
The Interplay between Glioblastoma and Its Microenvironment

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
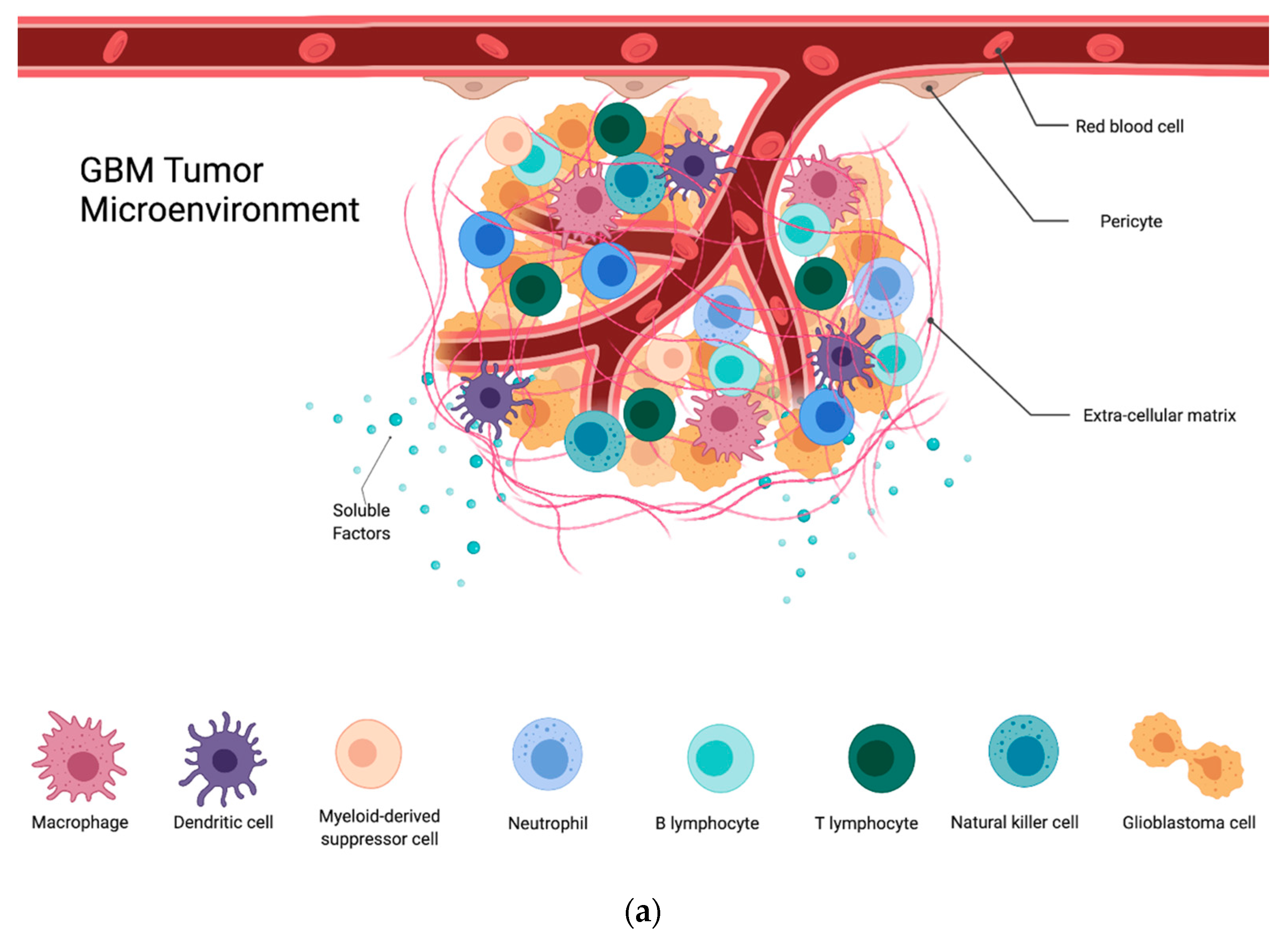
2. The Extracellular Matrix
3. The Blood–Brain Barrier
4. The Central Nervous System Resident Cells
5. GBM Cells and Glioma Stem Cells
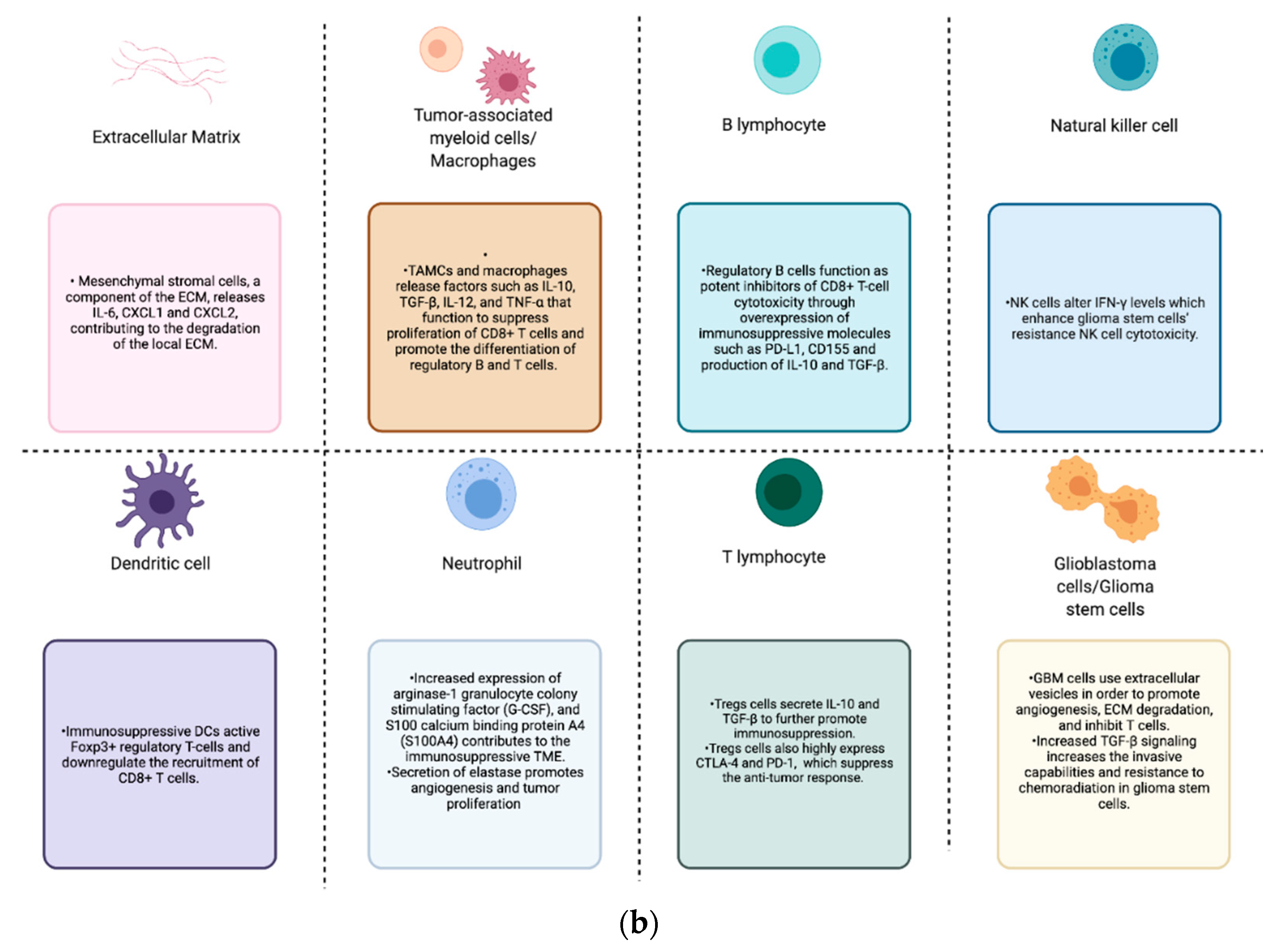
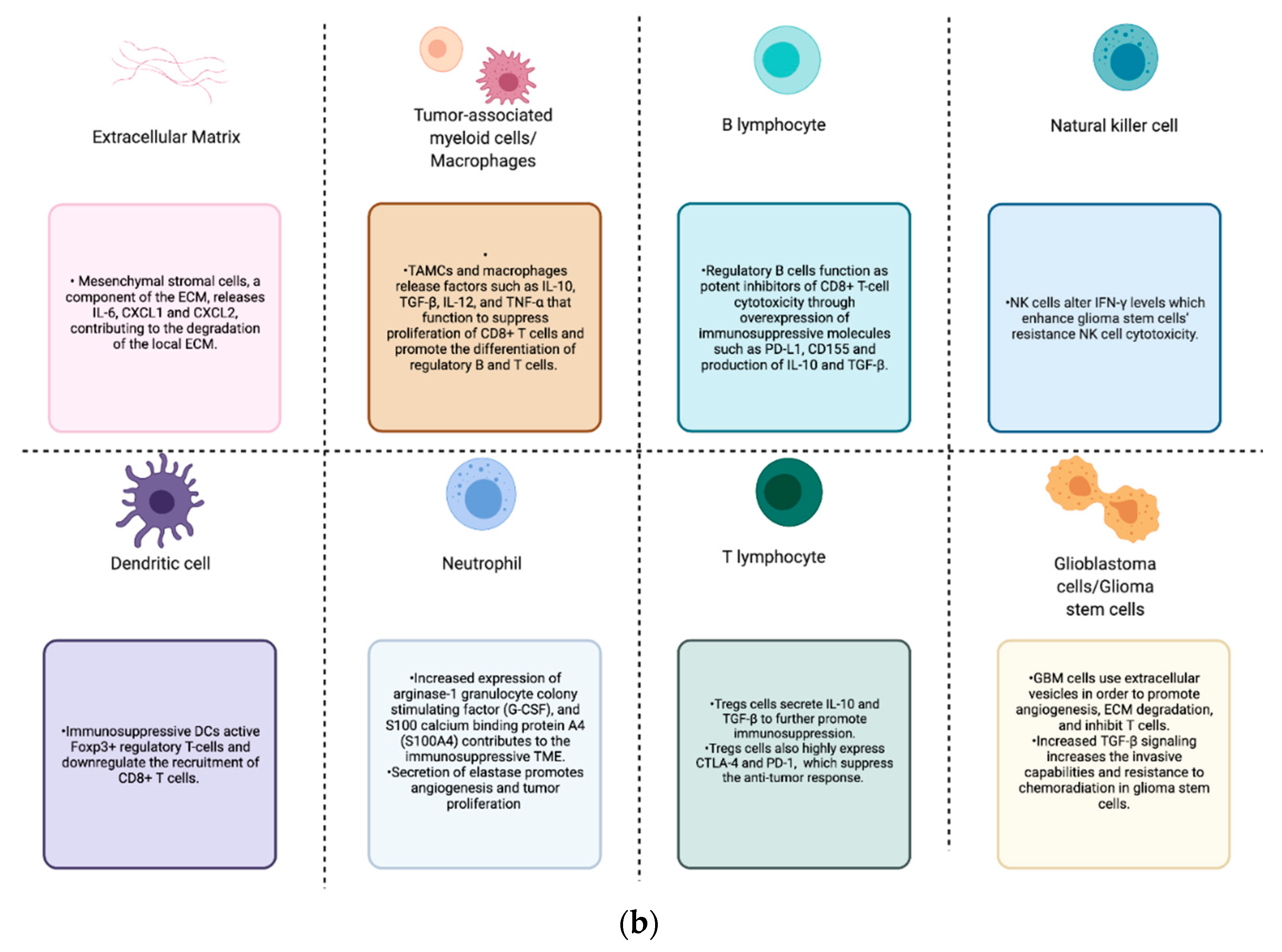
6. Immune Cells
6.1. Tumor-Associated Myeloid Cells
6.2. Tumor-Associated Neutrophils
6.3. Foxp3+ Regulatory T Cells
6.4. Natural Killers
6.5. Dendritic Cells
6.6. B Cells
7. Implications of the TME on Immunotherapies
8. Conclusions
Funding
Conflicts of Interest
References
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; ISBN 978-0-9944381-2-6. [Google Scholar]
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved Survival Time Trends for Glioblastoma Using the SEER 17 Population-Based Registries. J. Neurooncol. 2012, 107, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- DeCordova, S.; Shastri, A.; Tsolaki, A.G.; Yasmin, H.; Klein, L.; Singh, S.K.; Kishore, U. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front. Immunol. 2020, 11, 1402. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. An Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010, 17, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgaki, H.; Kleihues, P. The Definition of Primary and Secondary Glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Lam, D.; Enright, H.A.; Cadena, J.; Peters, S.K.G.; Sales, A.P.; Osburn, J.J.; Soscia, D.A.; Kulp, K.S.; Wheeler, E.K.; Fischer, N.O. Tissue-Specific Extracellular Matrix Accelerates the Formation of Neural Networks and Communities in a Neuron-Glia Co-Culture on a Multi-Electrode Array. Sci. Rep. 2019, 9, 4159. [Google Scholar] [CrossRef]
