
The Complex Interaction between P53 and miRNAs Joins New Awareness in Physiological Stress Responses



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
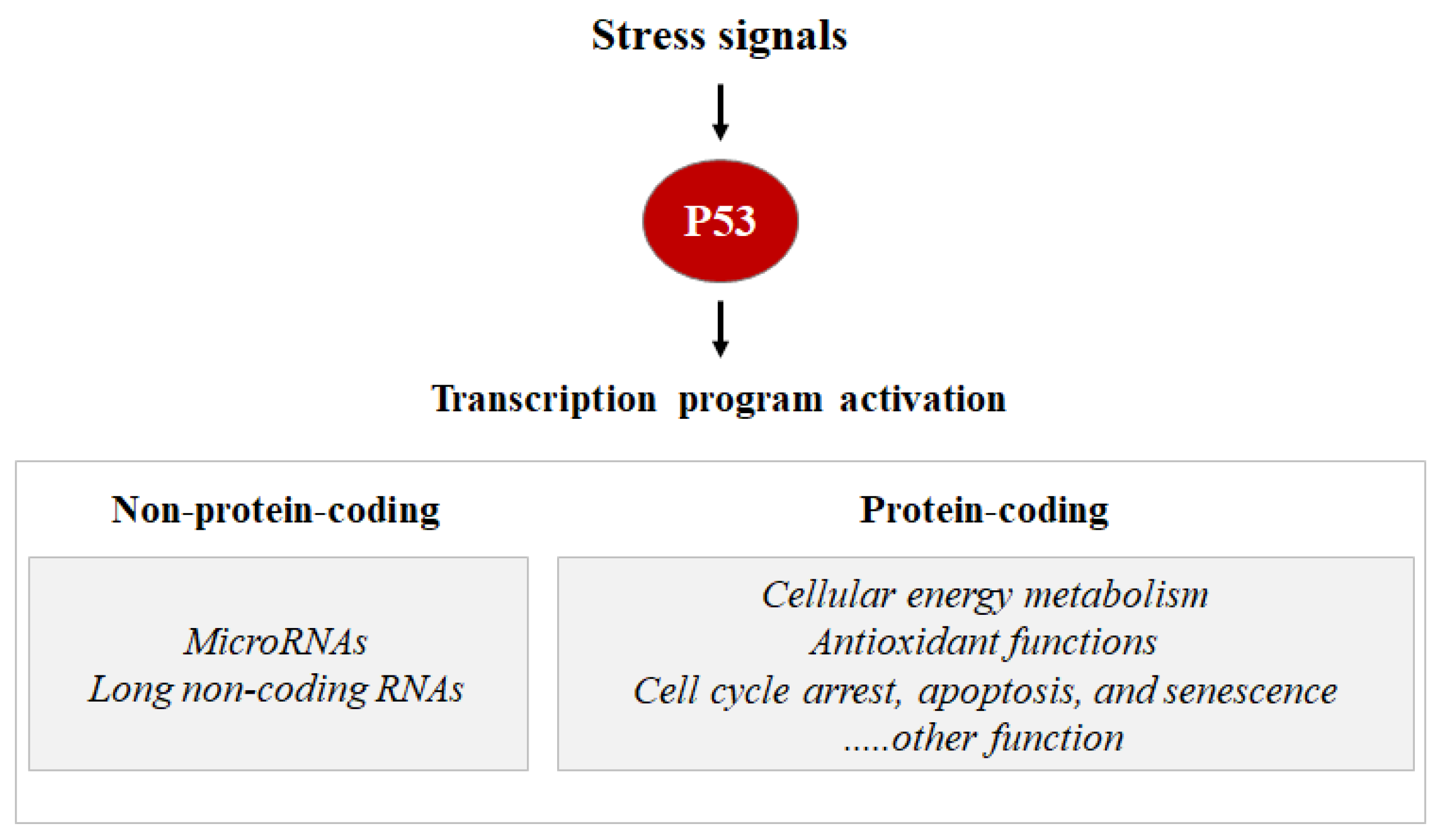
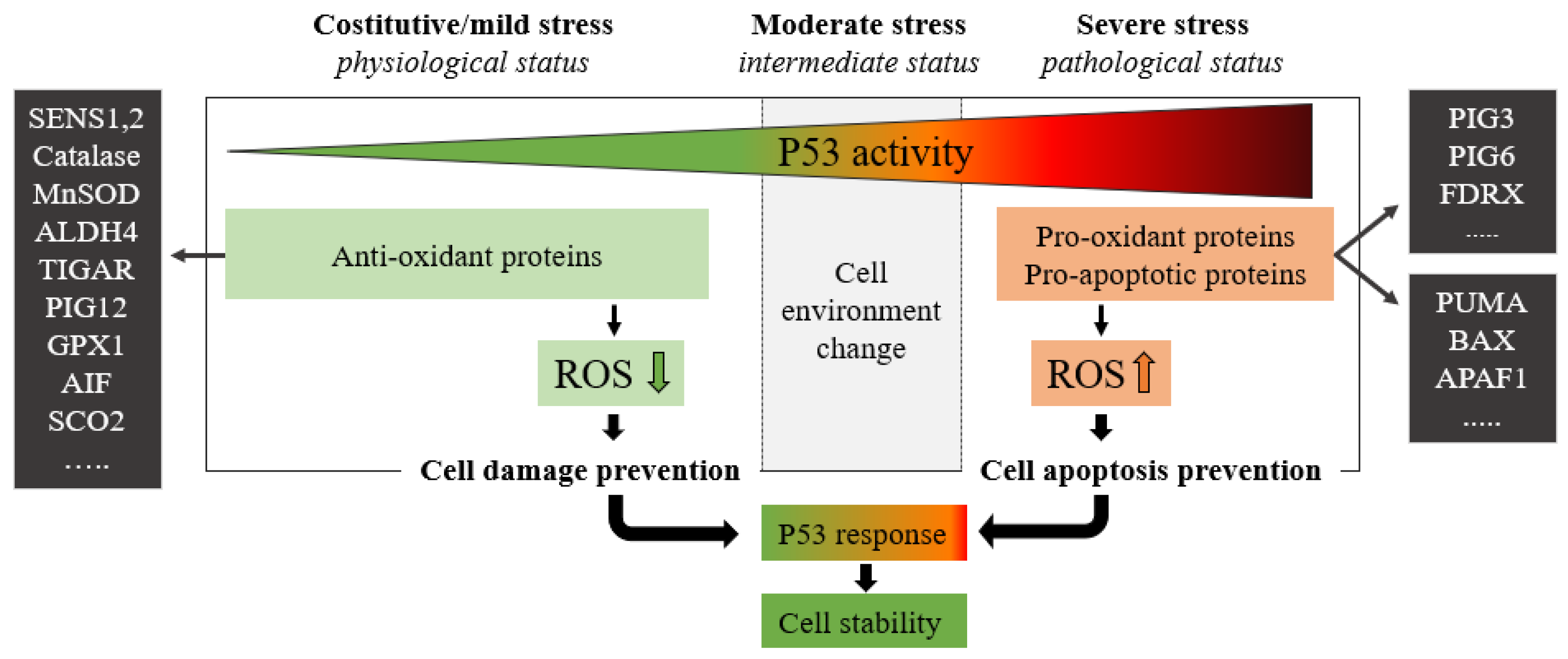
