
The Anti-Inflammatory Effect of the β1-Adrenergic Receptor Antagonist Metoprolol on High Glucose Treated Human Microvascular Retinal Endothelial Cells

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. HREC Culture and Treatments
2.3. Cell Viability
2.4. Tube Formation Assay
2.5. ROS Measurements
2.6. Western Blot Analysis
2.7. High-Content Screening (HCS) and Image Analysis
2.8. Extraction of Total RNA and Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
2.9. Statistical Analysis
3. Results
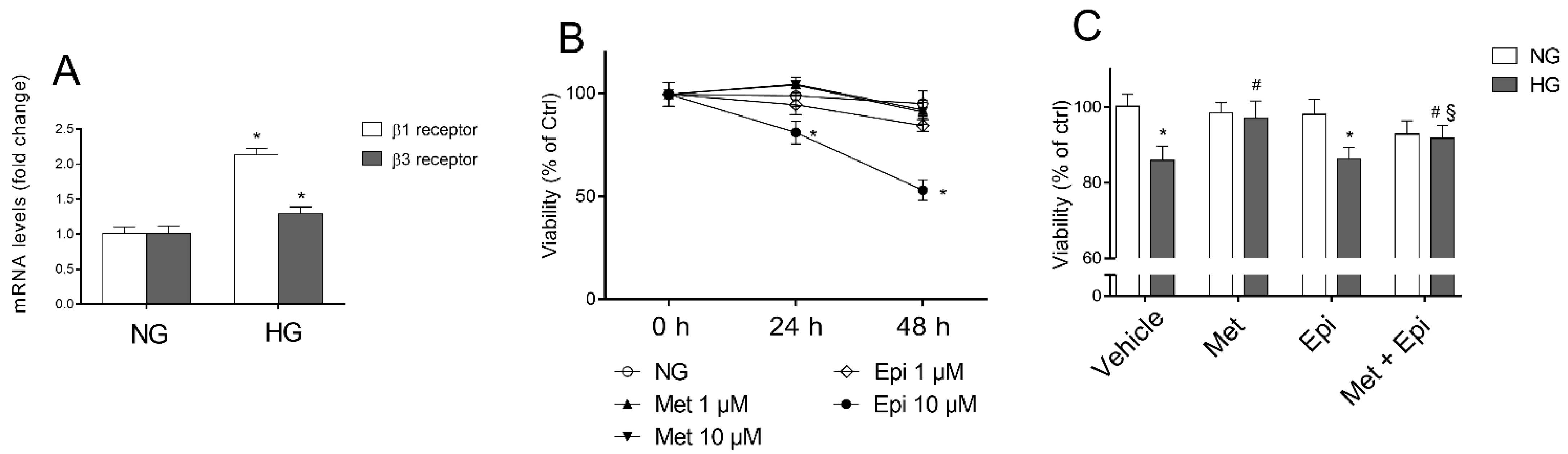
3.1. β1-Adrenergic Receptor Blockade Prevents HG-Induced Effects upon Metabolic Activity and Cell Proliferation in HREC
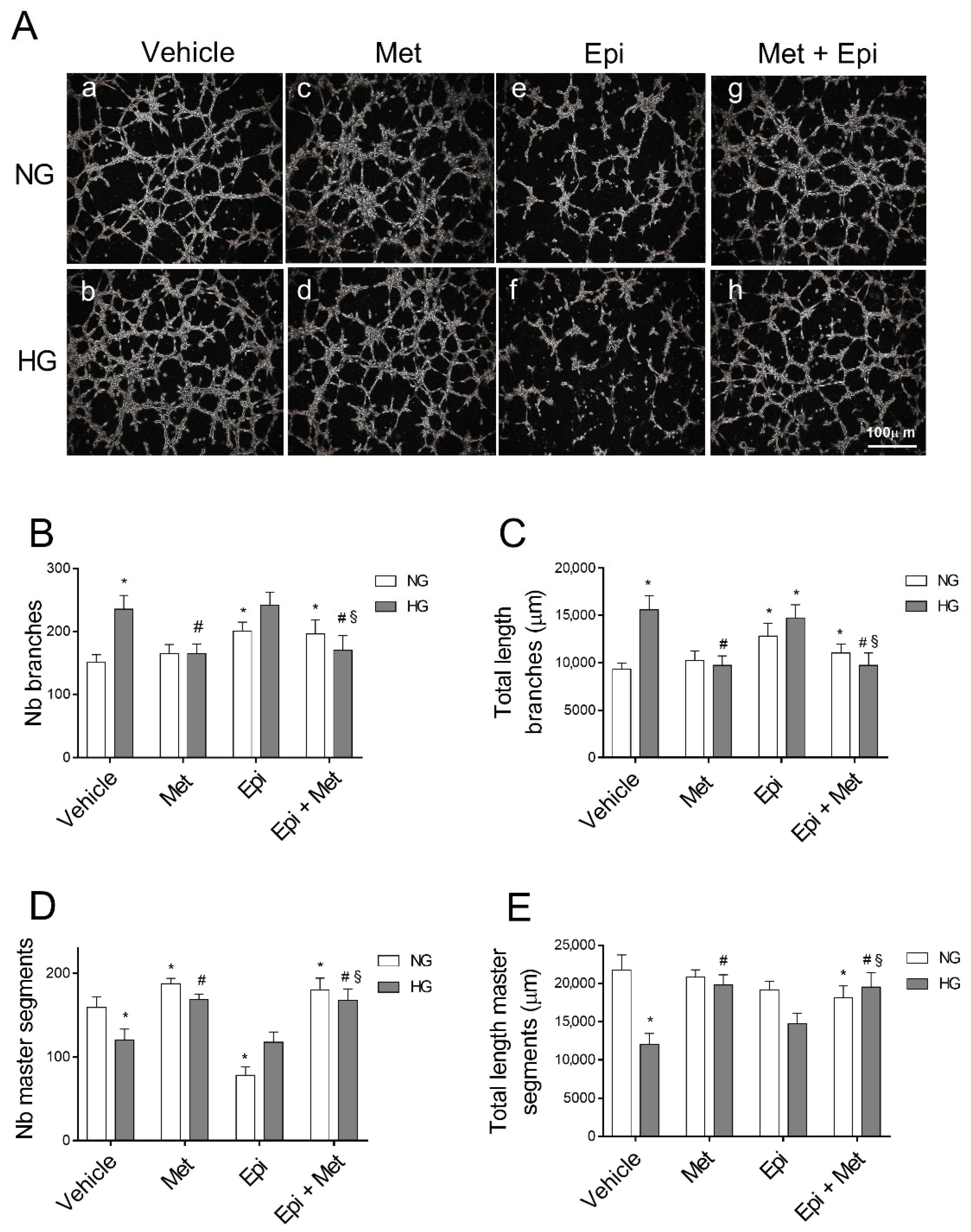
3.2. Metoprolol Counteracts the Increase in the Tube-like Structures of HREC Stimulated with HG
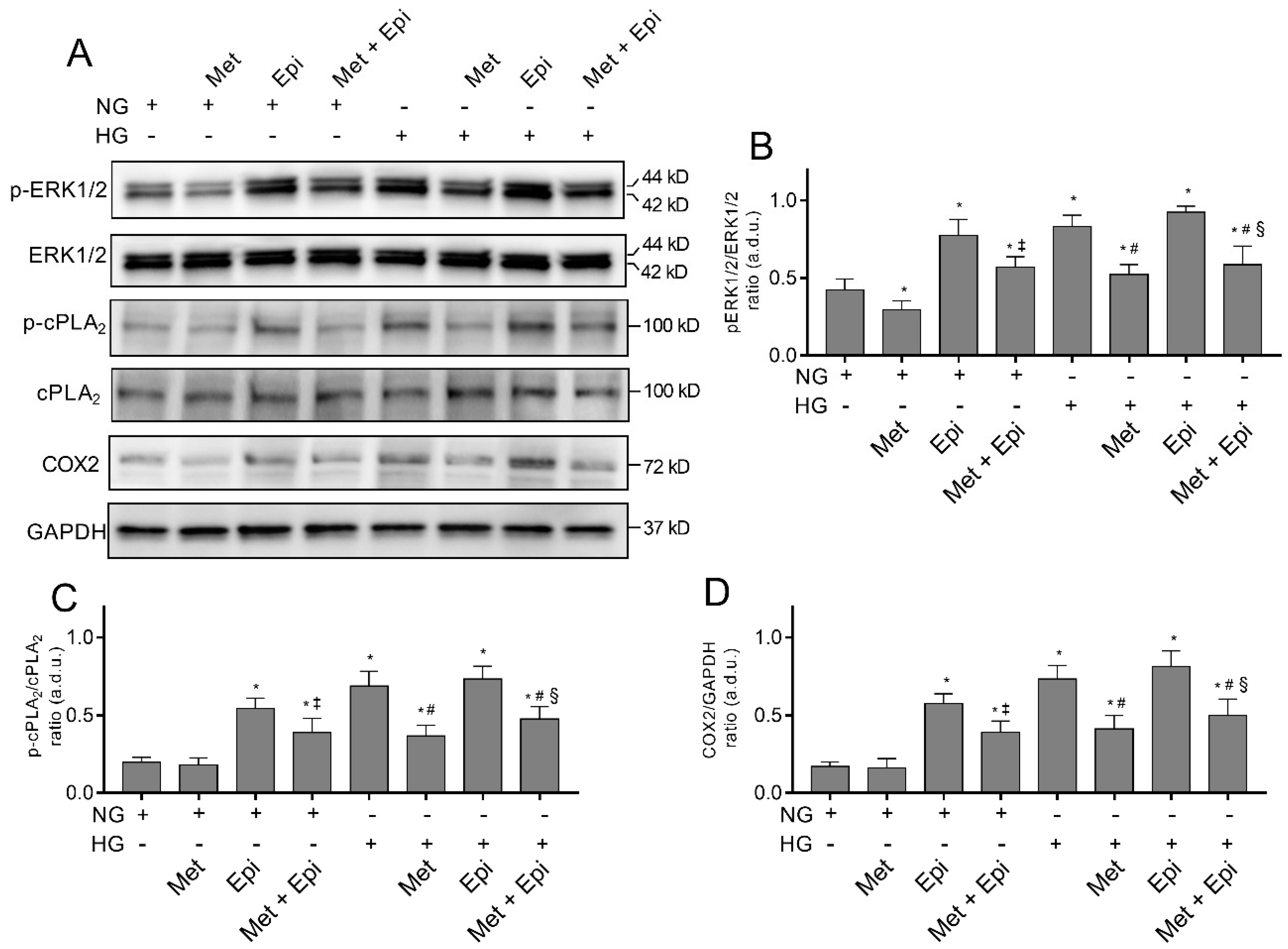
3.3. Metoprolol Down-Regulates the ERK1/2/cPLA2/COX2 Axis in HREC Treated with HG
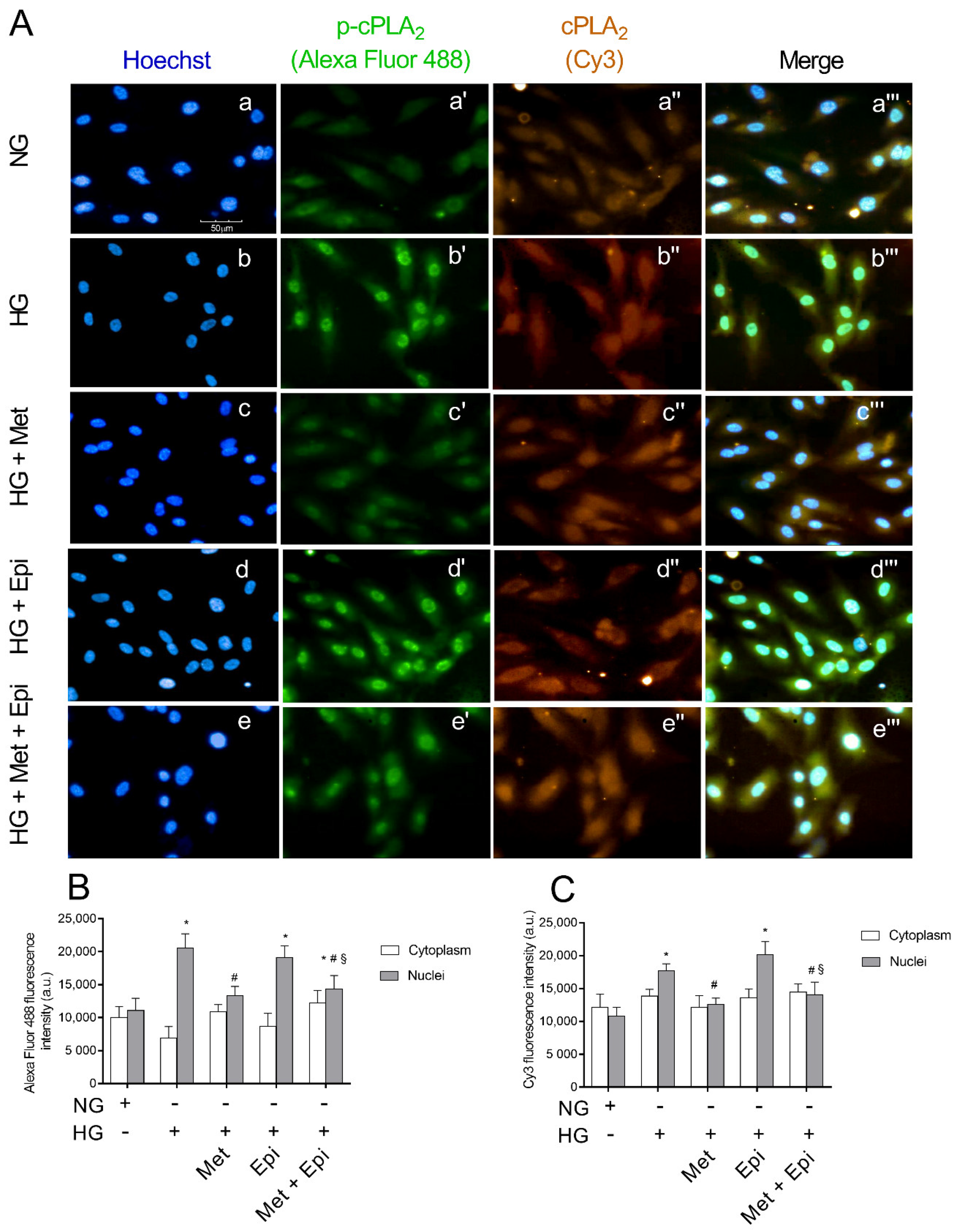
3.4. Metoprolol Reduced cPLA2 Nuclear Translocation in HREC Challenged with HG
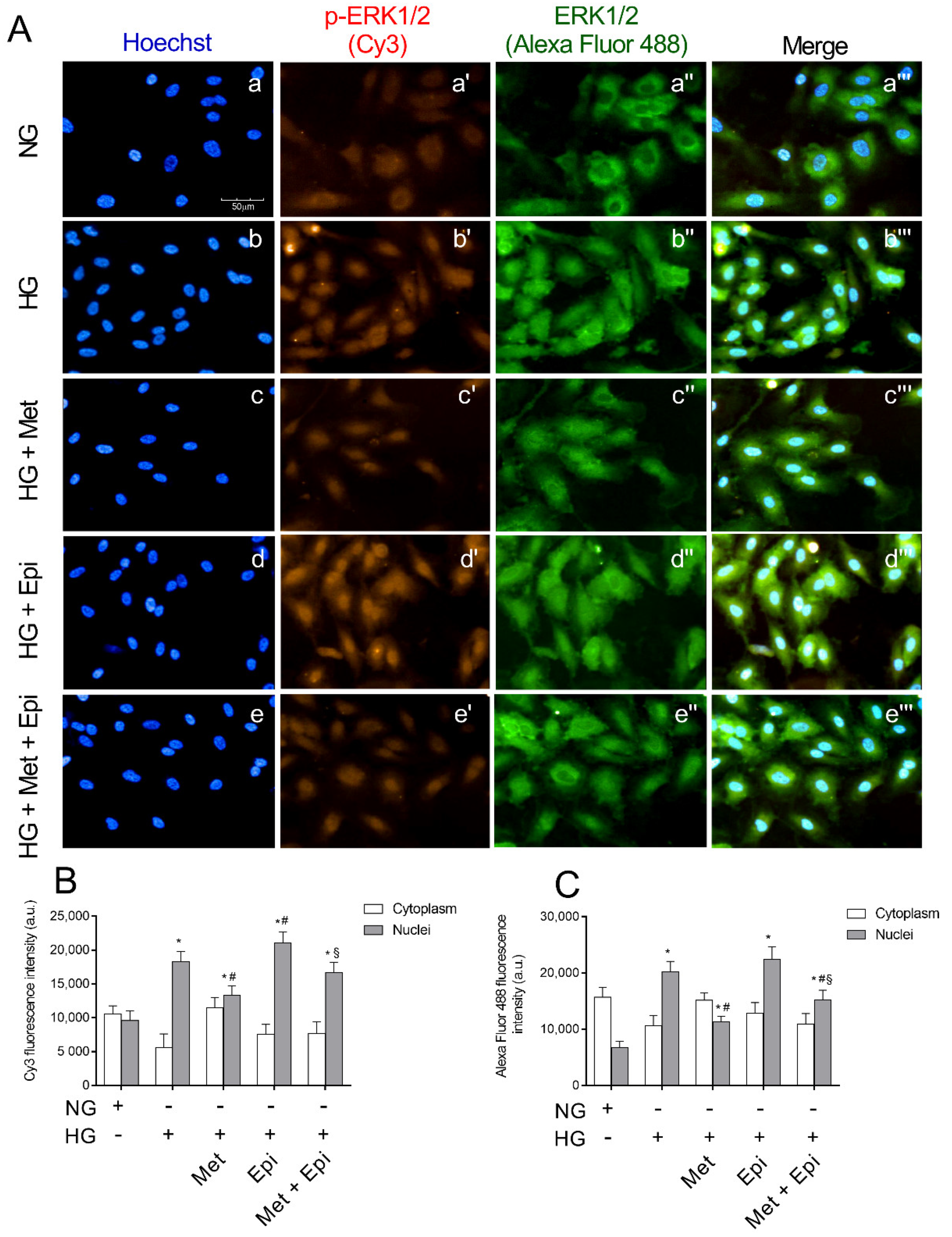
3.5. Metoprolol Reduced ERK 1/2 Phosphorylation in HREC Challenged with HG
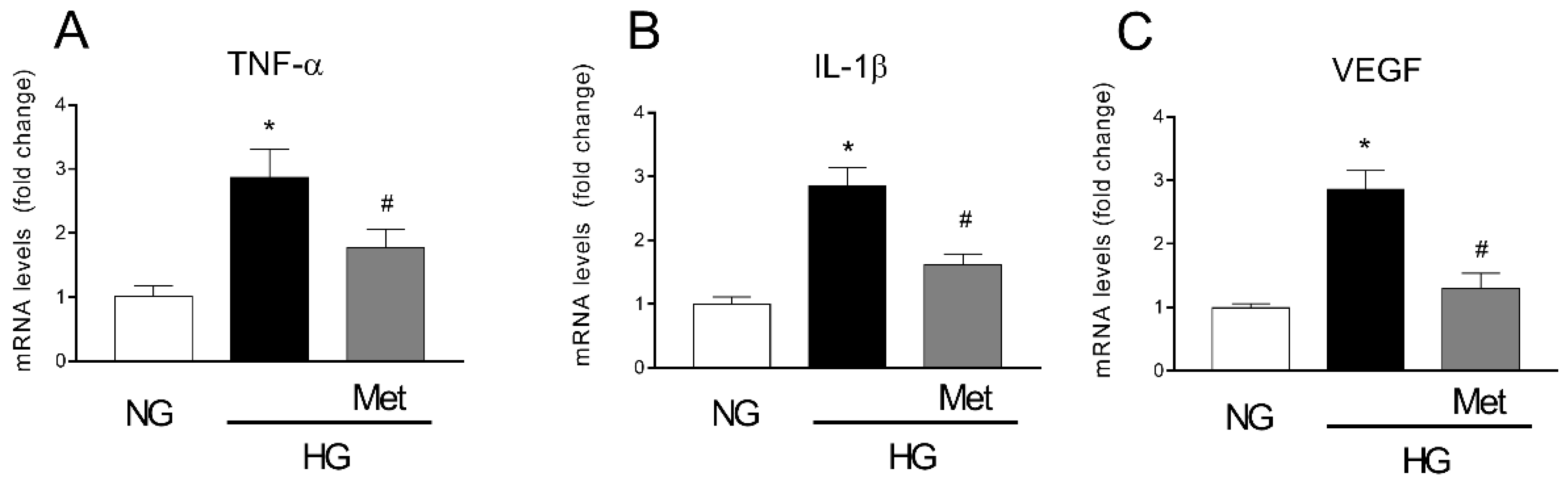
3.6. Metoprolol Down-Regulates the Release of TNF-α, VEGF, and IL-1b in HREC Treated with HG
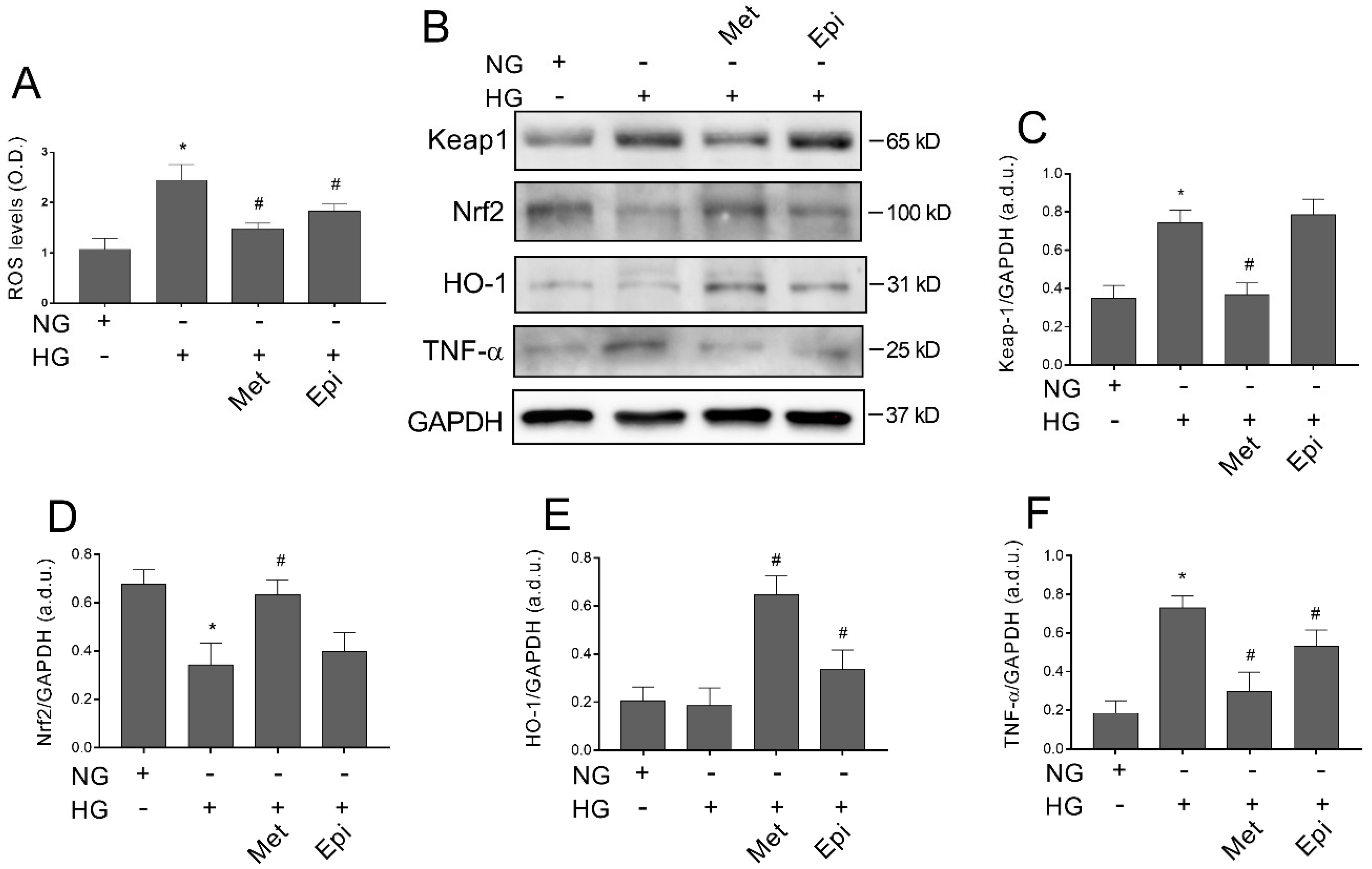
3.7. Metoprolol Counteracts Glucose-Induced ROS Accumulation by Activating the Keap1/Nrf2/HO-1 Pathway in HREC
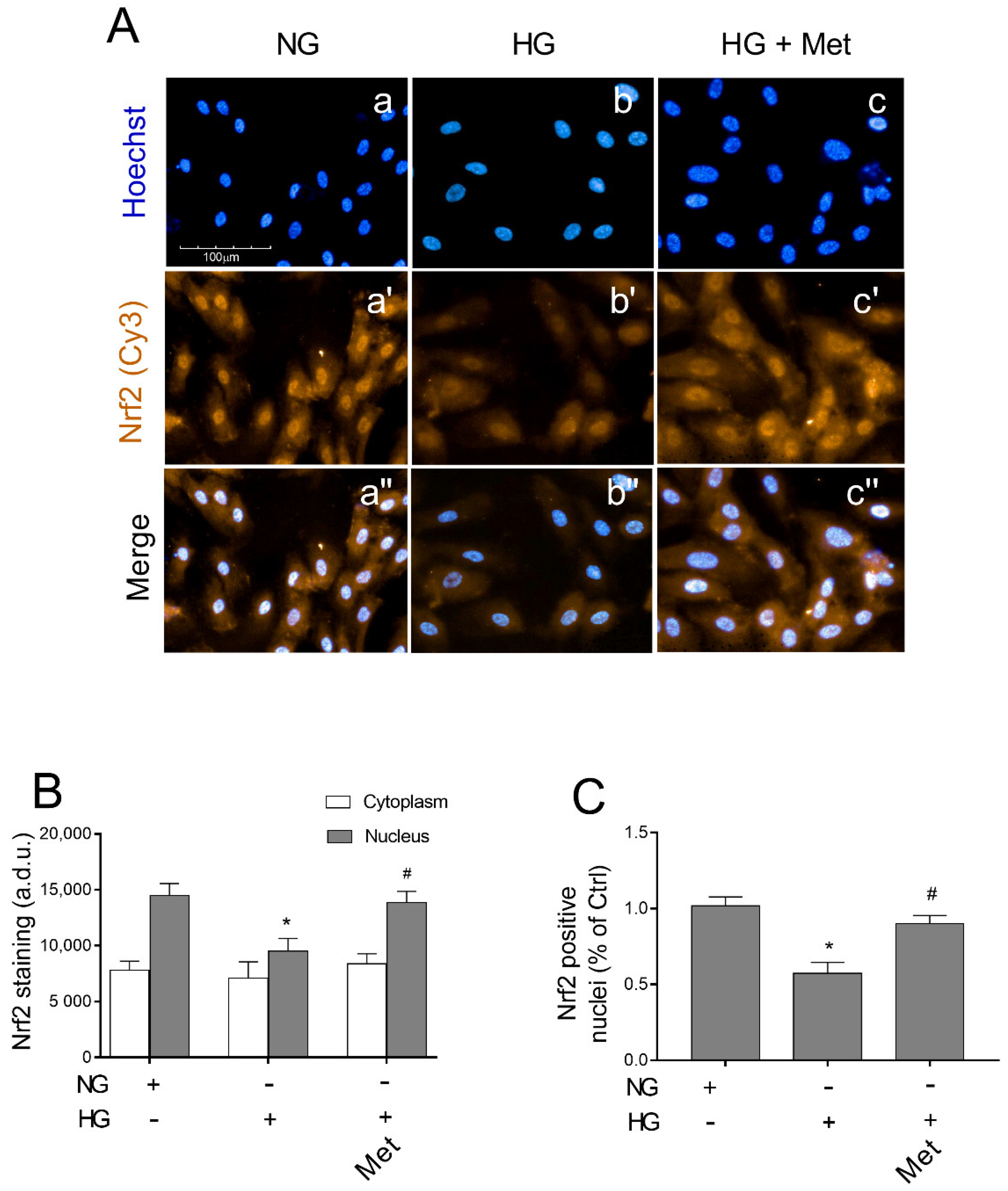
3.8. Metoprolol Induced Nuclear Nrf2 Nuclear Compartmentalization in HG-Treated HREC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Antonetti, D.A.; Silva, P.S.; Stitt, A.W. Current Understanding of the Molecular and Cellular Pathology of Diabetic Retinopathy. Nat. Rev. Endocrinol. 2021, 17, 195–206. [Google Scholar] [CrossRef]
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global Causes of Blindness and Distance Vision Impairment 1990–2020: A Systematic Review and Meta-Analysis. Lancet Glob. Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef]
- Rudraraju, M.; Narayanan, S.P.; Somanath, P.R. Regulation of Blood-Retinal Barrier Cell-Junctions in Diabetic Retinopathy. Pharmacol. Res. 2020, 161, 105115. [Google Scholar] [CrossRef]
