
The Long Non-Coding RNA GOMAFU in Schizophrenia: Function, Disease Risk, and Beyond



Abstract
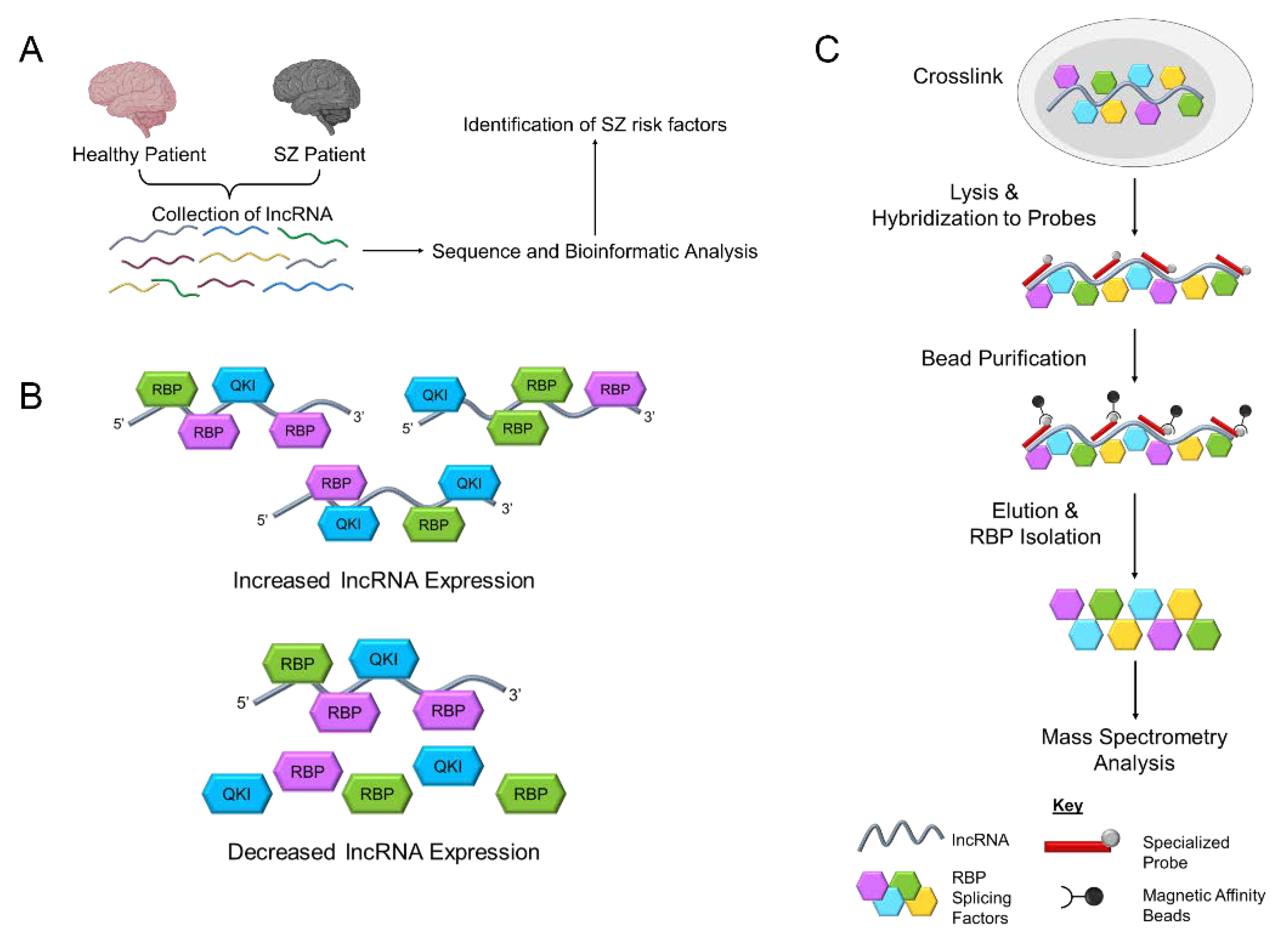
:1. Introduction
2. Schizophrenia, Candidate Genes, and Dysregulated Alternative Splicing during Neurodevelopment
3. Long Non-Coding RNAs in Schizophrenia
4. Discovery of GOMAFU and Disease-Associated Genetic Variants
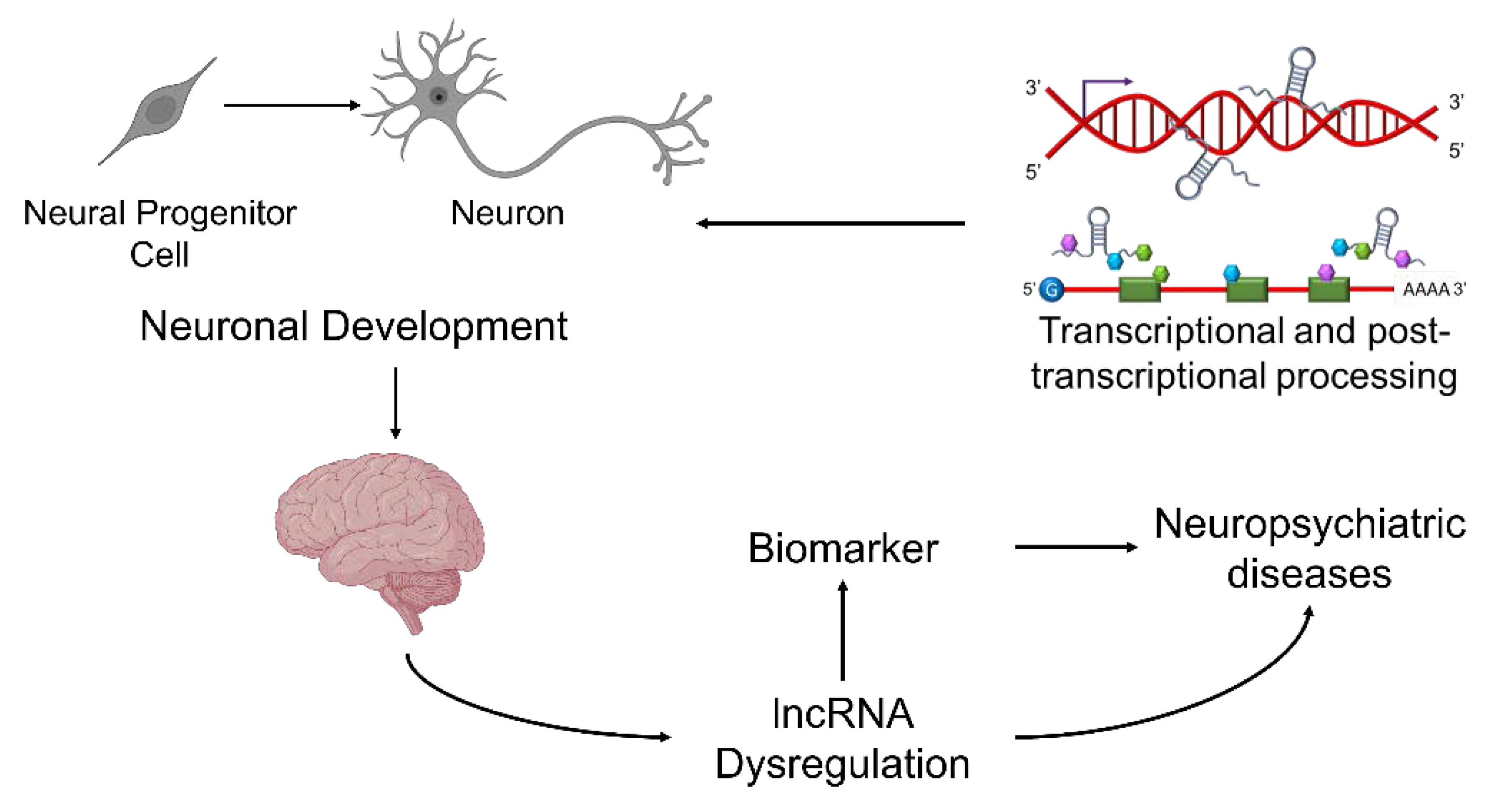
5. The lncRNA GOMAFU in Neuronal Development and Neurophysiological Function
6. Dysregulation of Human GOMAFU Is a Potential Risk Factor for Schizophrenia
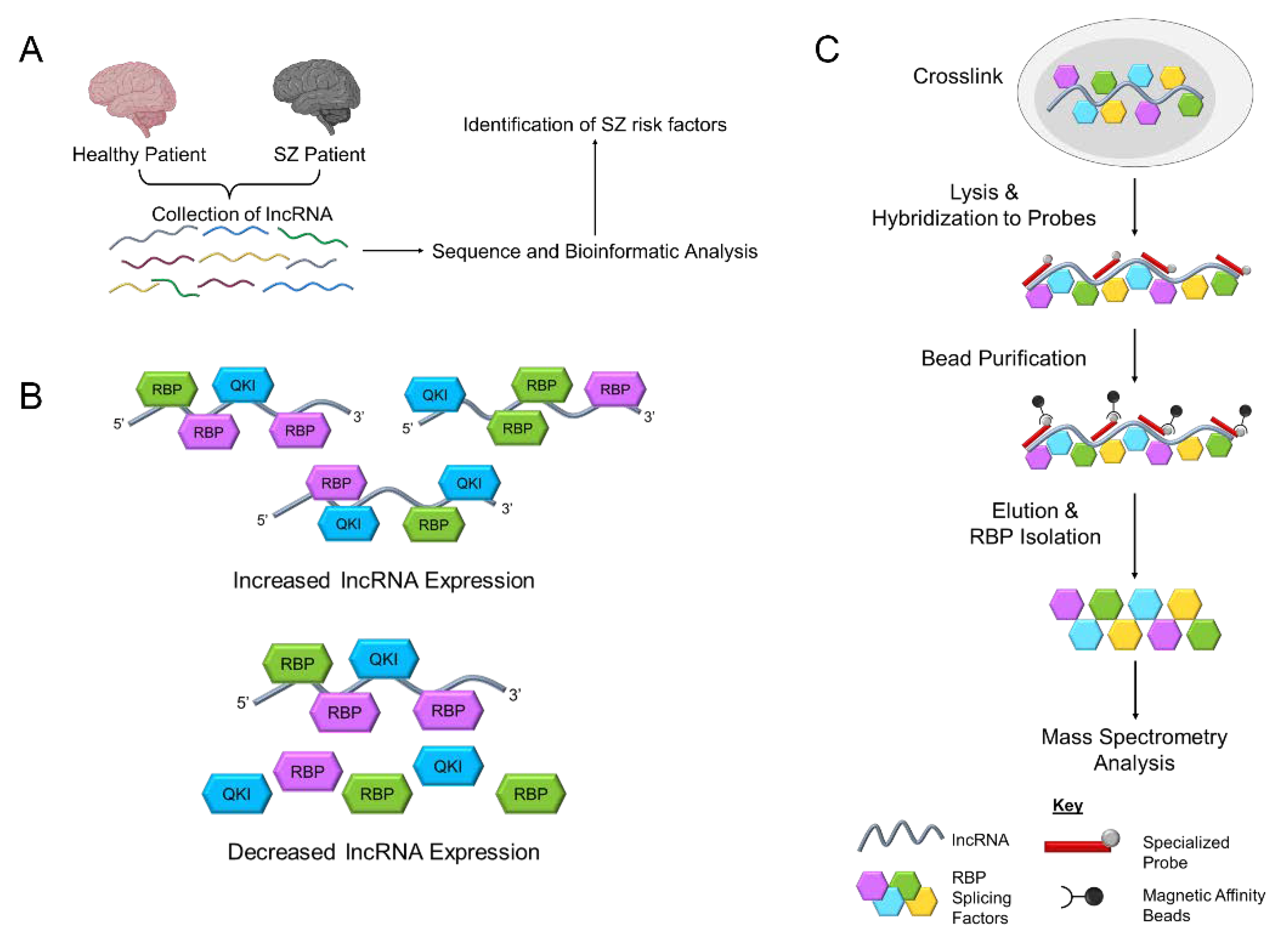
7. GOMAFU in the Peripheral Blood May Serve as a Biomarker for Diagnosis and Treatment for Schizophrenia
8. Mechanisms for GOMAFU to Regulate the Downstream Molecular Pathways in Schizophrenia
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattick, J.S. Non-coding RNAs: The architects of eukaryotic complexity. EMBO Rep. 2001, 2, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Aliperti, V.; Skonieczna, J.; Cerase, A. Long Non-Coding RNA (lncRNA) Roles in Cell Biology, Neurodevelopment and Neurological Disorders. Noncoding RNA 2021, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Dahariya, S.; Paddibhatla, I.; Kumar, S.; Raghuwanshi, S.; Pallepati, A.; Gutti, R.K. Long non-coding RNA: Classification, biogenesis and functions in blood cells. Mol. Immunol. 2019, 112, 82–92. [Google Scholar] [CrossRef]
- Zhang, K.; Shi, Z.M.; Chang, Y.N.; Hu, Z.M.; Qi, H.X.; Hong, W. The ways of action of long non-coding RNAs in cytoplasm and nucleus. Gene 2014, 547, 1–9. [Google Scholar] [CrossRef]
- Jarroux, J.; Morillon, A.; Pinskaya, M. History, Discovery, and Classification of lncRNAs. In Long Non Coding RNA Biology; Rao, M.R.S., Ed.; Springer: Singapore, 2017; pp. 1–46. [Google Scholar]
- Chen, L.L. Linking Long Noncoding RNA Localization and Function. Trends. Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef]
- van de Vondervoort, I.I.G.M.; Gordebeke, P.M.; Khoshab, N.; Tiesinga, P.H.E.; Buitelaar, J.K.; Kozicz, T.; Aschrafi, A.; Glennon, J.C. Long non-coding RNAs in neurodevelopmental disorders. Front. Mol. Neurosci. 2013, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Dykstra-Aiello, C.; Jickling, G.C.; Ander, B.P.; Shroff, N.; Zhan, X.; Liu, D.; Hull, H.; Orantia, M.; Stamova, B.S.; Sharp, F.R. Altered Expression of Long Noncoding RNAs in Blood After Ischemic Stroke and Proximity to Putative Stroke Risk Loci. Stroke 2016, 47, 2896–2903. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Wang, K.; Brunetti, T.; Xia, Y.; Jiao, C.; Dai, R.; Fitzgerald, D.; Thomas, A.; Jay, L.; Eckart, H.; et al. The DGCR5 long noncoding RNA may regulate expression of several schizophrenia-related genes. Sci. Transl. Med. 2018, 10, eaat6912. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Yu, Y.; Yang, W. Long noncoding RNA and its contribution to autism spectrum disorders. CNS Neurosci. Ther. 2017, 23, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Jablensky, A. The diagnostic concept of schizophrenia: Its history, evolution, and future prospects. Dialogues Clin. Neurosci. 2010, 12, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Kuperberg, G.; Heckers, S. Schizophrenia and cognitive function. Curr. Opin. Neurobiol. 2000, 10, 205–210. [Google Scholar] [CrossRef]
- Wang, Z.; Tong, Q.; Liao, H.; Rao, S.; Huang, X. Long non-coding RNAs in schizophrenia. Neurol. Psychiatry Brain Res. 2018, 30, 132–136. [Google Scholar] [CrossRef]
- Saha, S.; Chant, D.; Welham, J.; McGrath, J. A systematic review of the prevalence of schizophrenia. PLoS Med. 2005, 2, e141. [Google Scholar] [CrossRef]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Auquier, P.; Lançon, C.; Rouillon, F.; Lader, M. Mortality in schizophrenia. Pharmacoepidemiol. Drug Saf. 2007, 16, 1308–1312. [Google Scholar] [CrossRef]
- Iritani, S. What happens in the brain of schizophrenia patients? An investigation from the viewpoint of neuropathology. Nagoya J. Med. Sci. 2013, 75, 11–28. [Google Scholar]
- Sullivan, P.F.; Daly, M.J.; O’Donovan, M. Genetic architectures of psychiatric disorders: The emerging picture and its implications. Nat. Rev. Genet. 2012, 13, 537–551. [Google Scholar] [CrossRef]
- van Os, J.; Kenis, G.; Rutten, B.P.F. The environment and schizophrenia. Nature 2010, 468, 203–212. [Google Scholar] [CrossRef]
- Marín, O. Developmental timing and critical windows for the treatment of psychiatric disorders. Nat. Med. 2016, 22, 1229–1238. [Google Scholar] [CrossRef] [Green Version]
