Regenerating Motoneurons by Microglial Cells. Z. Zellforsch. Mikrosk. Anat. 1968, 85, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Gesase, A.P.; Kiyama, H. Morphological Changes and Expression of Protein Kinase CK2 Beta Subunit in the Microglia after Hypoglossal Nerve Transection. J. Neurocytol. 2000, 29, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Jinno, S.; Yamada, J. Using Comparative Anatomy in the Axotomy Model to Identify Distinct Roles for Microglia and Astrocytes in Synaptic Stripping. Neuron Glia Biol. 2011, 7, 55–66. [Google Scholar] [CrossRef]
- Graeber, M.B.; Bise, K.; Mehraein, P. Synaptic Stripping in the Human Facial Nucleus. Acta Neuropathol. 1993, 86, 179–181. [Google Scholar] [CrossRef]
- Spejo, A.B.; Oliveira, A.L.R. Synaptic Rearrangement Following Axonal Injury: Old and New Players. Neuropharmacology 2015, 96, 113–123. [Google Scholar] [CrossRef]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New Roles for the Synaptic Stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef]
- Perry, V.H.; O’Connor, V. The Role of Microglia in Synaptic Stripping and Synaptic Degeneration: A Revised Perspective. ASN Neuro 2010, 2, e00047. [Google Scholar] [CrossRef]
- Bendella, H.; Pavlov, S.P.; Grosheva, M.; Irintchev, A.; Angelova, S.K.; Merkel, D.; Sinis, N.; Kaidoglou, K.; Skouras, E.; Dunlop, S.A.; et al. Non-Invasive Stimulation of the Vibrissal Pad Improves Recovery of Whisking Function after Simultaneous Lesion of the Facial and Infraorbital Nerves in Rats. Exp. Brain Res. 2011, 212, 65–79. [Google Scholar] [CrossRef]
- Skouras, E.; Pavlov, S.; Bendella, H.; Angelov, D.N. Stimulation of Trigeminal Afferents Improves Motor Recovery after Facial Nerve Injury: Functional, Electrophysiological and Morphological Proofs. Adv. Anat. Embryol. Cell Biol. 2013, 213, 1–105. [Google Scholar] [CrossRef]
- Kitahara, T.; Kiryu, S.; Ohno, K.; Morita, N.; Kubo, T.; Kiyama, H. Up-regulation of ERK (MAP kinase) and MEK (MAP kinase kinase) Transcription After Rat Facial Nerve Transection. Neurosci. Res. 1994, 20, 275–280. [Google Scholar] [CrossRef]
- McNamara, R.K.; Jiang, Y.; Streit, W.J.; Lenox, R.H. Facial Motor Neuron Regeneration Induces a Unique Spatial and Temporal Pattern of Myristoylated Alanine-Rich C Kinase Substrate Expression. Neuroscience 2000, 97, 581–589. [Google Scholar] [CrossRef]
- Kirsch, M.; Terheggen, U.; Hofmann, H.-D. Ciliary Neurotrophic Factor Is an Early Lesion-Induced Retrograde Signal for Axotomized Facial Motoneurons. Mol. Cell. Neurosci. 2003, 24, 130–138. [Google Scholar] [CrossRef]
- Akazawa, C.; Tsuzuki, H.; Nakamura, Y.; Sasaki, Y.; Ohsaki, K.; Nakamura, S.; Arakawa, Y.; Kohsaka, S. The Upregulated Expression of Sonic Hedgehog in Motor Neurons after Rat Facial Nerve Axotomy. J. Neurosci. 2004, 24, 7923–7930. [Google Scholar] [CrossRef] [PubMed]
- Makwana, M.; Serchov, T.; Hristova, M.; Bohatschek, M.; Gschwendtner, A.; Kalla, R.; Liu, Z.; Heumann, R.; Raivich, G. Regulation and Function of Neuronal GTP-Ras in Facial Motor Nerve Regeneration. J. Neurochem. 2009, 108, 1453–1463. [Google Scholar] [CrossRef]
- Murphy, P.G.; Borthwick, L.S.; Johnston, R.S.; Kuchel, G.; Richardson, P.M. Nature of the Retrograde Signal from Injured Nerves That Induces Interleukin-6 MRNA in Neurons. J. Neurosci. 1999, 19, 3791–3800. [Google Scholar] [CrossRef]