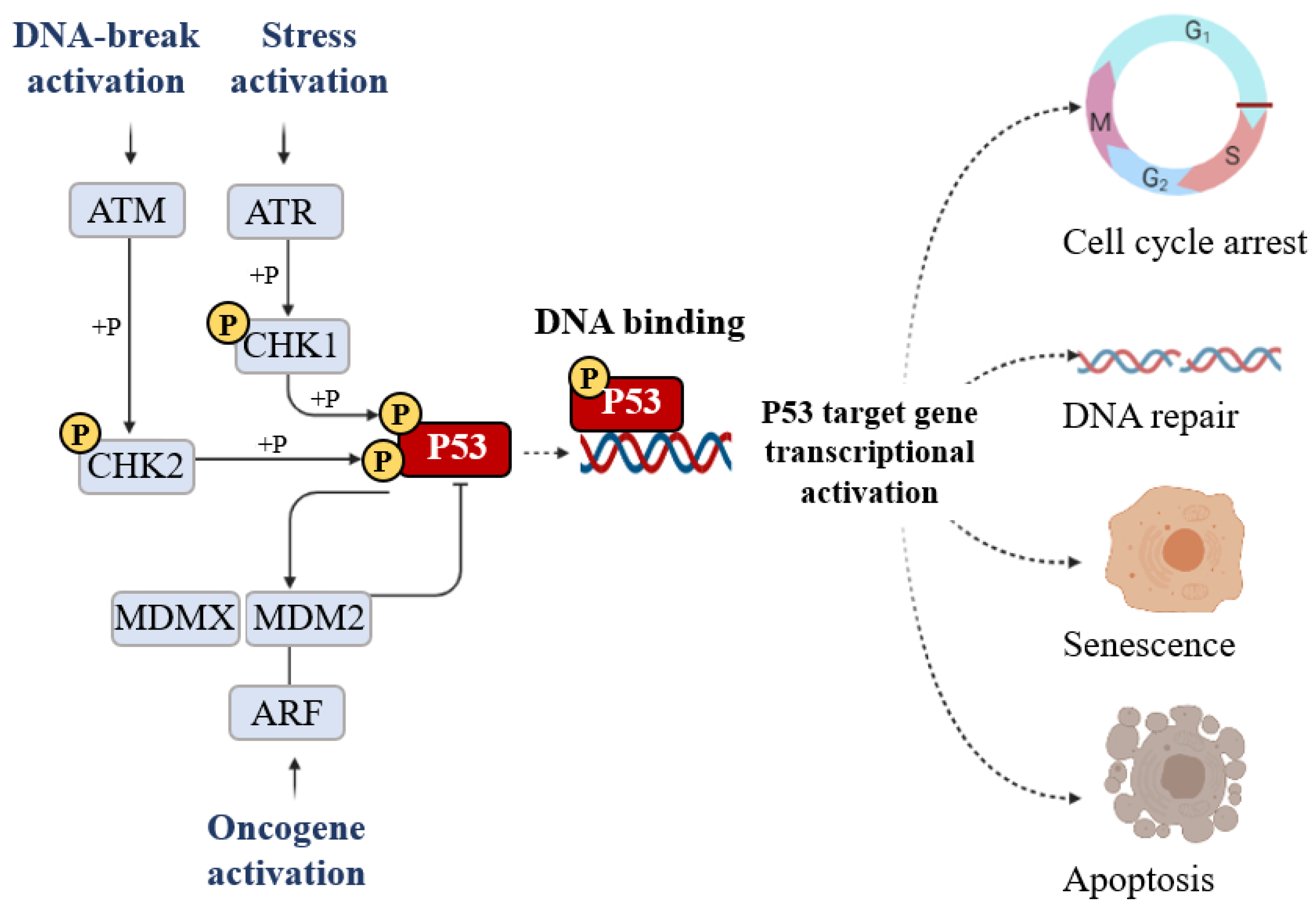
:1. P53 Is a Key Regulator of Cell Survival Pathways
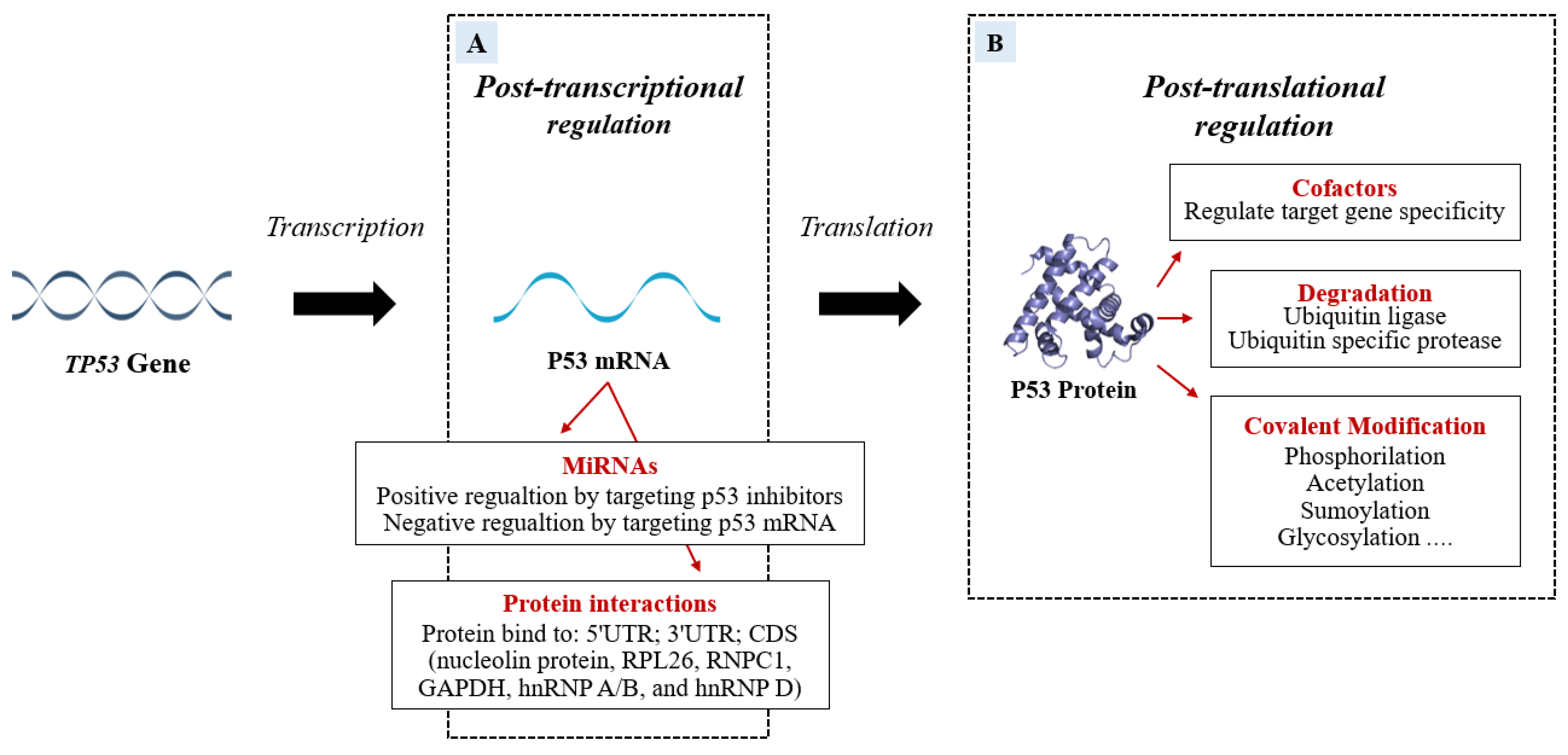
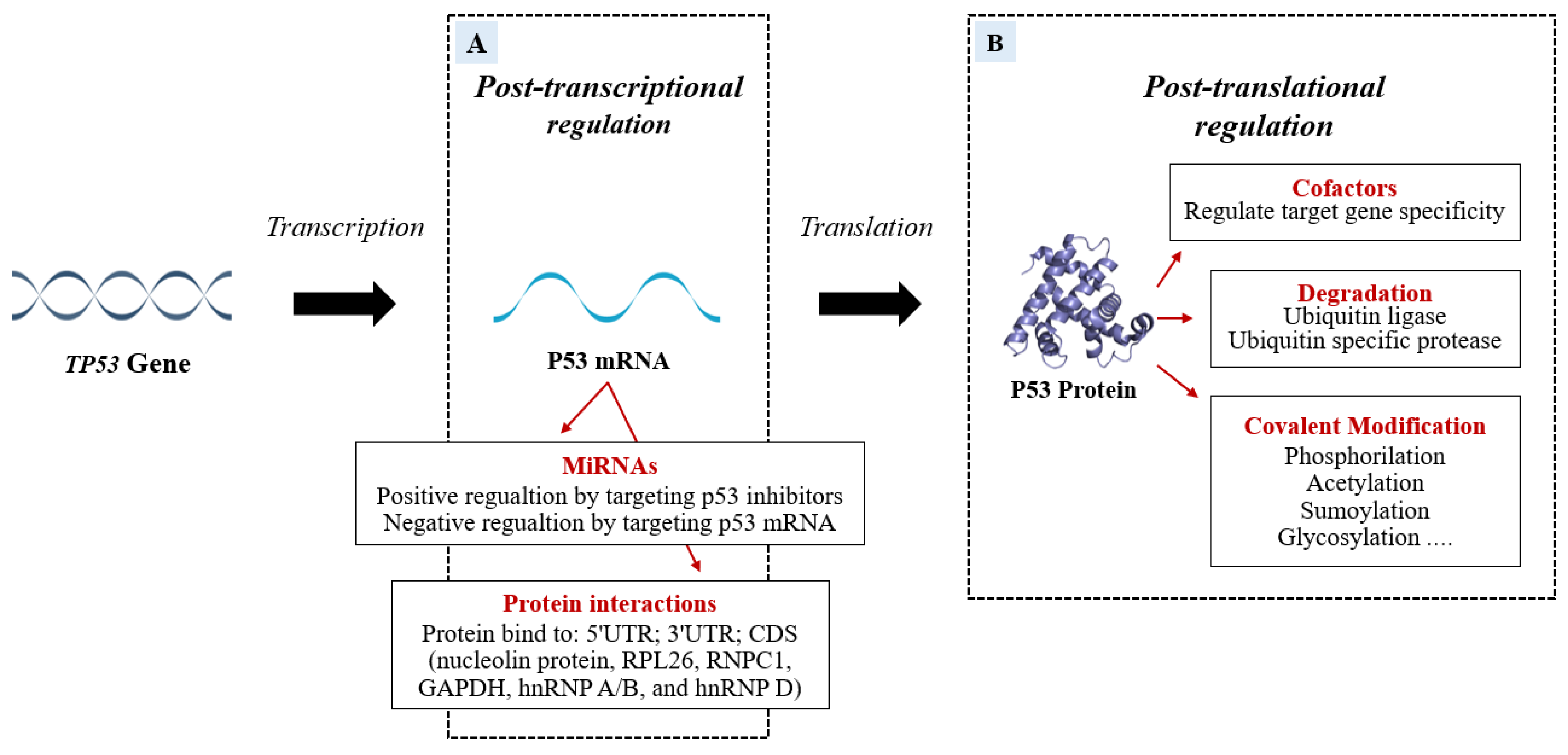
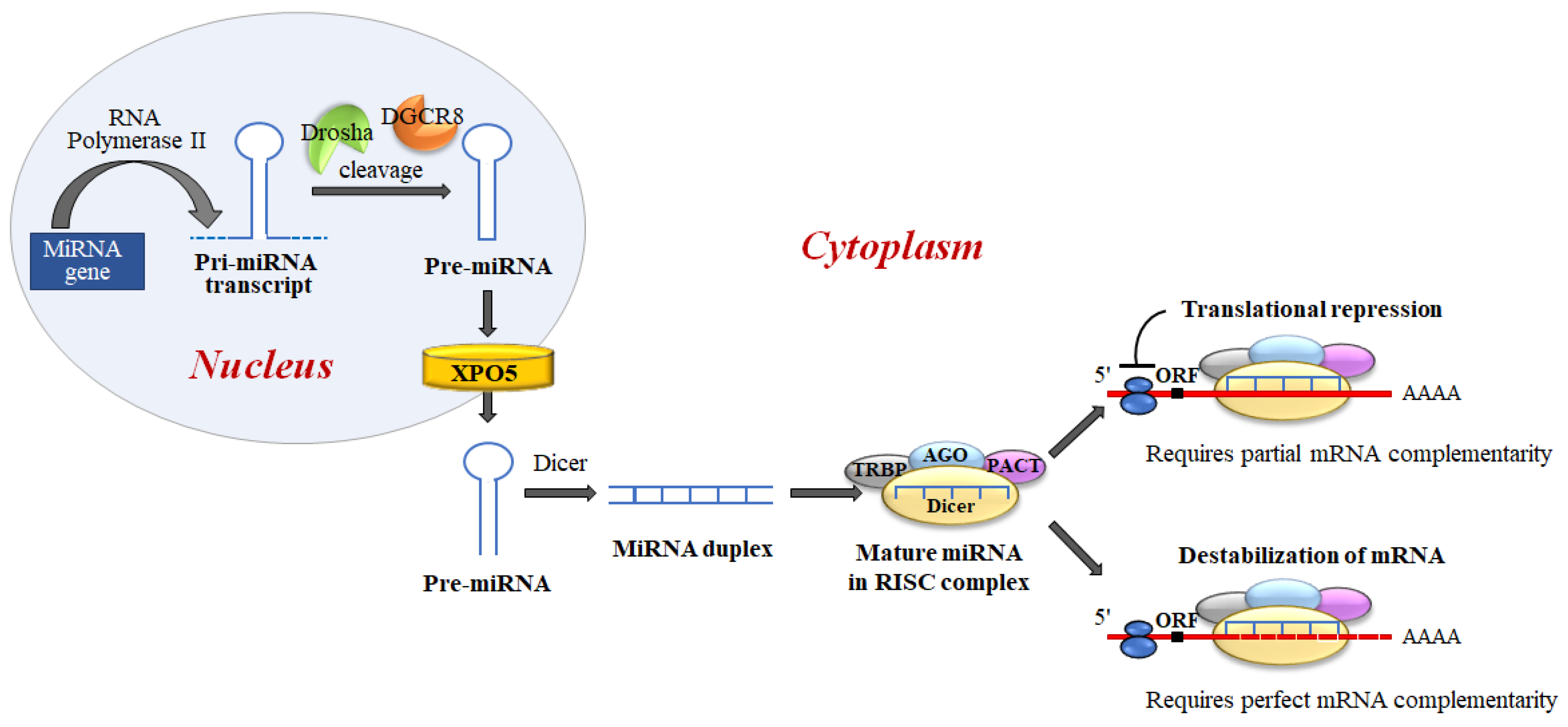
2. P53 Post-Transcriptional and Post-Translational Regulation