- Wang, W.; Lo, A. Diabetic Retinopathy: Pathophysiology and Treatments. IJMS 2018, 19, 1816. [Google Scholar] [CrossRef]
- Gonzalez, V.H.; Campbell, J.; Holekamp, N.M.; Kiss, S.; Loewenstein, A.; Augustin, A.J.; Ma, J.; Ho, A.C.; Patel, V.; Whitcup, S.M.; et al. Early and Long-Term Responses to Anti–Vascular Endothelial Growth Factor Therapy in Diabetic Macular Edema: Analysis of Protocol I Data. Am. J. Ophthalmol. 2016, 172, 72–79. [Google Scholar] [CrossRef]
- Casini, G.; Dal Monte, M.; Fornaciari, I.; Filippi, L.; Bagnoli, P. The β-Adrenergic System as a Possible New Target for Pharmacologic Treatment of Neovascular Retinal Diseases. Prog. Retin. Eye Res. 2014, 42, 103–129. [Google Scholar] [CrossRef]
- Laverty, R. Catecholamines: Role in Health and Disease. Drugs 1978, 16, 418–440. [Google Scholar] [CrossRef]
- Sorriento, D.; Santulli, G.; Del Giudice, C.; Anastasio, A.; Trimarco, B.; Iaccarino, G. Endothelial Cells Are Able to Synthesize and Release Catecholamines Both In Vitro and In Vivo. Hypertension 2012, 60, 129–136. [Google Scholar] [CrossRef]
- Steinle, J.J.; Chin, V.C.; Williams, K.P.; Panjala, S.R. Beta-Adrenergic Receptor Stimulation Modulates INOS Protein Levels through P38 and ERK1/2 Signaling in Human Retinal Endothelial Cells. Exp. Eye Res. 2008, 87, 30–34. [Google Scholar] [CrossRef]
- Steinle, J.J. Sympathetic Neurotransmission Modulates Expression of Inflammatory Markers in the Rat Retina. Exp. Eye Res. 2007, 84, 118–125. [Google Scholar] [CrossRef]
- Forrester, J.V.; Kuffova, L.; Delibegovic, M. The Role of Inflammation in Diabetic Retinopathy. Front. Immunol. 2020, 11, 583687. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nakatani, Y.; Atsumi, G.; Inoue, K.; Kudo, I. Regulatory Functions of Phospholipase A2. Crit. Rev. Immunol. 2017, 37, 121–179. [Google Scholar] [CrossRef] [PubMed]
- Lupo, G.; Motta, C.; Giurdanella, G.; Anfuso, C.D.; Alberghina, M.; Drago, F.; Salomone, S.; Bucolo, C. Role of Phospholipases A2 in Diabetic Retinopathy: In Vitro and in Vivo Studies. Biochem. Pharmacol. 2013, 86, 1603–1613. [Google Scholar] [CrossRef]
- Leslie, C.C. Cytosolic Phospholipase A2: Physiological Function and Role in Disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Kudo, I. Phospholipase A2. J. Biochem. 2002, 131, 285–292. [Google Scholar] [CrossRef]