- Kang, E.; Wen, Z.; Song, H.; Christian, K.M.; Ming, G.-L. Adult Neurogenesis and Psychiatric Disorders. Cold Spring Harb. Perspect. Biol. 2016, 8, a019026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apple, D.M.; Fonseca, R.S.; Kokovay, E. The role of adult neurogenesis in psychiatric and cognitive disorders. Brain Res. 2017, 1655, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Woo, T.-U.W. Neurobiology of schizophrenia onset. Curr. Top. Behav. Neurosci. 2014, 16, 267–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landek-Salgado, M.A.; Faust, T.E.; Sawa, A. Molecular substrates of schizophrenia: Homeostatic signaling to connectivity. Mol. Psychiatry 2016, 21, 10–28. [Google Scholar] [CrossRef] [Green Version]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.R.; Farh, K.-H.; Holmans, P.A.; Lee, P.; Bulik-Sullivan, B.; Collier, D.A.; Huang, H.; et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Chambers, J.S.; Perrone-Bizzozero, N.I. Altered Myelination of the Hippocampal Formation in Subjects with Schizophrenia and Bipolar Disorder. Neurochem. Res. 2004, 29, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Hakak, Y.; Walker John, R.; Li, C.; Wong Wing, H.; Davis Kenneth, L.; Buxbaum Joseph, D.; Haroutunian, V.; Fienberg Allen, A. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4746–4751. [Google Scholar] [CrossRef] [Green Version]
- Haroutunian, V.; Katsel, P.; Dracheva, S.; Davis, K.L. The Human Homolog of the QKI Gene Affected in the Severe Dysmyelination “Quaking” Mouse Phenotype: Downregulated in Multiple Brain Regions in Schizophrenia. Am. J. Psychiatry 2006, 163, 1834–1837. [Google Scholar] [CrossRef]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362, eaat8127. [Google Scholar] [CrossRef] [Green Version]
- Ellis, S.E.; Panitch, R.; West, A.B.; Arking, D.E. Transcriptome analysis of cortical tissue reveals shared sets of downregulated genes in autism and schizophrenia. Transl. Psychiatry 2016, 6, e817. [Google Scholar] [CrossRef] [Green Version]
- Hess, J.L.; Tylee, D.S.; Barve, R.; de Jong, S.; Ophoff, R.A.; Kumarasinghe, N.; Tooney, P.; Schall, U.; Gardiner, E.; Beveridge, N.J.; et al. Transcriptome-wide mega-analyses reveal joint dysregulation of immunologic genes and transcription regulators in brain and blood in schizophrenia. Schizophr. Res. 2016, 176, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross-Disorder Group of the Psychiatric Genomics Consortium; Lee, S.H.; Ripke, S.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; Goddard, M.E.; et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donovan, M.C.; Craddock, N.; Norton, N.; Williams, H.; Peirce, T.; Moskvina, V.; Nikolov, I.; Hamshere, M.; Carroll, L.; Georgieva, L.; et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 2008, 40, 1053–1055. [Google Scholar] [CrossRef]
- Li, Z.; Chen, J.; Yu, H.; He, L.; Xu, Y.; Zhang, D.; Yi, Q.; Li, C.; Li, X.; Shen, J.; et al. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat. Genet. 2017, 49, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Li, Z.; Xu, Q.; Wang, T.; Li, T.; Shen, J.; Zhang, F.; Chen, J.; Zhou, G.; Ji, W.; et al. Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nat. Genet. 2011, 43, 1224–1227. [Google Scholar] [CrossRef] [PubMed]
- Reble, E.; Dineen, A.; Barr, C.L. The contribution of alternative splicing to genetic risk for psychiatric disorders. Genes Brain Behav. 2018, 17, e12430. [Google Scholar] [CrossRef] [Green Version]
- Law, A.J.; Kleinman, J.E.; Weinberger, D.R.; Weickert, C.S. Disease-associated intronic variants in the ErbB4 gene are related to altered ErbB4 splice-variant expression in the brain in schizophrenia. Hum. Mol. Genet. 2007, 16, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Nakata, K.; Lipska Barbara, K.; Hyde Thomas, M.; Ye, T.; Newburn Erin, N.; Morita, Y.; Vakkalanka, R.; Barenboim, M.; Sei, Y.; Weinberger Daniel, R.; et al. DISC1 splice variants are upregulated in schizophrenia and associated with risk polymorphisms. Proc. Natl. Acad. Sci. USA 2009, 106, 15873–15878. [Google Scholar] [CrossRef] [Green Version]
- Aberg, K.; Saetre, P.; Lindholm, E.; Ekholm, B.; Pettersson, U.; Adolfsson, R.; Jazin, E. Human QKI, a new candidate gene for schizophrenia involved in myelination. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141, 84–90. [Google Scholar] [CrossRef]
- Wu, J.I.; Reed, R.B.; Grabowski, P.J.; Artzt, K. Function of quaking in myelination: Regulation of alternative splicing. Proc. Natl. Acad. Sci. USA 2002, 99, 4233–4238. [Google Scholar] [CrossRef] [Green Version]
- Glatt, S.J.; Cohen, O.S.; Faraone, S.V.; Tsuang, M.T. Dysfunctional gene splicing as a potential contributor to neuropsychiatric disorders. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 156, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, T.; Manabe, T. Aberrant regulation of alternative pre-mRNA splicing in schizophrenia. Neurochem. Int. 2010, 57, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Holste, D.; Kreiman, G.; Burge, C.B. Variation in alternative splicing across human tissues. Genome Biol. 2004, 5, R74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdon, A.; Scuderi, S.; Capauto, D.; Abyzov, A.; Vaccarino, F.M. PsychENCODE and beyond: Transcriptomics and epigenomics of brain development and organoids. Neuropsychopharmacology 2021, 46, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Norton, N.; Moskvina, V.; Morris, D.W.; Bray, N.J.; Zammit, S.; Williams, N.M.; Williams, H.J.; Preece, A.C.; Dwyer, S.; Wilkinson, J.C.; et al. Evidence that interaction between neuregulin 1 and its receptor erbB4 increases susceptibility to schizophrenia. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2006, 141, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Silberberg, G.; Darvasi, A.; Pinkas-Kramarski, R.; Navon, R. The involvement of ErbB4 with schizophrenia: Association and expression studies. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2006, 141, 142–148. [Google Scholar] [CrossRef]