3. P53 Transcriptional Activation, and miRNA-mRNA Regulatory Network
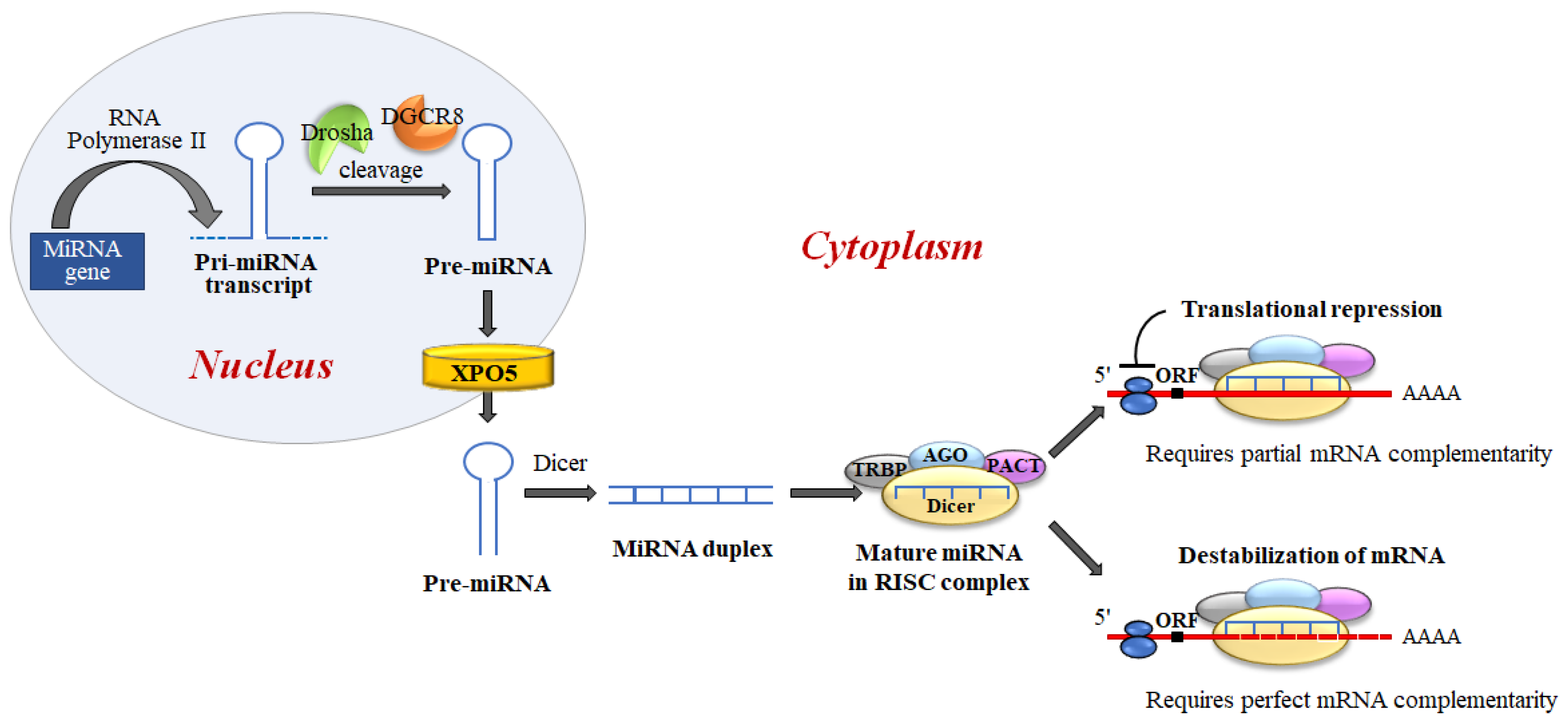
4. MiRNA Interacts with P53 at Multiple Levels
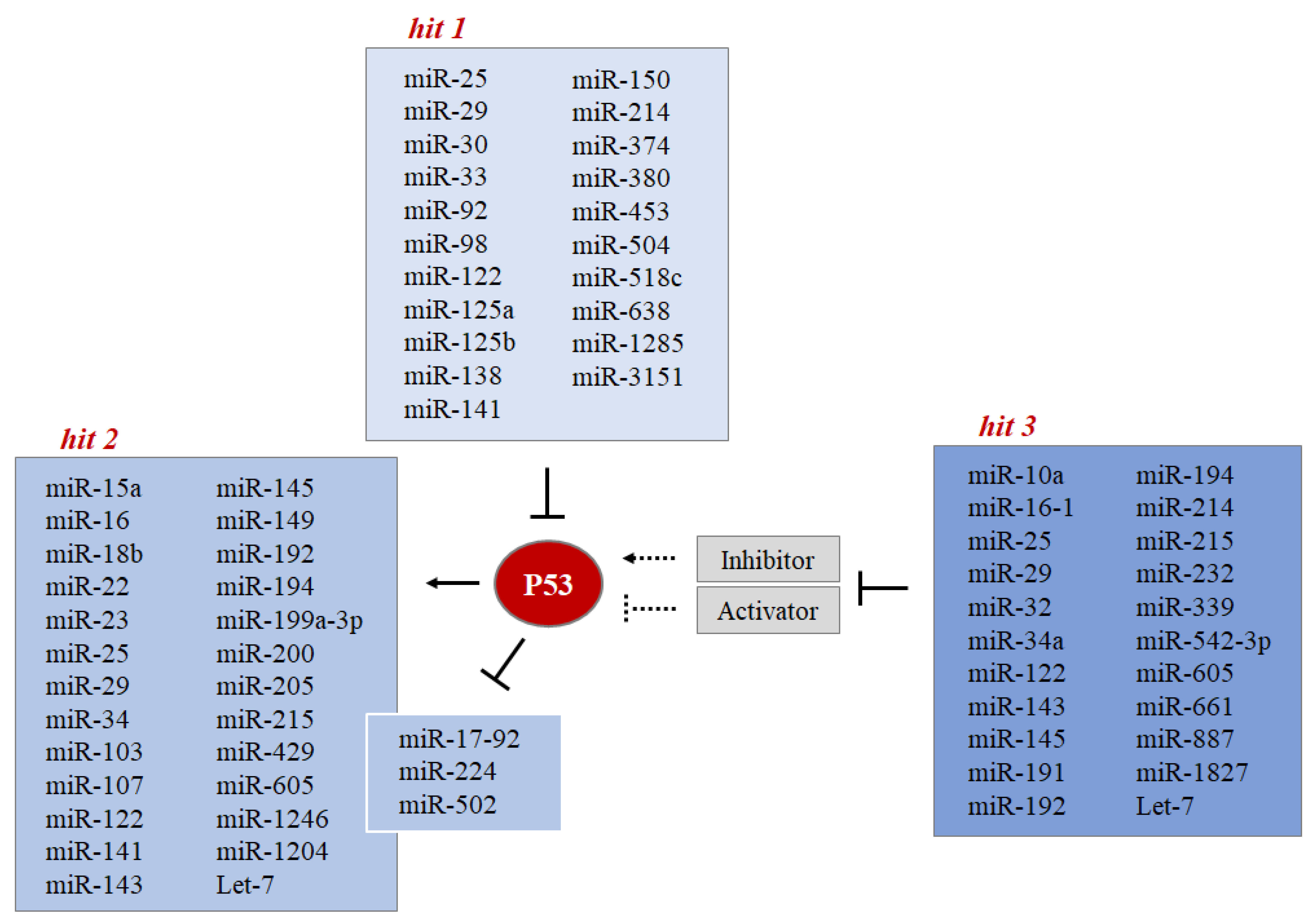
- Hit 1 MiRNAs directly control the level of P53 protein.
- Hit 2 P53 controls the expression level of some miRNAs through the regulation of their transcription or biogenesis.
- Hit 3 MiRNAs indirectly control the level of P53 protein by targeting its regulators (such as MDMs).