- Barros, C.S.; Franco, S.J.; Müller, U. Extracellular Matrix: Functions in the Nervous System. Cold Spring Harb. Perspect. Biol. 2011, 3, a005108. [Google Scholar] [CrossRef] [Green Version]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular Matrix Assembly: A Multiscale Deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Wang, C.; Sinha, S.; Jiang, X.; Murphy, L.; Fitch, S.; Wilson, C.; Grant, G.; Yang, F. Matrix Stiffness Modulates Patient-Derived Glioblastoma Cell Fates in Three-Dimensional Hydrogels. Tissue Eng. Part A 2020, 27, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Belousov, A.; Titov, S.; Shved, N.; Garbuz, M.; Malykin, G.; Gulaia, V.; Kagansky, A.; Kumeiko, V. The Extracellular Matrix and Biocompatible Materials in Glioblastoma Treatment. Front. Bioeng. Biotechnol. 2019, 7, 341. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kumar, S. CD44-Mediated Adhesion to Hyaluronic Acid Contributes to Mechanosensing and Invasive Motility. Mol. Cancer Res. 2014, 12, 1416–1429. [Google Scholar] [CrossRef] [Green Version]
- Hossain, A.; Gumin, J.; Gao, F.; Figueroa, J.; Shinojima, N.; Takezaki, T.; Priebe, W.; Villarreal, D.; Kang, S.-G.; Joyce, C.; et al. Mesenchymal Stem Cells Isolated from Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/Gp130/STAT3 Pathway. Stem. Cells 2015, 33, 2400–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrobono, D.; Giacomelli, C.; Marchetti, L.; Martini, C.; Trincavelli, M.L. High Adenosine Extracellular Levels Induce Glioblastoma Aggressive Traits Modulating the Mesenchymal Stromal Cell Secretome. Int. J. Mol. Sci. 2020, 21, 7706. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Liu, S.; Lal, B.; Wei, S.; Wang, S.; Zhan, D.; Zhang, H.; Lee, R.S.; Gao, P.; Lopez-Bertoni, H.; et al. Extracellular Matrix Protein Tenascin C Increases Phagocytosis Mediated by CD47 Loss of Function in Glioblastoma. Cancer Res. 2019, 79, 2697–2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, C.; Wang, L.; Chen, Y. A Comprehensive Review in Improving Delivery of Small-Molecule Chemotherapeutic Agents Overcoming the Blood-Brain/Brain Tumor Barriers for Glioblastoma Treatment. Drug Deliv. 2019, 26, 551–565. [Google Scholar] [CrossRef]
- Bhowmik, A.; Khan, R.; Ghosh, M.K. Blood Brain Barrier: A Challenge for Effectual Therapy of Brain Tumors. BioMed Res. Int. 2015, 2015, e320941. [Google Scholar] [CrossRef] [Green Version]
- Harder, B.G.; Blomquist, M.R.; Wang, J.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A.; Loftus, J.C.; Tran, N.L. Developments in Blood-Brain Barrier Penetrance and Drug Repurposing for Improved Treatment of Glioblastoma. Front. Oncol. 2018, 8, 462. [Google Scholar] [CrossRef] [Green Version]
- Wolburg, H.; Noell, S.; Fallier-Becker, P.; Mack, A.F.; Wolburg-Buchholz, K. The Disturbed Blood-Brain Barrier in Human Glioblastoma. Mol Asp. Med. 2012, 33, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.G.; Campanati, L.; Righy, C.; D’Andrea-Meira, I.; Spohr, T.C.L. de S. e; Porto-Carreiro, I.; Pereira, C.M.; Balça-Silva, J.; Kahn, S.A.; DosSantos, M.F.; et al. Gliomas and the Vascular Fragility of the Blood Brain Barrier. Front. Cell Neurosci. 