- Donath, M.Y. Targeting Inflammation in the Treatment of Type 2 Diabetes: Time to Start. Nat. Rev. Drug Discov. 2014, 13, 465–476. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1–Nrf2 System in Cancers: Stress Response and Anabolic Metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 Pathway: Mechanisms of Activation and Dysregulation in Cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct Cysteine Residues in Keap1 Are Required for Keap1-Dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Mol. Cell Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef]
- Chen, X.; Qi, J.; Wu, Q.; Jiang, H.; Wang, J.; Chen, W.; Mao, A.; Zhu, M. High Glucose Inhibits Vascular Endothelial Keap1/Nrf2/ARE Signal Pathway via Downregulation of Monomethyltransferase SET8 Expression. Acta Biochim. Biophys. Sin. 2020, 52, 506–516. [Google Scholar] [CrossRef]
- Chen, G.; Chen, X.; Niu, C.; Huang, X.; An, N.; Sun, J.; Huang, S.; Ye, W.; Li, S.; Shen, Y.; et al. Baicalin Alleviates Hyperglycemia-Induced Endothelial Impairment via Nrf2. J. Endocrinol. 2019, 240, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Giurdanella, G.; Anfuso, C.D.; Olivieri, M.; Lupo, G.; Caporarello, N.; Eandi, C.M.; Drago, F.; Bucolo, C.; Salomone, S. Aflibercept, Bevacizumab and Ranibizumab Prevent Glucose-Induced Damage in Human Retinal Pericytes in Vitro, through a PLA2/COX-2/VEGF-A Pathway. Biochem. Pharmacol. 2015, 96, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Lupo, G.; Cambria, M.T.; Olivieri, M.; Rocco, C.; Caporarello, N.; Longo, A.; Zanghì, G.; Salmeri, M.; Foti, M.C.; Anfuso, C.D. Anti-angiogenic Effect of Quercetin and Its 8-methyl Pentamethyl Ether Derivative in Human Microvascular Endothelial Cells. J. Cell. Mol. Med. 2019, 23, 6565–6577. [Google Scholar] [CrossRef]
- Giurdanella, G.; Lupo, G.; Gennuso, F.; Conti, F.; Furno, D.L.; Mannino, G.; Anfuso, C.D.; Drago, F.; Salomone, S.; Bucolo, C. Activation of the VEGF-A/ERK/PLA2 Axis Mediates Early Retinal Endothelial Cell Damage Induced by High Glucose: New Insight from an In Vitro Model of Diabetic Retinopathy. IJMS 2020, 21, 7528. [Google Scholar] [CrossRef]