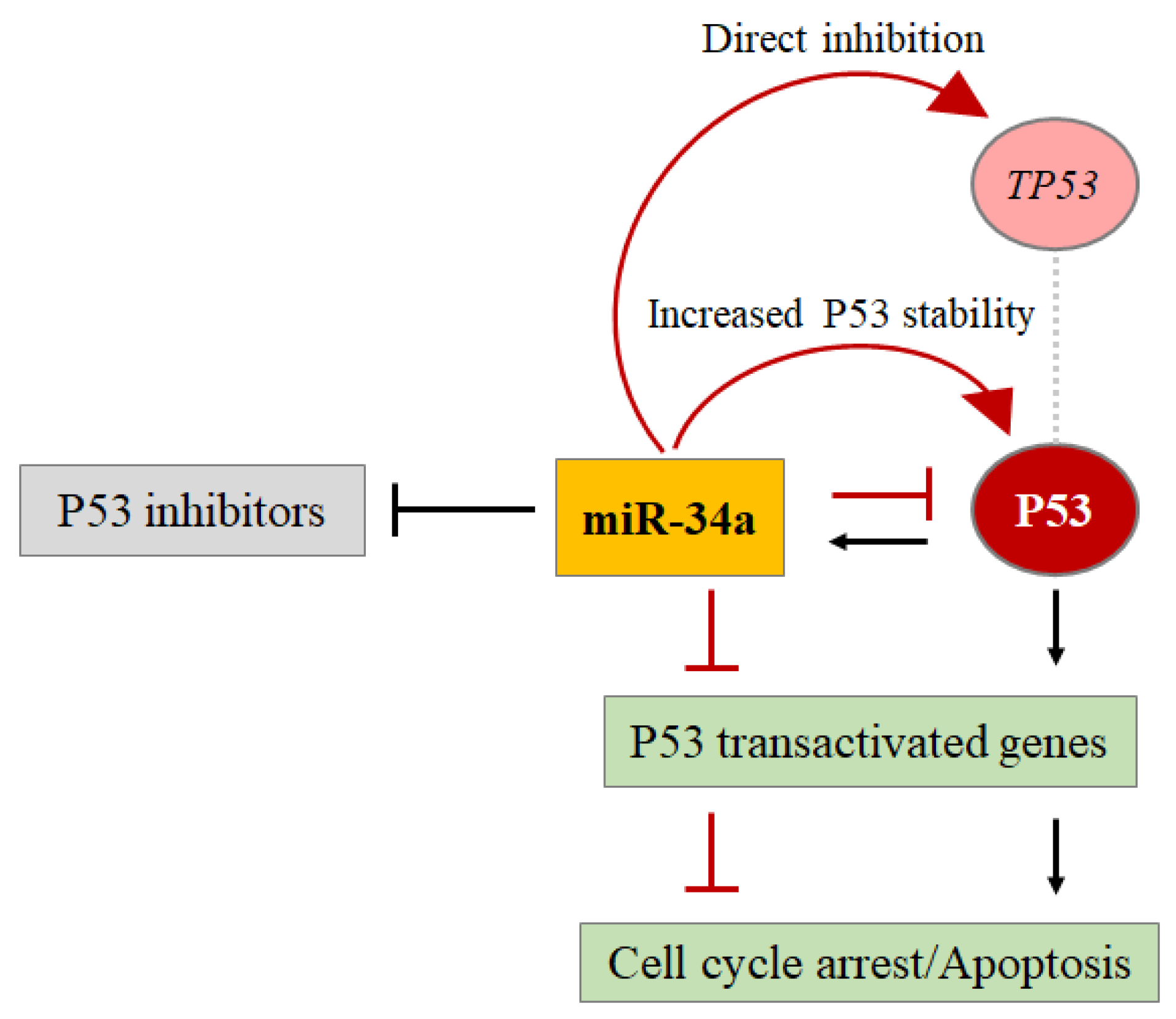
4.1. MiRNAs Directly Control the P53 Protein Level (Hit 1)
4.2. P53 Controls the Expression Level of Some miRNAs through the Regulation of Their Transcription or Biogenesis (Hit 2)
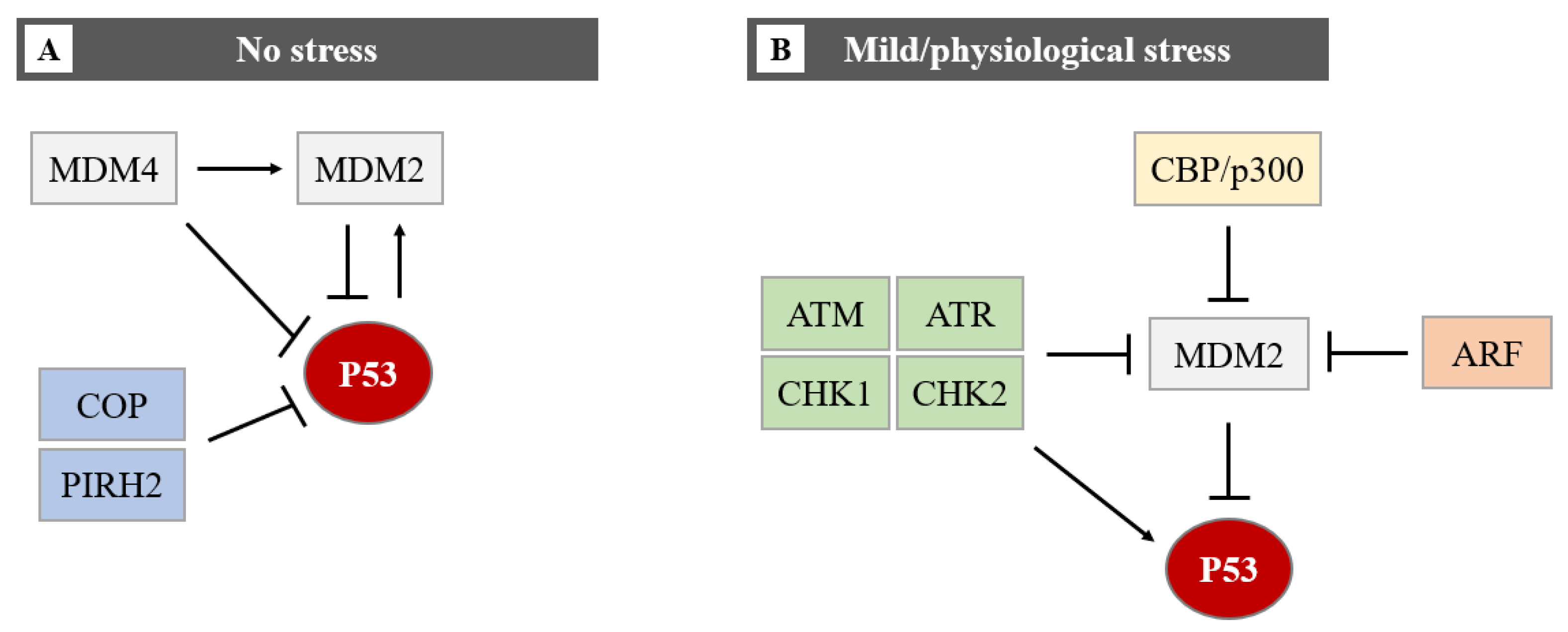
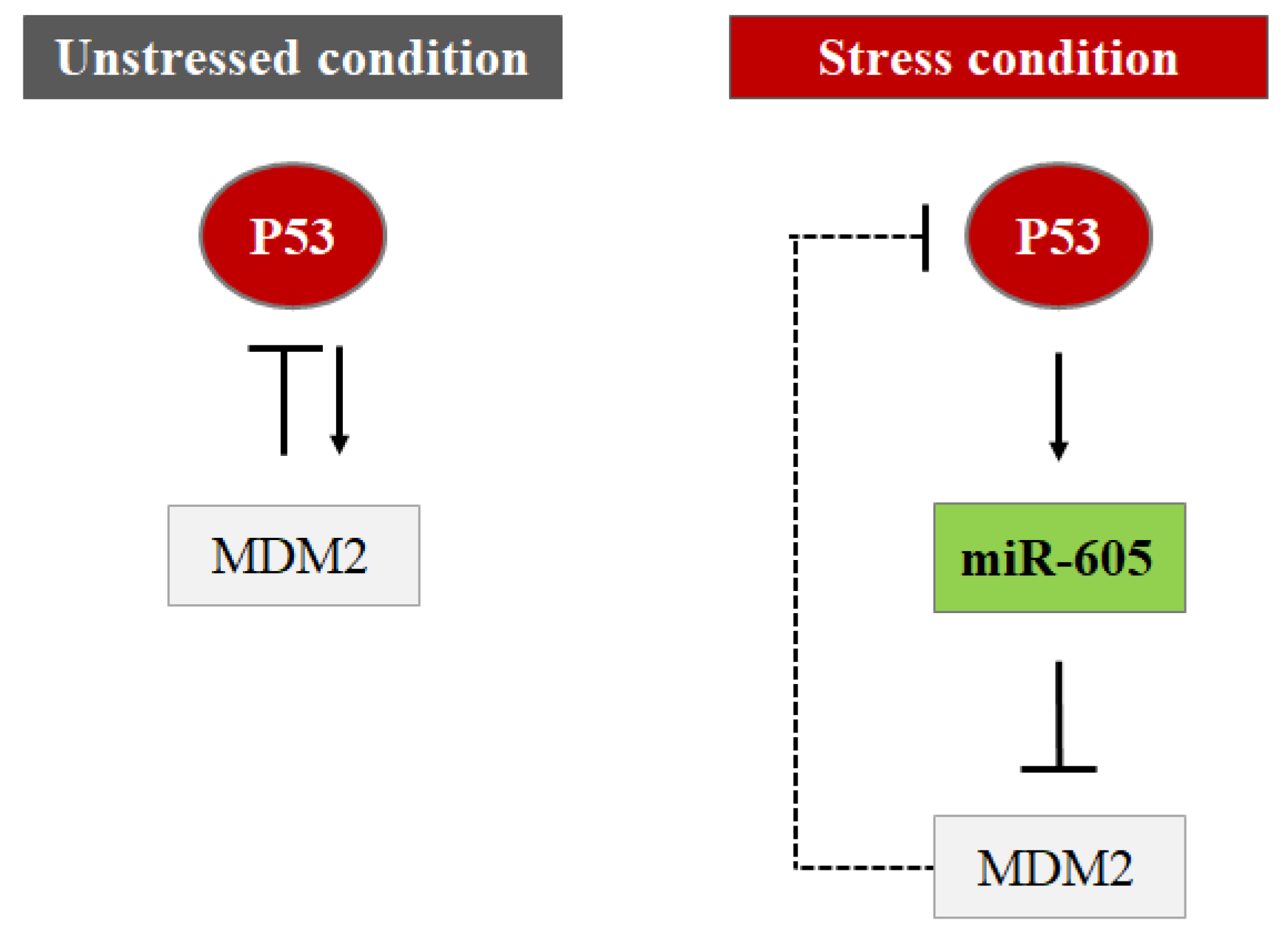
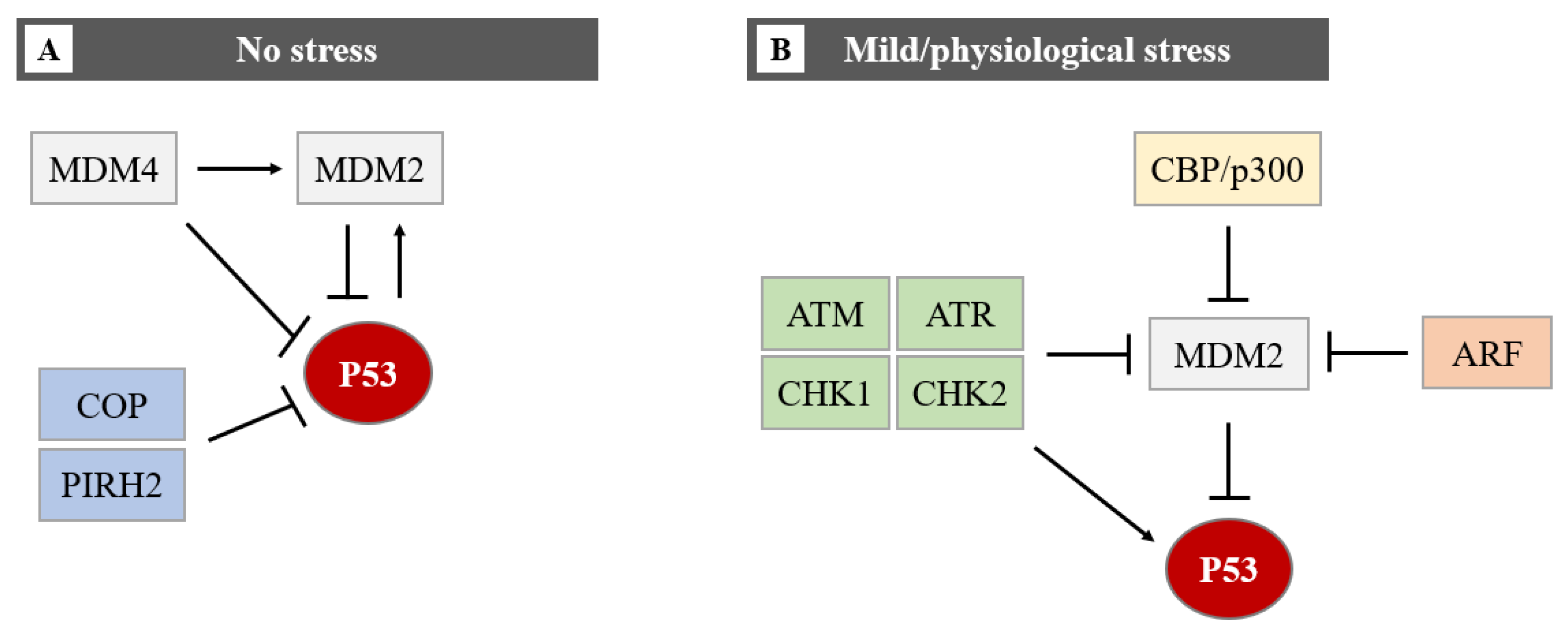
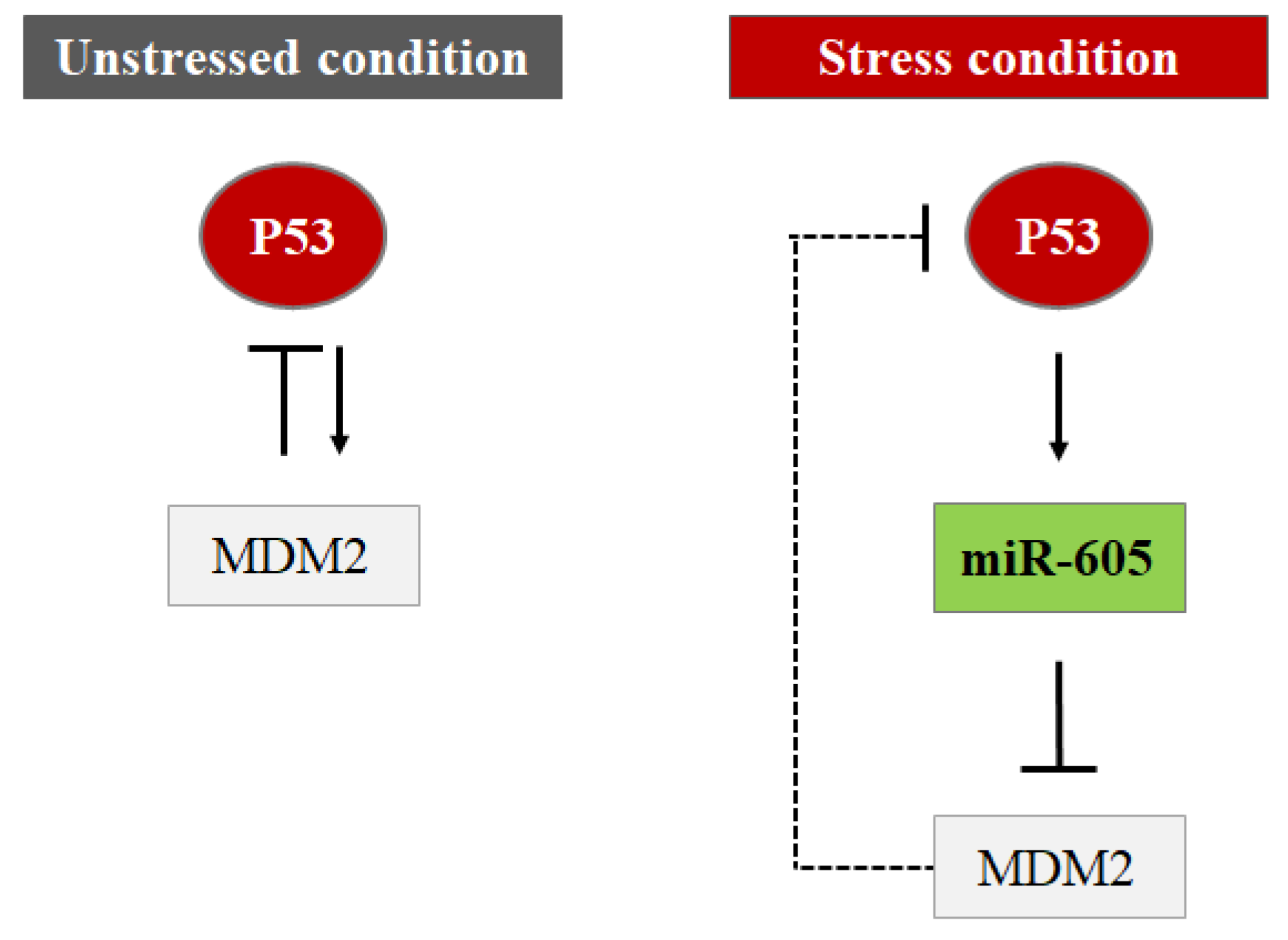
4.3. MiRNAs Indirectly Control the P53 Protein Level by Targeting Its Regulators (Hit 3)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Donehower, L.A. Phosphatases Reverse P53-Mediated Cell Cycle Checkpoints. Proc. Natl. Acad. Sci. USA 2014, 111, 7172–7173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting P53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, C.A.; Attardi, L.D. P53 at a Glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Liu, J.; Feng, Z. The Regulation of Cellular Metabolism by Tumor Suppressor P53. Cell Biosci. 2013, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, J.A.; Espinosa, J.M. The Impact of Post-Transcriptional Regulation in the P53 Network. Brief. Funct. Genom. 2013, 12, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-P53 Pathway Revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef]
- Hu, W.; Feng, Z.; Levine, A.J. The Regulation of Multiple P53 Stress Responses Is Mediated through MDM2. Genes Cancer 2012, 3, 199–208. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. New Insights into P53 Activation. Cell Res. 2010, 20, 614–621. [Google Scholar] [CrossRef]
- Shi, D.; Gu, W. Dual Roles of MDM2 in the Regulation of P53: Ubiquitination Dependent and Ubiquitination Independent Mechanisms of MDM2 Repression of P53 Activity. Genes Cancer 2012, 3, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, C.; Zhao, Y.; Feng, Z. MicroRNA Control of P53: MicroRNAs and P53. J. Cell. Biochem. 2017, 118, 7–14. [Google Scholar] [CrossRef]
- Bizzarri, A.R.; Cannistraro, S. Direct Interaction of MiRNA and CircRNA with the Oncosuppressor P53: An Intriguing Perspective in Cancer Research. Cancers 2021, 13, 6108. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. Ubiquitination, Phosphorylation and Acetylation: The Molecular Basis for P53 Regulation. Curr. Opin. Cell Biol. 2003, 15, 164–171. [Google Scholar] [CrossRef]
- Loughery, J.; Meek, D. Switching on P53: An Essential Role for Protein Phosphorylation? BioDiscovery 2013, 8, e8946. [Google Scholar] [CrossRef]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical Role for P53-Serine 15 Phosphorylation in Stimulating Transactivation at P53-Responsive Promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef] [Green Version]
- Maclaine, N.J.; Hupp, T.R. The Regulation of P53 by Phosphorylation: A Model for How Distinct Signals Integrate into the P53 Pathway. Aging 2009, 1, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Chernov, M.V.; Ramana, C.V.; Adler, V.V.; Stark, G.R. Stabilization and Activation of P53 Are Regulated Independently by Different Phosphorylation Events. Proc. Natl. Acad. Sci. USA 1998, 95, 2284–2289. [Google Scholar] [CrossRef] [Green Version]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a Consensus Binding Site for P53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef]