2014, 8, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momeny, M.; Moghaddaskho, F.; Gortany, N.K.; Yousefi, H.; Sabourinejad, Z.; Zarrinrad, G.; Mirshahvaladi, S.; Eyvani, H.; Barghi, F.; Ahmadinia, L.; et al. Blockade of Vascular Endothelial Growth Factor Receptors by Tivozanib Has Potential Anti-Tumour Effects on Human Glioblastoma Cells. Sci. Rep. 2017, 7, 44075. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Chen, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; et al. Hypoxia Promotes Glioma-Associated Macrophage Infiltration via Periostin and Subsequent M2 Polarization by Upregulating TGF-Beta and M-CSFR. Oncotarget 2016, 7, 80521–80542. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, T.; Yang, N.; Xu, S.; Li, X.; Wang, D. Hypoxia and Macrophages Promote Glioblastoma Invasion by the CCL4-CCR5 Axis. Oncol. Rep. 2016, 36, 3522–3528. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-Associated Reactive Astrocytes Aid the Evolution of Immunosuppressive Environment in Glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [Green Version]
- Iser, I.C.; Lenz, G.; Wink, M.R. EMT-like Process in Glioblastomas and Reactive Astrocytes. Neurochem. Int. 2019, 122, 139–143. [Google Scholar] [CrossRef]
- Niklasson, M.; Bergström, T.; Jarvius, M.; Sundström, A.; Nyberg, F.; Haglund, C.; Larsson, R.; Westermark, B.; Segerman, B.; Segerman, A. Mesenchymal Transition and Increased Therapy Resistance of Glioblastoma Cells Is Related to Astrocyte Reactivity. J. Pathol. 2019, 249, 295–307. [Google Scholar] [CrossRef]