- Rishal, I.; Fainzilber, M. Retrograde Signaling in Axonal Regeneration. Exp. Neurol. 2010, 223, 5–10. [Google Scholar] [CrossRef]
- Streit, W.J.; Dumoulin, F.L.; Raivich, G.; Kreutzberg, G.W. Calcitonin Gene-Related Peptide Increases in Rat Facial Motoneurons after Peripheral Nerve Transection. Neurosci. Lett. 1989, 101, 143–148. [Google Scholar] [CrossRef]
- Volonté, C.; Amadio, S.; Cavaliere, F.; D’Ambrosi, N.; Vacca, F.; Bernardi, G. Extracellular ATP and Neurodegeneration. Curr. Drug Targets-CNS Neurol. Disord. 2003, 2, 403–412. [Google Scholar] [CrossRef]
- Fields, R.D.; Burnstock, G. Purinergic Signalling in Neuron-Glia Interactions. Nat. Rev. Neurosci. 2006, 7, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T.; Wang, M.; Li, W. Regulation of Microglia by Ionotropic Glutamatergic and GABAergic Neurotransmission. Neuron Glia Biol. 2011, 7, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.; Dale, N. Activity-Dependent Release of Adenosine: A Critical Re-Evaluation of Mechanism. Curr. Neuropharmacol. 2008, 6, 329–337. [Google Scholar] [CrossRef]
- Luo, P.; Chu, S.-F.; Zhang, Z.; Xia, C.-Y.; Chen, N.-H. Fractalkine/CX3CR1 Is Involved in the Cross-Talk between Neuron and Glia in Neurological Diseases. Brain Res. Bull. 2019, 146, 12–21. [Google Scholar] [CrossRef]
- Lyons, A.; Downer, E.J.; Crotty, S.; Nolan, Y.M.; Mills, K.H.G.; Lynch, M.A. CD200 Ligand Receptor Interaction Modulates Microglial Activation in Vivo and in Vitro: A Role for IL-4. J. Neurosci. 2007, 27, 8309–8313. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, K.; Gonzalez, P.; Acarin, L. The Immune Inhibitory Complex CD200/CD200R Is Developmentally Regulated in the Mouse Brain. J. Comp. Neurol. 2012, 520, 2657–2675. [Google Scholar] [CrossRef] [PubMed]
- Patkó, T.; Vassias, I.; Vidal, P.P.; De Waele, C. Modulation of the Voltage-Gated Sodium- and Calcium-Dependent Potassium Channels in Rat Vestibular and Facial Nuclei after Unilateral Labyrinthectomy and Facial Nerve Transsection: An In Situ Hybridization Study. Neuroscience 2003, 117, 265–280. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hoppe, D.; Gottmann, K.; Banati, R.; Kreutzberg, G. Cultured Microglial Cells Have a Distinct Pattern of Membrane Channels Different from Peritoneal Macrophages. J. Neurosci. Res. 1990, 26, 278–287. [Google Scholar] [CrossRef]
- Korotzer, A.R.; Cotman, C.W. Voltage-Gated Currents Expressed by Rat Microglia in Culture. Glia 1992, 6, 81–88. [Google Scholar] [CrossRef]
- Nörenberg, W.; Illes, P.; Gebicke-Haerter, P.J. Sodium Channel in Isolated Human Brain Macrophages (Microglia). Glia 1994, 10, 165–172. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakajima, K.; Ishijima, T. Events Occurring in the Axotomized Facial Nucleus. Cells 2022, 11, 2068. https://doi.org/10.3390/cells11132068
Nakajima K, Ishijima T. Events Occurring in the Axotomized Facial Nucleus. Cells. 2022; 11(13):2068. https://doi.org/10.3390/cells11132068
Chicago/Turabian StyleNakajima, Kazuyuki, and Takashi Ishijima. 2022. "Events Occurring in the Axotomized Facial Nucleus" Cells 11, no. 13: 2068. https://doi.org/10.3390/cells11132068
APA StyleNakajima, K., & Ishijima, T. (2022). Events Occurring in the Axotomized Facial Nucleus. Cells, 11(13), 2068. https://doi.org/10.3390/cells11132068