- Mannino, G.; Longo, A.; Gennuso, F.; Anfuso, C.D.; Lupo, G.; Giurdanella, G.; Giuffrida, R.; Lo Furno, D. Effects of High Glucose Concentration on Pericyte-Like Differentiated Human Adipose-Derived Mesenchymal Stem Cells. IJMS 2021, 22, 4604. [Google Scholar] [CrossRef] [PubMed]
- Anfuso, C.D.; Giurdanella, G.; Motta, C.; Muriana, S.; Lupo, G.; Ragusa, N.; Alberghina, M. PKCα-MAPK/ERK-Phospholipase A2 Signaling Is Required for Human Melanoma-Enhanced Brain Endothelial Cell Proliferation and Motility. Microvasc. Res. 2009, 78, 338–357. [Google Scholar] [CrossRef]
- Anfuso, C.D.; Longo, A.; Distefano, A.; Amorini, A.M.; Salmeri, M.; Zanghì, G.; Giallongo, C.; Giurdanella, G.; Lupo, G. Uveal Melanoma Cells Elicit Retinal Pericyte Phenotypical and Biochemical Changes in an in Vitro Model of Coculture. IJMS 2020, 21, 5557. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, T.; Gong, C.; Sheng, Y.; Lu, B.; Zhou, L.; Ji, L.; Wang, Z. Erianin Inhibits High Glucose-Induced Retinal Angiogenesis via Blocking ERK1/2-Regulated HIF-1α-VEGF/VEGFR2 Signaling Pathway. Sci. Rep. 2016, 6, 34306. [Google Scholar] [CrossRef]
- Tan, A.; Li, T.; Ruan, L.; Yang, J.; Luo, Y.; Li, L.; Wu, X. Knockdown of Malat1 Alleviates High-Glucose-Induced Angiogenesis through Regulating MiR-205-5p/VEGF-A Axis. Exp. Eye Res. 2021, 207, 108585. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Y.; Xie, T.; Zhan, P.; Zou, J.; Nie, X.; Shao, J.; Zhuang, M.; Tan, C.; Tan, J.; et al. Prostaglandin E2/EP2 Receptor Signalling Pathway Promotes Diabetic Retinopathy in a Rat Model of Diabetes. Diabetologia 2019, 62, 335–348. [Google Scholar] [CrossRef]
- Giurdanella, G.; Lazzara, F.; Caporarello, N.; Lupo, G.; Anfuso, C.D.; Eandi, C.M.; Leggio, G.M.; Drago, F.; Bucolo, C.; Salomone, S. Sulodexide Prevents Activation of the PLA2/COX-2/VEGF Inflammatory Pathway in Human Retinal Endothelial Cells by Blocking the Effect of AGE/RAGE. Biochem. Pharmacol. 2017, 142, 145–154. [Google Scholar] [CrossRef]
- Álvarez-Diduk, R.; Galano, A. Adrenaline and Noradrenaline: Protectors against Oxidative Stress or Molecular Targets? J. Phys. Chem. B 2015, 119, 3479–3491. [Google Scholar] [CrossRef]