- Kruse, J.-P.; Gu, W. Modes of P53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Advani, V.M.; Ivanov, P. Translational Control under Stress: Reshaping the Translatome. BioEssays 2019, 41, 1900009. [Google Scholar] [CrossRef]
- Hershey, J.W.B.; Sonenberg, N.; Mathews, M.B. Principles of Translational Control. Cold Spring Harb. Perspect. Biol. 2019, 11, a032607. [Google Scholar] [CrossRef]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. MiRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.-S.; Phillips, M.D.; Betel, D.; Mu, P.; Ventura, A.; Siepel, A.C.; Chen, K.C.; Lai, E.C. Widespread Regulatory Activity of Vertebrate MicroRNA* Species. RNA 2011, 17, 312–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olejniczak, M.; Kotowska-Zimmer, A.; Krzyzosiak, W. Stress-Induced Changes in MiRNA Biogenesis and Functioning. Cell. Mol. Life Sci. 2018, 75, 177–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Paul, P.; Chakraborty, A.; Sarkar, D.; Langthasa, M.; Rahman, M.; Bari, M.; Singha, R.K.S.; Malakar, A.K.; Chakraborty, S. Interplay between MiRNAs and Human Diseases. J. Cell. Physiol. 2018, 233, 2007–2018. [Google Scholar] [CrossRef]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Brodersen, P.; Voinnet, O. Revisiting the Principles of MicroRNA Target Recognition and Mode of Action. Nat. Rev. Mol. Cell Biol. 2009, 10, 141–148. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of MicroRNA Processing by P53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef]
- Leung, A.K.L.; Sharp, P.A. MicroRNA Functions in Stress Responses. Mol. Cell 2010, 40, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Emde, A.; Hornstein, E. MiRNAs at the Interface of Cellular Stress and Disease. EMBO J. 2014, 33, 1428–1437. [Google Scholar] [CrossRef] [Green Version]
- Emde, A.; Eitan, C.; Liou, L.; Libby, R.T.; Rivkin, N.; Magen, I.; Reichenstein, I.; Oppenheim, H.; Eilam, R.; Silvestroni, A.; et al. Dysregulated MiRNA Biogenesis Downstream of Cellular Stress and ALS-Causing Mutations: A New Mechanism for ALS. EMBO J. 2015, 34, 2633–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Zhang, C.; Wu, R.; Hu, W. Tumor Suppressor P53 Meets MicroRNAs. J. Mol. Cell Biol. 2011, 3, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Kültz, D. Molecular and Evolutionary Basis of the Cellular Stress Response. Annu. Rev. Physiol. 2005, 67, 225–257. [Google Scholar] [CrossRef] [PubMed]
- Basile, F.; Capaccia, C.; Zampini, D.; Biagetti, T.; Diverio, S.; Guelfi, G. Omics Insights into Animal Resilience and Stress Factors. Animals 2020, 11, 47. [Google Scholar] [CrossRef]
- Liddicoat, B.J.; Chalk, A.M.; Walkley, C.R. ADAR1, Inosine and the Immune Sensing System: Distinguishing Self from Non-Self. WIREs RNA 2016, 7, 157–172. [Google Scholar] [CrossRef]
- Le, M.T.N.; Teh, C.; Shyh-Chang, N.; Xie, H.; Zhou, B.; Korzh, V.; Lodish, H.F.; Lim, B. MicroRNA-125b Is a Novel Negative Regulator of P53. Genes Dev. 2009, 23, 862–876. [Google Scholar] [CrossRef] [Green Version]