- Stefansson, H.; Sigurdsson, E.; Steinthorsdottir, V.; Bjornsdottir, S.; Sigmundsson, T.; Ghosh, S.; Brynjolfsson, J.; Gunnarsdottir, S.; Ivarsson, O.; Chou, T.T.; et al. Neuregulin 1 and susceptibility to schizophrenia. Am. J. Hum. Genet. 2002, 71, 877–892. [Google Scholar] [CrossRef] [Green Version]
- Veikkolainen, V.; Vaparanta, K.; Halkilahti, K.; Iljin, K.; Sundvall, M.; Elenius, K. Function of ERBB4 is determined by alternative splicing. Cell Cycle 2011, 10, 2647–2657. [Google Scholar] [CrossRef] [Green Version]
- Millar, J.K.; Wilson-Annan, J.C.; Anderson, S.; Christie, S.; Taylor, M.S.; Semple, C.A.M.; Devon, R.S.; Clair, D.M.S.; Muir, W.J.; Blackwood, D.H.R.; et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 2000, 9, 1415–1423. [Google Scholar] [CrossRef] [Green Version]
- Fazzari, P.; Paternain, A.V.; Valiente, M.; Pla, R.; Luján, R.; Lloyd, K.; Lerma, J.; Marín, O.; Rico, B. Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature 2010, 464, 1376–1380. [Google Scholar] [CrossRef]
- Joshi, D.; Fullerton, J.M.; Weickert, C.S. Elevated ErbB4 mRNA is related to interneuron deficit in prefrontal cortex in schizophrenia. J. Psychiatr. Res. 2014, 53, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vullhorst, D.; Neddens, J.; Karavanova, I.; Tricoire, L.; Petralia, R.S.; McBain, C.J.; Buonanno, A. Selective expression of ErbB4 in interneurons, but not pyramidal cells, of the rodent hippocampus. J. Neurosci. 2009, 29, 12255–12264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flames, N.; Long, J.E.; Garratt, A.N.; Fischer, T.M.; Gassmann, M.; Birchmeier, C.; Lai, C.; Rubenstein, J.L.R.; Marín, O. Short- and Long-Range Attraction of Cortical GABAergic Interneurons by Neuregulin-1. Neuron 2004, 44, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Brandon, N.J.; Sawa, A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat. Rev. Neurosci. 2011, 12, 707–722. [Google Scholar] [CrossRef]
- Chubb, J.E.; Bradshaw, N.J.; Soares, D.C.; Porteous, D.J.; Millar, J.K. The DISC locus in psychiatric illness. Mol. Psychiatry 2008, 13, 36–64. [Google Scholar] [CrossRef]
- Ishizuka, K.; Paek, M.; Kamiya, A.; Sawa, A. A Review of Disrupted-in-Schizophrenia-1 (disc1): Neurodevelopment, Cognition, and Mental Conditions. Biol. Psychiatry 2006, 59, 1189–1197. [Google Scholar] [CrossRef]
- Porteous, D.J.; Thomson, P.; Brandon, N.J.; Millar, J.K. The Genetics and Biology of Disc1—An Emerging Role in Psychosis and Cognition. Biol. Psychiatry 2006, 60, 123–131. [Google Scholar] [CrossRef]
- Kamiya, A.; Kubo, K.-i.; Tomoda, T.; Takaki, M.; Youn, R.; Ozeki, Y.; Sawamura, N.; Park, U.; Kudo, C.; Okawa, M.; et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 2005, 7, 1167–1178. [Google Scholar] [CrossRef]
- Shohat, S.; Ben-David, E.; Shifman, S. Varying Intolerance of Gene Pathways to Mutational Classes Explain Genetic Convergence across Neuropsychiatric Disorders. Cell Rep. 2017, 18, 2217–2227. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.M.; Moran, J.L.; Fromer, M.; Ruderfer, D.; Solovieff, N.; Roussos, P.; O’Dushlaine, C.; Chambert, K.; Bergen, S.E.; Kähler, A.; et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014, 506, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Genovese, G.; Fromer, M.; Stahl, E.A.; Ruderfer, D.M.; Chambert, K.; Landén, M.; Moran, J.L.; Purcell, S.M.; Sklar, P.; Sullivan, P.F.; et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci. 2016, 19, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.J.; O’Donovan, M.C. The genetics of neuropsychiatric disorders. Brain Neurosci. Adv. 2019, 2, 2398212818799271. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.-W.; Luo, T.; Zou, S.-S.; Wu, A.-S. The Role of Long Noncoding RNAs in Central Nervous System and Neurodegenerative Diseases. Front. Behav. Neurosci. 2018, 12, 175. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zuo, X.; Deng, H.; Liu, X.; Liu, L.; Ji, A. Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res. Bull. 2013, 97, 69–80. [Google Scholar] [CrossRef]
- Cao, J. The functional role of long non-coding RNAs and epigenetics. Biol. Proc. Online 2014, 16, 11. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Yang, L.; Chen, L.-L. The Diversity of Long Noncoding RNAs and Their Generation. Trends Genet. 2017, 33, 540–552. [Google Scholar] [CrossRef]