- Hammes, H.-P.; Lin, J.; Renner, O.; Shani, M.; Lundqvist, A.; Betsholtz, C.; Brownlee, M.; Deutsch, U. Pericytes and the Pathogenesis of Diabetic Retinopathy. Diabetes 2002, 51, 3107–3112. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.H.; Jeong, H.S.; Kim, K.J.; Han, M.H.; Lee, E.H.; Lee, K.; Cho, C.H. B-Adrenergic Receptor Agonists Attenuate Pericyte Loss in Diabetic Retinas through Akt Activation. FASEB J. 2018, 32, 2324–2338. [Google Scholar] [CrossRef]
- Nassiri, S.; Houshmand, G.; Feghhi, M.; Kheirollah, A.; Bahadoram, M.; Nassiri, N. Effect of Periocular Injection of Celecoxib and Propranolol on Ocular Level of Vascular Endothelial Growth Factor in a Diabetic Mouse Model. Int. J. Ophthalmol. 2016, 9, 821. [Google Scholar] [CrossRef]
- Cammalleri, M.; Locri, F.; Catalani, E.; Filippi, L.; Cervia, D.; Dal Monte, M.; Bagnoli, P. The Beta Adrenergic Receptor Blocker Propranolol Counteracts Retinal Dysfunction in a Mouse Model of Oxygen Induced Retinopathy: Restoring the Balance between Apoptosis and Autophagy. Front. Cell. Neurosci. 2017, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Nourinia, R.; Rezaei Kanavi, M.; Kaharkaboudi, A.; Taghavi, S.I.; Aldavood, S.J.; Darjatmoko, S.R.; Wang, S.; Gurel, Z.; Lavine, J.A.; Safi, S.; et al. Ocular Safety of Intravitreal Propranolol and Its Efficacy in Attenuation of Choroidal Neovascularization. Invest. Ophthalmol. Vis. Sci. 2015, 56, 8228. [Google Scholar] [CrossRef]
- Dal Monte, M.; Casini, G.; la Marca, G.; Isacchi, B.; Filippi, L.; Bagnoli, P. Eye Drop Propranolol Administration Promotes the Recovery of Oxygen-Induced Retinopathy in Mice. Exp. Eye Res. 2013, 111, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Dal Monte, M.; Cammalleri, M.; Mattei, E.; Filippi, L.; Bagnoli, P. Protective Effects of 1/2 Adrenergic Receptor Deletion in a Model of Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 59–73. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ristori, C.; Filippi, L.; Monte, M.D.; Martini, D.; Cammalleri, M.; Fortunato, P.; Bagnoli, P. Role of the Adrenergic System in a Mouse Model of Oxygen-Induced Retinopathy: Antiangiogenic Effects of β-adrenoreceptor blockade. Investig. Ophthalmol. Vis. Sci. 2011, 52, 16. [Google Scholar] [CrossRef] [PubMed]