- Otsuka, K.; Ochiya, T. Genetic Networks Lead and Follow Tumor Development: MicroRNA Regulation of Cell Cycle and Apoptosis in the P53 Pathways. BioMed Res. Int. 2014, 2014, 749724. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Chan, C.S.; Wu, R.; Zhang, C.; Sun, Y.; Song, J.S.; Tang, L.H.; Levine, A.J.; Feng, Z. Negative Regulation of Tumor Suppressor P53 by MicroRNA MiR-504. Mol. Cell 2010, 38, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.F.; Lal, A. MicroRNAs, Wild-Type and Mutant P53: More Questions than Answers. RNA Biol. 2012, 9, 781–791. [Google Scholar] [CrossRef]
- Park, S.-Y.; Lee, J.H.; Ha, M.; Nam, J.-W.; Kim, V.N. MiR-29 MiRNAs Activate P53 by Targeting P85α and CDC42. Nat. Struct. Mol. Biol. 2009, 16, 23–29. [Google Scholar] [CrossRef]
- Suh, S.-S.; Yoo, J.Y.; Nuovo, G.J.; Jeon, Y.-J.; Kim, S.; Lee, T.J.; Kim, T.; Bakacs, A.; Alder, H.; Kaur, B.; et al. MicroRNAs/TP53 Feedback Circuitry in Glioblastoma Multiforme. Proc. Natl. Acad. Sci. USA 2012, 109, 5316–5321. [Google Scholar] [CrossRef] [Green Version]
- Guelfi, G.; Iaboni, M.; Sansone, A.; Capaccia, C.; Santoro, M.M.; Diverio, S. Extracellular Circulating MiRNAs as Stress-Related Signature to Search and Rescue Dogs. Sci. Rep. 2022, 12, 3213. [Google Scholar] [CrossRef]
- Liao, J.-M.; Cao, B.; Zhou, X.; Lu, H. New Insights into P53 Functions through Its Target MicroRNAs. J. Mol. Cell Biol. 2014, 6, 206–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isik, M.; Blackwell, T.K.; Berezikov, E. MicroRNA Mir-34 Provides Robustness to Environmental Stress Response via the DAF-16 Network in C. Elegans. Sci. Rep. 2016, 6, 36766. [Google Scholar] [CrossRef]
- Navarro, F.; Lieberman, J. MiR-34 and P53: New Insights into a Complex Functional Relationship. PLoS ONE 2015, 10, e0132767. [Google Scholar] [CrossRef] [PubMed]
- Andolina, D.; Di Segni, M.; Accoto, A.; Lo Iacono, L.; Borreca, A.; Ielpo, D.; Berretta, N.; Perlas, E.; Puglisi-Allegra, S.; Ventura, R. MicroRNA-34 Contributes to the Stress-Related Behavior and Affects 5-HT Prefrontal/GABA Amygdalar System through Regulation of Corticotropin-Releasing Factor Receptor 1. Mol. Neurobiol. 2018, 55, 7401–7412. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, V.; Jung, P.; Verdoodt, B.; Lodygin, D.; Epanchintsev, A.; Menssen, A.; Meister, G.; Hermeking, H. Differential Regulation of MicroRNAs by P53 Revealed by Massively Parallel Sequencing: MiR-34a Is a P53 Target That Induces Apoptosis and G1-Arrest. Cell Cycle 2007, 6, 1586–1593. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Sun, Q.; Zhang, Z.; Ge, S.; Han, Z.-G.; Chen, W.-T. Loss of MicroRNA-143/145 Disturbs Cellular Growth and Apoptosis of Human Epithelial Cancers by Impairing the MDM2-P53 Feedback Loop. Oncogene 2013, 32, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wu, S.; Muhammad, S.; Ren, Q.; Sun, C. MiR-103/107 Promote ER Stress-Mediated Apoptosis via Targeting the Wnt3a/β-Catenin/ATF6 Pathway in Preadipocytes. J. Lipid Res. 2018, 59, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Yao, G.; Yin, M.; Lü, M.; Tian, H.; Liu, L.; Lian, J.; Huang, X.; Sun, F. Transcriptional Cooperation between P53 and NF-ΚB P65 Regulates MicroRNA-224 Transcription in Mouse Ovarian Granulosa Cells. Mol. Cell. Endocrinol. 2013, 370, 119–129. [Google Scholar] [CrossRef]