- Ørom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar] [CrossRef] [Green Version]
- Knauss, J.L.; Sun, T. Regulatory mechanisms of long noncoding RNAs in vertebrate central nervous system development and function. Neuroscience 2013, 235, 200–214. [Google Scholar] [CrossRef] [Green Version]
- Nie, L.; Wu, H.-J.; Hsu, J.-M.; Chang, S.-S.; Labaff, A.M.; Li, C.-W.; Wang, Y.; Hsu, J.L.; Hung, M.-C. Long non-coding RNAs: Versatile master regulators of gene expression and crucial players in cancer. Am. J. Transl. Res. 2012, 4, 127–150. [Google Scholar]
- Roberts, T.C.; Morris, K.V.; Wood, M.J. The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Philos. Trans. R Soc. Lond. B Biol. Sci. 2014, 369, 20130507. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA binding protein quaking regulates formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilusz, J.E. Long noncoding RNAs: Re-writing dogmas of RNA processing and stability. Biochim. Biophys Acta 2016, 1859, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Sunwoo, H.; Dinger, M.E.; Wilusz, J.E.; Amaral, P.P.; Mattick, J.S.; Spector, D.L. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 2009, 19, 347–359. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.A.; Valenstein, M.L.; Yario, T.A.; Tycowski, K.T.; Steitz, J.A. Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENbeta noncoding RNAs. Proc. Natl. Acad. Sci. USA 2012, 109, 19202–19207. [Google Scholar] [CrossRef] [Green Version]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3′ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Wilusz, J.E.; JnBaptiste, C.K.; Lu, L.Y.; Kuhn, C.D.; Joshua-Tor, L.; Sharp, P.A. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev. 2012, 26, 2392–2407. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell. 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Kornienko, A.E.; Guenzl, P.M.; Barlow, D.P.; Pauler, F.M. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, J.T.Y.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Dey, B.K.; Mueller, A.C.; Dutta, A. Long non-coding RNAs as emerging regulators of differentiation, development, and disease. Transcription 2014, 5, e944014. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, I.A.; Mehler, M.F. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci. 2012, 13, 528–541. [Google Scholar] [CrossRef] [Green Version]
- Cabili, M.N.; Dunagin, M.C.; McClanahan, P.D.; Biaesch, A.; Padovan-Merhar, O.; Regev, A.; Rinn, J.L.; Raj, A. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015, 16, 20. [Google Scholar] [CrossRef] [Green Version]
- Tsagakis, I.; Douka, K.; Birds, I.; Aspden, J.L. Long non-coding RNAs in development and disease: Conservation to mechanisms. J. Pathol. 2020, 250, 480–495. [Google Scholar] [CrossRef] [Green Version]
- Bridges, M.C.; Daulagala, A.C.; Kourtidis, A. LNCcation: lncRNA localization and function. J. Cell Biol. 2021, 220, e202009045. [Google Scholar] [CrossRef]
- Vance, K.W.; Ponting, C.P. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends. Genet. 2014, 30, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- McHugh, C.A.; Chen, C.-K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Arun, G.; Mao, Y.S.; Lazar, Z.; Hung, G.; Bhattacharjee, G.; Xiao, X.; Booth, C.J.; Wu, J.; Zhang, C.; et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012, 2, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 2009, 33, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, T.; Subhash, S.; Vaid, R.; Enroth, S.; Uday, S.; Reinius, B.; Mitra, S.; Mohammed, A.; James, A.R.; Hoberg, E.; et al. MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 2015, 6, 7743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sas-Nowosielska, H.; Magalska, A. Long Noncoding RNAs-Crucial Players Organizing the Landscape of the Neuronal Nucleus. Int. J. Mol. Sci. 2021, 22, 3478. [Google Scholar] [CrossRef] [PubMed]
- Ip, J.Y.; Nakagawa, S. Long non-coding RNAs in nuclear bodies. Dev. Growth Differ. 2012, 54, 44–54. [Google Scholar] [CrossRef]
- Noh, J.H.; Kim, K.M.; McClusky, W.G.; Abdelmohsen, K.; Gorospe, M. Cytoplasmic functions of long noncoding RNAs. Wiley Interdiscip. Rev. RNA 2018, 9, e1471. [Google Scholar] [CrossRef]
- Quan, Z.; Zheng, D.; Qing, H. Regulatory Roles of Long Non-Coding RNAs in the Central Nervous System and Associated Neurodegenerative Diseases. Front. Cell Neurosci. 2017, 11, 175. [Google Scholar] [CrossRef]