- Safi, S.Z.; Qvist, R.; Yan, G.O.S.; Ismail, I.S.B. Differential Expression and Role of Hyperglycemia Induced Oxidative Stress in Epigenetic Regulation of Β1, Β2 and Β3-Adrenergic Receptors in Retinal Endothelial Cells. BMC Med. Genom. 2014, 7, 29. [Google Scholar] [CrossRef]
- Phipps, J.A.; Wilkinson-Berka, J.L.; Fletcher, E.L. Retinal Dysfunction in Diabetic Ren-2 Rats Is Ameliorated by Treatment with Valsartan but Not Atenolol. Invest. Ophthalmol. Vis. Sci. 2007, 48, 927. [Google Scholar] [CrossRef][Green Version]
- Wilkinsonberka, J.; Tan, G.; Jaworski, K.; Ninkovic, S. Valsartan but Not Atenolol Improves Vascular Pathology in Diabetic Ren-2 Rat Retina. Am. J. Hypertens. 2007, 20, 423–430. [Google Scholar] [CrossRef][Green Version]
- Yan, L.; Dong, Y.; Qing, T.; Deng, Y.; Han, X.; Shi, W.; Li, J.; Gao, F.; Zhang, X.; Tian, Y.; et al. Metoprolol Rescues Endothelial Progenitor Cell Dysfunction in Diabetes. PeerJ 2020, 8, e9306. [Google Scholar] [CrossRef] [PubMed]
- Wiktorowska-Owczarek, A.; Namiecińska, M.; Berezińska, M.; Nowak, J.Z. Characteristics of Adrenaline-Driven Receptor-Mediated Signals in Human Microvessel-Derived Endothelial Cells. Pharm. Rep. 2008, 60, 950–956. [Google Scholar]
- Warne, T.; Moukhametzianov, R.; Baker, J.G.; Nehmé, R.; Edwards, P.C.; Leslie, A.G.W.; Schertler, G.F.X.; Tate, C.G. The Structural Basis for Agonist and Partial Agonist Action on a β(1)-Adrenergic Receptor. Nature 2011, 469, 241–244. [Google Scholar] [CrossRef]
- Ji, Y.; Chen, S.; Li, K.; Xiao, X.; Zheng, S.; Xu, T. The Role of β-Adrenergic Receptor Signaling in the Proliferation of Hemangioma-Derived Endothelial Cells. Cell Div. 2013, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Yang, X.; Xu, G.; Bi, J.; Lv, R.; Huo, R. Propranolol Suppresses HUVEC Viability, Migration, VEGF Expression, and Promotes Apoptosis by Downregulation of MiR-4295. J. Cell. Biochem. 2019, 120, 6614–6623. [Google Scholar] [CrossRef]
- Andreeva, A.; Piontek, J.; Blasig, I.; Utepbergenov, D. Assembly of Tight Junction Is Regulated by the Antagonism of Conventional and Novel Protein Kinase C Isoforms. Int. J. Biochem. Cell Biol. 2005, 38, 222–223. [Google Scholar] [CrossRef]