- Hermeking, H. MicroRNAs in the P53 Network: Micromanagement of Tumour Suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Krell, J.; Stebbing, J.; Frampton, A.E.; Carissimi, C.; Harding, V.; De Giorgio, A.; Fulci, V.; Macino, G.; Colombo, T.; Castellano, L. The Role of TP53 in MiRNA Loading onto AGO2 and in Remodelling the MiRNA–MRNA Interaction Network. Lancet 2015, 385, S15. [Google Scholar] [CrossRef] [Green Version]
- Mudhasani, R.; Zhu, Z.; Hutvagner, G.; Eischen, C.M.; Lyle, S.; Hall, L.L.; Lawrence, J.B.; Imbalzano, A.N.; Jones, S.N. Loss of MiRNA Biogenesis Induces P19Arf-P53 Signaling and Senescence in Primary Cells. J. Cell Biol. 2008, 181, 1055–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J.; Hu, W.; Feng, Z. The P53 Pathway: What Questions Remain to Be Explored? Cell Death Differ. 2006, 13, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, Y.; Pilpel, Y.; Oren, M. MicroRNAs and Alu Elements in the P53-Mdm2-Mdm4 Regulatory Network. J. Mol. Cell Biol. 2014, 6, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Liu, J.; Wang, X.; Feng, Z. The Regulation of the P53/MDM2 Feedback Loop by MicroRNAs. RNA Dis. 2015, 2, e502. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Tan, C.; Yue, X.; Zhao, Y.; Peng, J.; Wang, X.; Laddha, S.V.; Chan, C.S.; Zheng, S.; et al. MicroRNA-1827 Represses MDM2 to Positively Regulate Tumor Suppressor P53 and Suppress Tumorigenesis. Oncotarget 2016, 7, 8783–8796. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Lin, H.; Luo, X.; Luo, X.; Wang, Z. MiR-605 Joins P53 Network to Form a P53:MiR-605:Mdm2 Positive Feedback Loop in Response to Stress. EMBO J. 2011, 30, 524–532. [Google Scholar] [CrossRef]
- Hoffman, Y.; Bublik, D.R.; Pilpel, Y.; Oren, M. MiR-661 Downregulates Both Mdm2 and Mdm4 to Activate P53. Cell Death Differ. 2014, 21, 302–309. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capaccia, C.; Diverio, S.; Zampini, D.; Guelfi, G. The Complex Interaction between P53 and miRNAs Joins New Awareness in Physiological Stress Responses. Cells 2022, 11, 1631. https://doi.org/10.3390/cells11101631
Capaccia C, Diverio S, Zampini D, Guelfi G. The Complex Interaction between P53 and miRNAs Joins New Awareness in Physiological Stress Responses. Cells. 2022; 11(10):1631. https://doi.org/10.3390/cells11101631
Chicago/Turabian StyleCapaccia, Camilla, Silvana Diverio, Danilo Zampini, and Gabriella Guelfi. 2022. "The Complex Interaction between P53 and miRNAs Joins New Awareness in Physiological Stress Responses" Cells 11, no. 10: 1631. https://doi.org/10.3390/cells11101631
APA StyleCapaccia, C., Diverio, S., Zampini, D., & Guelfi, G. (2022). The Complex Interaction between P53 and miRNAs Joins New Awareness in Physiological Stress Responses. Cells, 11(10), 1631. https://doi.org/10.3390/cells11101631