- Giovarelli, M.; Bucci, G.; Ramos, A.; Bordo, D.; Wilusz, C.J.; Chen, C.-Y.; Puppo, M.; Briata, P.; Gherzi, R. H19 long noncoding RNA controls the mRNA decay promoting function of KSRP. Proc. Natl. Acad. Sci. USA 2014, 111, E5023–E5028. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kopp, F.; Chang, T.-C.; Sataluri, A.; Chen, B.; Sivakumar, S.; Yu, H.; Xie, Y.; Mendell, J.T. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell 2016, 164, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legnini, I.; Morlando, M.; Mangiavacchi, A.; Fatica, A.; Bozzoni, I. A feedforward regulatory loop between HuR and the long noncoding RNA linc-MD1 controls early phases of myogenesis. Mol. Cell 2014, 53, 506–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Qureshi, I.A.; Gokhan, S.; Dinger, M.E.; Li, G.; Mattick, J.S.; Mehler, M.F. Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci. 2010, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, J.A.; Wolvetang, E.J.; Mattick, J.S.; Rinn, J.L.; Barry, G. Mechanisms of Long Non-coding RNAs in Mammalian Nervous System Development, Plasticity, Disease, and Evolution. Neuron 2015, 88, 861–877. [Google Scholar] [CrossRef] [Green Version]
- Kadakkuzha, B.M.; Liu, X.-A.; McCrate, J.; Shankar, G.; Rizzo, V.; Afinogenova, A.; Young, B.; Fallahi, M.; Carvalloza, A.C.; Raveendra, B.; et al. Transcriptome analyses of adult mouse brain reveal enrichment of lncRNAs in specific brain regions and neuronal populations. Front. Cell Neurosci. 2015, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 2016, 17, 67. [Google Scholar] [CrossRef] [Green Version]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [Green Version]
- Clark, B.S.; Blackshaw, S. Long non-coding RNA-dependent transcriptional regulation in neuronal development and disease. Front. Genet. 2014, 5, 164. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Pedrosa, E.; Shah, A.; Hrabovsky, A.; Maqbool, S.; Zheng, D.; Lachman, H.M. RNA-Seq of human neurons derived from iPS cells reveals candidate long non-coding RNAs involved in neurogenesis and neuropsychiatric disorders. PLoS ONE 2011, 6, e23356. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, K.C.; Frith, M.C.; Mattick, J.S. Rapid evolution of noncoding RNAs: Lack of conservation does not mean lack of function. Trends Genet. 2006, 22, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Batista, P.J.; Chang, H.Y. Long noncoding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponjavic, J.; Oliver, P.L.; Lunter, G.; Ponting, C.P. Genomic and transcriptional co-localization of protein-coding and long non-coding RNA pairs in the developing brain. PLoS Genet. 2009, 5, e1000617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, B.S.; Blackshaw, S. Understanding the Role of lncRNAs in Nervous System Development. Adv. Exp. Med. Biol. 2017, 1008, 253–282. [Google Scholar] [CrossRef] [Green Version]
- Parikshak, N.N.; Swarup, V.; Belgard, T.G.; Irimia, M.; Ramaswami, G.; Gandal, M.J.; Hartl, C.; Leppa, V.; Ubieta, L.d.l.T.; Huang, J.; et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540, 423–427. [Google Scholar] [CrossRef]
- D’haene, E.; Jacobs, E.Z.; Volders, P.-J.; De Meyer, T.; Menten, B.; Vergult, S. Identification of long non-coding RNAs involved in neuronal development and intellectual disability. Sci. Rep. 2016, 6, 28396. [Google Scholar] [CrossRef] [Green Version]
- Talkowski, M.E.; Maussion, G.; Crapper, L.; Rosenfeld, J.A.; Blumenthal, I.; Hanscom, C.; Chiang, C.; Lindgren, A.; Pereira, S.; Ruderfer, D.; et al. Disruption of a large intergenic noncoding RNA in subjects with neurodevelopmental disabilities. Am. J. Hum. Genet. 2012, 91, 1128–1134. [Google Scholar] [CrossRef] [Green Version]
- Labonté, B.; Abdallah, K.; Maussion, G.; Yerko, V.; Yang, J.; Bittar, T.; Quessy, F.; Golden, S.A.; Navarro, L.; Checknita, D.; et al. Regulation of impulsive and aggressive behaviours by a novel lncRNA. Mol. Psychiatry 2021, 26, 3751–3764. [Google Scholar] [CrossRef]
- Cohen, O.S.; McCoy, S.Y.; Middleton, F.A.; Bialosuknia, S.; Zhang-James, Y.; Liu, L.; Tsuang, M.T.; Faraone, S.V.; Glatt, S.J. Transcriptomic analysis of postmortem brain identifies dysregulated splicing events in novel candidate genes for schizophrenia. Schizophr. Res. 2012, 142, 188–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, E.; Bagheri-Hosseinabadi, Z.; De Toma, I.; Jafarisani, M.; Sadeghi, I. The importance of long non-coding RNAs in neuropsychiatric disorders. Mol. Asp. Med. 2019, 70, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Wei, Z.; Chang, X.; Liu, Y.; Gur, R.E.; Sleiman, P.M.A.; Hakonarson, H. The Long Noncoding RNA Landscape in Amygdala Tissues from Schizophrenia Patients. EBioMedicine 2018, 34, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, J.; Liu, X.; Ma, L.; Xu, Y.; Ren, Y. A preliminary analysis of LncRNA biomarkers for schizophrenia. Epigenomics 2021, 13, 1443–1458. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.-Q.; Hu, H.-L.; Ye, N.; Shen, Y.; Xu, Q. Genetic variants in long non-coding RNA MIAT contribute to risk of paranoid schizophrenia in a Chinese Han population. Schizophr. Res. 2015, 166, 125–130. [Google Scholar] [CrossRef]
- Rao, S.; Tian, L.; Cao, H.; Baranova, A.; Zhang, F. Involvement of the long intergenic non-coding RNA LINC00461 in schizophrenia. BMC Psychiatry 2022, 22, 59. [Google Scholar] [CrossRef]