- Harhaj, N.S.; Felinski, E.A.; Wolpert, E.B.; Sundstrom, J.M.; Gardner, T.W.; Antonetti, D.A. VEGF Activation of Protein Kinase C Stimulates Occludin Phosphorylation and Contributes to Endothelial Permeability. Invest. Ophthalmol. Vis. Sci. 2006, 47, 5106. [Google Scholar] [CrossRef]
- Shi, Y.; Vanhoutte, P.M. Macro- and Microvascular Endothelial Dysfunction in Diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef]
- Kowluru, R.A. Role of Interleukin-1 in the Pathogenesis of Diabetic Retinopathy. Br. J. Ophthalmol. 2004, 88, 1343–1347. [Google Scholar] [CrossRef]
- Yao, Y.; Li, R.; Du, J.; Li, X.; Zhao, L.; Long, L.; Li, D.; Lu, S. Tumor Necrosis Factor-α and Diabetic Retinopathy: Review and Meta-Analysis. Clin. Chim. Acta 2018, 485, 210–217. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing Mitochondrial Superoxide Production Blocks Three Pathways of Hyperglycaemic Damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Calderon, G.D.; Juarez, O.H.; Hernandez, G.E.; Punzo, S.M.; De la Cruz, Z.D. Oxidative Stress and Diabetic Retinopathy: Development and Treatment. Eye 2017, 31, 1122–1130. [Google Scholar] [CrossRef]
- Luo, P.; Wang, M.-H. Eicosanoids, β-Cell Function, and Diabetes. Prostaglandins Other Lipid Mediat. 2011, 95, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dobrian, A.D.; Lieb, D.C.; Cole, B.K.; Taylor-Fishwick, D.A.; Chakrabarti, S.K.; Nadler, J.L. Functional and Pathological Roles of the 12- and 15-Lipoxygenases. Prog. Lipid Res. 2011, 50, 115–131. [Google Scholar] [CrossRef]
- Huang, H.; Al-Shabrawey, M.; Wang, M.-H. Cyclooxygenase- and Cytochrome P450-Derived Eicosanoids in Stroke. Prostaglandins Other Lipid Mediat. 2016, 122, 45–53. [Google Scholar] [CrossRef]
- Li, Y.-B.; Han, J.-Y.; Jiang, W.; Wang, J. Selenium Inhibits High Glucose-Induced Cyclooxygenase-2 and P-Selectin Expression in Vascular Endothelial Cells. Mol. Biol. Rep. 2011, 38, 2301–2306. [Google Scholar] [CrossRef]
- Muzaffar, S.; Shukla, N.; Lobo, C.; Angelini, G.D.; Jeremy, J.Y. Iloprost Inhibits Superoxide Formation and Gp91 phox Expression Induced by the Thromboxane A2 Analogue U46619, 8-Isoprostane F2α, Prostaglandin F2α, Cytokines and Endotoxin in the Pig Pulmonary Artery: Superoxide and Eicosanoids in Pig Pulmonary Artery. Br. J. Pharmacol. 2004, 141, 488–496. [Google Scholar] [CrossRef]