- Blackshaw, S.; Harpavat, S.; Trimarchi, J.; Cai, L.; Huang, H.; Kuo, W.P.; Weber, G.; Lee, K.; Fraioli, R.E.; Cho, S.-H.; et al. Genomic analysis of mouse retinal development. PLoS Biol. 2004, 2, E247. [Google Scholar] [CrossRef]
- Rapicavoli, N.A.; Poth, E.M.; Blackshaw, S. The long noncoding RNA RNCR2 directs mouse retinal cell specification. BMC Dev. Biol. 2010, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Sone, M.; Hayashi, T.; Tarui, H.; Agata, K.; Takeichi, M.; Nakagawa, S. The mRNA-like noncoding RNA Gomafu constitutes a novel nuclear domain in a subset of neurons. J. Cell Sci. 2007, 120, 2498–2506. [Google Scholar] [CrossRef] [Green Version]
- Tsuiji, H.; Yoshimoto, R.; Hasegawa, Y.; Furuno, M.; Yoshida, M.; Nakagawa, S. Competition between a noncoding exon and introns: Gomafu contains tandem UACUAAC repeats and associates with splicing factor-1. Genes Cells 2011, 16, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, A.; Hasegawa, Y.; Ishida, K.; Yanaka, K.; Nakagawa, S. Formation of nuclear bodies by the lncRNA Gomafu-associating proteins Celf3 and SF1. Genes Cells Devoted Mol. Cell. Mech. 2014, 19, 704–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spector, D.L.; Lamond, A.I. Nuclear speckles. Cold Spring Harb. Perspect. Biol. 2011, 3, a000646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galganski, L.; Urbanek, M.O.; Krzyzosiak, W.J. Nuclear speckles: Molecular organization, biological function and role in disease. Nucleic Acids Res. 2017, 45, 10350–10368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Shao, C.; Wu, Q.J.; Chen, G.; Zhou, J.; Yang, B.; Li, H.; Gou, L.T.; Zhang, Y.; Wang, Y.; et al. NEAT1 scaffolds RNA-binding proteins and the Microprocessor to globally enhance pri-miRNA processing. Nat. Struct. Mol. Biol. 2017, 24, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Ozaki, K.; Sato, H.; Mizuno, H.; Susumu, S.; Takahashi, A.; Miyamoto, Y.; Ikegawa, S.; Kamatani, N.; Hori, M.; et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J. Hum. Genet. 2006, 51, 1087–1099. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; He, X.; Zhu, X.; Pang, S.; Yan, B. Promoter polymorphisms in the lncRNA-MIAT gene associated with acute myocardial infarction in Chinese Han population: A case-control study. Biosci. Rep. 2020, 40, BSR20191203. [Google Scholar] [CrossRef] [Green Version]
- Aprea, J.; Prenninger, S.; Dori, M.; Ghosh, T.; Monasor, L.S.; Wessendorf, E.; Zocher, S.; Massalini, S.; Alexopoulou, D.; Lesche, M.; et al. Transcriptome sequencing during mouse brain development identifies long non-coding RNAs functionally involved in neurogenic commitment. EMBO J. 2013, 32, 3145–3160. [Google Scholar] [CrossRef]
- Barry, G.; Briggs, J.A.; Vanichkina, D.P.; Poth, E.M.; Beveridge, N.J.; Ratnu, V.S.; Nayler, S.P.; Nones, K.; Hu, J.; Bredy, T.W.; et al. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol. Psychiatry 2014, 19, 486–494. [Google Scholar] [CrossRef] [Green Version]
- Spadaro, P.A.; Flavell, C.R.; Widagdo, J.; Ratnu, V.S.; Troup, M.; Ragan, C.; Mattick, J.S.; Bredy, T.W. Long Noncoding RNA-Directed Epigenetic Regulation of Gene Expression Is Associated with Anxiety-like Behavior in Mice. Biol. Psychiatry 2015, 78, 848–859. [Google Scholar] [CrossRef] [Green Version]
- Mackay, D.S.; Boskovska, O.B.; Knopf, H.L.S.; Lampi, K.J.; Shiels, A. A nonsense mutation in CRYBB1 associated with autosomal dominant cataract linked to human chromosome 22q. Am. J. Hum. Genet. 2002, 71, 1216–1221. [Google Scholar] [CrossRef] [Green Version]
- Leng, X.-Y.; Wang, S.; Cao, N.-Q.; Qi, L.-B.; Yan, Y.-B. The N-Terminal Extension of βB1-Crystallin Chaperones β-Crystallin Folding and Cooperates with αA-Crystallin. Biochemistry 2014, 53, 2464–2473. [Google Scholar] [CrossRef] [PubMed]
- Ip, J.Y.; Sone, M.; Nashiki, C.; Pan, Q.; Kitaichi, K.; Yanaka, K.; Abe, T.; Takao, K.; Miyakawa, T.; Blencowe, B.J.; et al. Gomafu lncRNA knockout mice exhibit mild hyperactivity with enhanced responsiveness to the psychostimulant methamphetamine. Sci. Rep. 2016, 6, 27204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhu, L.; Guan, F.; Yan, Z.; Liu, D.; Han, W.; Chen, T. Relationship between schizophrenia and changes in the expression of the long non-coding RNAs Meg3, Miat, Neat1 and Neat2. J. Psychiatr. Res. 2018, 106, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, S.; Liu, C.; Knowles, J.A.; Vaccarino, F.M.; Farnham, P.J.; Crawford, G.E.; Jaffe, A.E.; Pinto, D.; Dracheva, S.; Geschwind, D.H.; et al. The PsychENCODE project. Nat. Neurosci. 2015, 18, 1707–1712. [Google Scholar] [CrossRef] [Green Version]
- Chung, D.W.; Volk, D.W.; Arion, D.; Zhang, Y.; Sampson, A.R.; Lewis, D.A. Dysregulated ErbB4 Splicing in Schizophrenia: Selective Effects on Parvalbumin Expression. Am. J. Psychiatry 2016, 173, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-J.; Zhang, M.; Yin, D.-M.; Wen, L.; Ting, A.; Wang, P.; Lu, Y.-S.; Zhu, X.-H.; Li, S.-J.; Wu, C.-Y.; et al. ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 21818–21823. [Google Scholar] [CrossRef] [Green Version]