- Capone, C.; Faraco, G.; Anrather, J.; Zhou, P.; Iadecola, C. Cyclooxygenase 1–Derived Prostaglandin E 2 and EP1 Receptors Are Required for the Cerebrovascular Dysfunction Induced by Angiotensin II. Hypertension 2010, 55, 911–917. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence (5′–3′) | Amplicon (bp) | Accession n. |
|---|---|---|---|
| VEGFA | Fw: ATCTTCAAGCCATCCTGTGTGC | 121 | NM_001025366.3 |
| Rv: GAGGTTTGATCCGCATAATCTG | |||
| IL-1β | Fw: AGCTACGAATCTCCGACCAC | 186 | NM_000576.3 |
| Rv: CGTTATCCCATGTGTCGAAGAA | |||
| TNF-α | Fw: AGCCCATGTTGTAGCAAA CC | 134 | NM_000594.4 |
| Rv: TGAGGTACAGGCCCTCTGAT | |||
| 18S rRNA | Fw: TAAGTCCCTGCCCTTTGTACACA | 69 | NR 146119 |
| Rv: GATCCGAGGGCCTCACTAAAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giurdanella, G.; Longo, A.; Distefano, A.; Olivieri, M.; Cristaldi, M.; Cosentino, A.; Agafonova, A.; Caporarello, N.; Lupo, G.; Anfuso, C.D. The Anti-Inflammatory Effect of the β1-Adrenergic Receptor Antagonist Metoprolol on High Glucose Treated Human Microvascular Retinal Endothelial Cells. Cells 2022, 11, 51. https://doi.org/10.3390/cells11010051
Giurdanella G, Longo A, Distefano A, Olivieri M, Cristaldi M, Cosentino A, Agafonova A, Caporarello N, Lupo G, Anfuso CD. The Anti-Inflammatory Effect of the β1-Adrenergic Receptor Antagonist Metoprolol on High Glucose Treated Human Microvascular Retinal Endothelial Cells. Cells. 2022; 11(1):51. https://doi.org/10.3390/cells11010051
Chicago/Turabian StyleGiurdanella, Giovanni, Anna Longo, Alfio Distefano, Melania Olivieri, Martina Cristaldi, Alessia Cosentino, Aleksandra Agafonova, Nunzia Caporarello, Gabriella Lupo, and Carmelina Daniela Anfuso. 2022. "The Anti-Inflammatory Effect of the β1-Adrenergic Receptor Antagonist Metoprolol on High Glucose Treated Human Microvascular Retinal Endothelial Cells" Cells 11, no. 1: 51. https://doi.org/10.3390/cells11010051
APA StyleGiurdanella, G., Longo, A., Distefano, A., Olivieri, M., Cristaldi, M., Cosentino, A., Agafonova, A., Caporarello, N., Lupo, G., & Anfuso, C. D. (2022). The Anti-Inflammatory Effect of the β1-Adrenergic Receptor Antagonist Metoprolol on High Glucose Treated Human Microvascular Retinal Endothelial Cells. Cells, 11(1), 51. https://doi.org/10.3390/cells11010051