- Ting, A.K.; Chen, Y.; Wen, L.; Yin, D.-M.; Shen, C.; Tao, Y.; Liu, X.; Xiong, W.-C.; Mei, L. Neuregulin 1 promotes excitatory synapse development and function in GABAergic interneurons. J. Neurosci. 2011, 31, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Lu, Y.-S.; Zhu, X.-H.; Li, X.-M.; Woo, R.-S.; Chen, Y.-J.; Yin, D.-M.; Lai, C.; Terry Alvin, V.; Vazdarjanova, A.; et al. Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc. Natl. Acad. Sci. USA 2010, 107, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Volk, D.W.; Eggan, S.M.; Mirnics, K.; Pierri, J.N.; Sun, Z.; Sampson, A.R.; Lewis, D.A. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J. Neurosci. 2003, 23, 6315–6326. [Google Scholar] [CrossRef]
- Chen, S.; Sun, X.; Niu, W.; Kong, L.; He, M.; Li, W.; Zhong, A.; Lu, J.; Zhang, L. Aberrant Expression of Long Non-Coding RNAs in Schizophrenia Patients. Med. Sci. Monit. 2016, 22, 3340–3351. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Cui, Y.; Li, X.; Wang, B.; Na, L.; Shi, J.; Wang, L.; Qiu, L.; Zhang, K.; Liu, G.; et al. A co-expression network analysis reveals lncRNA abnormalities in peripheral blood in early-onset schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 63, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rao, S.; Xu, Y.; Zhang, F.; Wang, Z.; Zhao, X. Changes in the level of Long Non-Coding RNA Gomafu gene expression in schizophrenia patients before and after antipsychotic medication. Schizophr. Res. 2018, 195, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Badrlou, E.; Ghafouri-Fard, S.; Omrani, M.D.; Neishabouri, S.M.; Arsang-Jang, S.; Taheri, M.; Pouresmaeili, F. Expression of BDNF-Associated lncRNAs in Treatment-Resistant Schizophrenia Patients. J. Mol. Neurosci. 2021, 71, 2249–2259. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.J. Molecular defects in the dysmyelinating mutant quaking. J. Neurosci. Res. 1998, 51, 417–422. [Google Scholar] [CrossRef]
- Zhao, L.; Tian, D.; Xia, M.; Macklin, W.B.; Feng, Y. Rescuing qkV dysmyelination by a single isoform of the selective RNA-binding protein QKI. J. Neurosci. 2006, 26, 11278–11286. [Google Scholar] [CrossRef]
- Sidman, R.L.; Dickie, M.M.; Appel, S.H. Mutant Mice (Quaking and Jimpy) with Deficient Myelination in the Central Nervous System. Science 1964, 144, 309. [Google Scholar] [CrossRef]
- Chen, T.; Richard, S. Structure-Function Analysis of Qk1: A Lethal Point Mutation in Mouse quaking Prevents Homodimerization. Mol. Cell. Biol. 1998, 18, 4863–4871. [Google Scholar] [CrossRef] [Green Version]
- Chénard, C.A.; Richard, S. New implications for the QUAKING RNA binding protein in human disease. J. Neurosci. Res. 2008, 86, 233–242. [Google Scholar] [CrossRef]
- Biedermann, B.; Hotz, H.-R.; Ciosk, R. The Quaking family of RNA-binding proteins: Coordinators of the cell cycle and differentiation. Cell Cycle 2010, 9, 1929–1933. [Google Scholar] [CrossRef]
- Mandler, M.D.; Ku, L.; Feng, Y. A cytoplasmic quaking I isoform regulates the hnRNP F/H-dependent alternative splicing pathway in myelinating glia. Nucleic Acids Res. 2014, 42, 7319–7329. [Google Scholar] [CrossRef]
- Wu, J.; Zhou, L.; Tonissen, K.; Tee, R.; Artzt, K. The quaking I-5 protein (QKI-5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J. Biol. Chem. 1999, 274, 29202–29210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tian, D.; Ku, L.; Osterhout, D.J.; Feng, Y. The Selective RNA-binding Protein Quaking I (QKI) Is Necessary and Sufficient for Promoting Oligodendroglia Differentiation. J. Biol. Chem. 2007, 282, 23553–23560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larocque, D.; Fragoso, G.; Huang, J.; Mushynski, W.E.; Loignon, M.; Richard, S.; Almazan, G. The QKI-6 and QKI-7 RNA Binding Proteins Block Proliferation and Promote Schwann Cell Myelination. PLoS ONE 2009, 4, e5867. [Google Scholar] [CrossRef] [PubMed]
- Darbelli, L.; Richard, S. Emerging functions of the Quaking RNA-binding proteins and link to human diseases. Wiley Interdiscip. Rev. RNA 2016, 7, 399–412. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Li, D.; Feng, Y. Destabilization and mislocalization of myelin basic protein mRNAs in quaking dysmyelination lacking the QKI RNA-binding proteins. J. Neurosci. 2000, 20, 4944–4953. [Google Scholar] [CrossRef] [Green Version]
- Suiko, T.; Kobayashi, K.; Aono, K.; Kawashima, T.; Inoue, K.; Ku, L.; Feng, Y.; Koike, C. Expression of Quaking RNA-Binding Protein in the Adult and Developing Mouse Retina. PLoS ONE 2016, 11, e0156033. [Google Scholar] [CrossRef]
- Teplova, M.; Hafner, M.; Teplov, D.; Essig, K.; Tuschl, T.; Patel, D.J. Structure-function studies of STAR family Quaking proteins bound to their in vivo RNA target sites. Genes Dev. 2013, 27, 928–940. [Google Scholar] [CrossRef] [Green Version]
- Aberg, K.; Saetre, P.; Jareborg, N.; Jazin, E. Human QKI, a potential regulator of mRNA expression of human oligodendrocyte-related genes involved in schizophrenia. Proc Natl. Acad. Sci. USA 2006, 103, 7482–7487. [Google Scholar] [CrossRef] [Green Version]
- Lauriat, T.L.; Shiue, L.; Haroutunian, V.; Verbitsky, M.; Ares, M., Jr.; Ospina, L.; McInnes, L.A. Developmental expression profile of quaking, a candidate gene for schizophrenia, and its target genes in human prefrontal cortex and hippocampus shows regional specificity. J. Neurosci. Res. 2008, 86, 785–796. [Google Scholar] [CrossRef]
- Lindholm, E.; Åberg, K.; Ekholm, B.; Pettersson, U.; Adolfsson, R.; Jazin, E.E. Reconstruction of ancestral haplotypes in a 12-generation schizophrenia pedigree. Psychiatr. Genet. 2004, 14, 1–8. [Google Scholar] [CrossRef]
- Lindholm, E.; Ekholm, B.; Shaw, S.; Jalonen, P.; Johansson, G.; Pettersson, U.; Sherrington, R.; Adolfsson, R.; Jazin, E. A schizophrenia-susceptibility locus at 6q25, in one of the world’s largest reported pedigrees. Am. J. Hum. Genet. 2001, 69, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.Z.; Kondo, T.; Murata, T.; Ebersole, T.A.; Nishi, T.; Tada, K.; Ushio, Y.; Yamamura, K.; Abe, K. Expression of Hqk encoding a KH RNA binding protein is altered in human glioma. Jpn. J. Cancer Res. 2002, 93, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.M.; Vangompel, M.J.W.; Sametsky, E.A.; Clark, M.F.; Savage, J.C.; Disterhoft, J.F.; Kohtz, J.D. Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat. Neurosci. 2009, 12, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.-Y.; Johnson, R.; Stanton, L.W. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 2012, 31, 522–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parasramka, M.A.; Maji, S.; Matsuda, A.; Yan, I.K.; Patel, T. Long non-coding RNAs as novel targets for therapy in hepatocellular carcinoma. Pharmacol. Ther. 2016, 161, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.B.; Hezroni, H.; Goldrich, M.J.; Ulitsky, I. Regulation of Neuroregeneration by Long Noncoding RNAs. Mol. Cell 2018, 72, 553–567.e555. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Chang, H.Y. ChIRP-MS: RNA-Directed Proteomic Discovery. In X-Chromosome Inactivation: Methods and Protocols; Sado, T., Ed.; Springer: New York, NY, USA, 2018; pp. 37–45. [Google Scholar]
- Chu, C.; Zhang, Q.C.; Da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Chang, H.Y. Systematic Discovery of Xist RNA Binding Proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Spitale, R.C.; Chang, H.Y. Technologies to probe functions and mechanisms of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015, 22, 29–35. [Google Scholar] [CrossRef]
- Simon, M.D.; Wang, C.I.; Kharchenko, P.V.; West, J.A.; Chapman, B.A.; Alekseyenko, A.A.; Borowsky, M.L.; Kuroda, M.I.; Kingston, R.E. The genomic binding sites of a noncoding RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20497–20502. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.D. Capture hybridization analysis of RNA targets (CHART). Curr. Protoc. Mol. Biol. 2013, 101, 21–25. [Google Scholar] [CrossRef]
- Yang, Y.; Wen, L.; Zhu, H. Unveiling the hidden function of long non-coding RNA by identifying its major partner-protein. Cell Biosci. 2015, 5, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
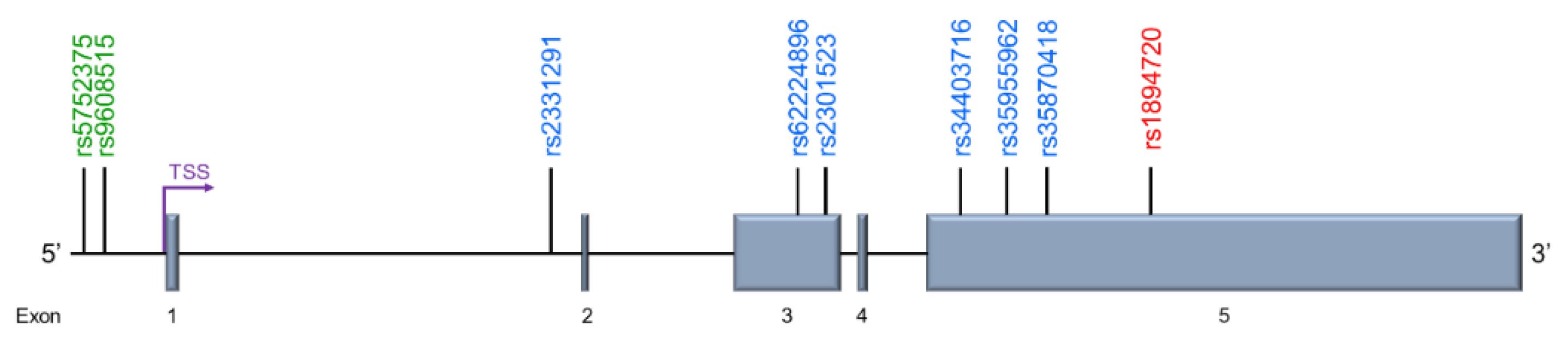
| Disease | dbSNP | Alternative Identification | SNP Position | Chromosomal Location | Allele | Ref. |
|---|---|---|---|---|---|---|
| Acute Myocardial Infarction | rs5752375 | g.4063 T>C | −938 | 26656544 | T/C | [137] |
| rs9608515 | g.4137 T>C | −864 | 26656618 | T/C | ||
| Myocardial Infarction | rs2331291 | - | 5376 | 26662857 | C/T | [136] |
| rs62224896 | Exon 3 8813 | 8851 | 26666332 | G/A | ||
| rs2301523 | - | 9224 | 26666705 | G/A | ||
| rs34403716 | Exon 5 11,093 | 11,132 | 26668613 | G/A | ||
| rs35955962 | Exon 5 11,741 | 11,780 | 26669261 | G/A | ||
| rs35870418 | Exon 5 12,311 | 12,350 | 26669831 | C/T | ||
| Paranoid Schizophrenia | rs1894720 | - | 13,780 | 26671261 | G/T | [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakutansky, P.M.; Feng, Y. The Long Non-Coding RNA GOMAFU in Schizophrenia: Function, Disease Risk, and Beyond. Cells 2022, 11, 1949. https://doi.org/10.3390/cells11121949
Zakutansky PM, Feng Y. The Long Non-Coding RNA GOMAFU in Schizophrenia: Function, Disease Risk, and Beyond. Cells. 2022; 11(12):1949. https://doi.org/10.3390/cells11121949
Chicago/Turabian StyleZakutansky, Paul M., and Yue Feng. 2022. "The Long Non-Coding RNA GOMAFU in Schizophrenia: Function, Disease Risk, and Beyond" Cells 11, no. 12: 1949. https://doi.org/10.3390/cells11121949
APA StyleZakutansky, P. M., & Feng, Y. (2022). The Long Non-Coding RNA GOMAFU in Schizophrenia: Function, Disease Risk, and Beyond. Cells, 11(12), 1949. https://doi.org/10.3390/cells11121949