- Al-kharboosh, R.; ReFaey, K.; Lara-Velazquez, M.; Grewal, S.S.; Imitola, J.; Quiñones-Hinojosa, A. Inflammatory Mediators in Glioma Microenvironment Play a Dual Role in Gliomagenesis and Mesenchymal Stem Cell Homing: Implication for Cellular Therapy. Mayo Clin. Proc. Innov. Qual. Outcomes 2020, 4, 443–459. [Google Scholar] [CrossRef]
- Maas, S.L.N.; Abels, E.R.; Van De Haar, L.L.; Zhang, X.; Morsett, L.; Sil, S.; Guedes, J.; Sen, P.; Prabhakar, S.; Hickman, S.E.; et al. Glioblastoma Hijacks Microglial Gene Expression to Support Tumor Growth. J. Neuroinflamm. 2020, 17, 120. [Google Scholar] [CrossRef]
- Gillespie, S.; Monje, M. An Active Role for Neurons in Glioma Progression: Making Sense of Scherer’s Structures. Neuro Oncol. 2018, 20, 1292–1299. [Google Scholar] [CrossRef]
- Johung, T.; Monje, M. Neuronal Activity in the Glioma Microenvironment. Curr. Opin. Neurobiol. 2017, 47, 156–161. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groblewska, M.; Litman-Zawadzka, A.; Mroczko, B. The Role of Selected Chemokines and Their Receptors in the Development of Gliomas. Int. J. Mol. Sci. 2020, 21, 3704. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 Directly Enhanced Endothelial Cell Survival, Proliferation, and Matrix Metalloproteinases Production and Regulated Angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mou, L.; Pan, Y.; Feng, C.; Zhang, J.; Li, J. CXCL8 Promotes Glioma Progression By Activating The JAK/STAT1/HIF-1α/Snail Signaling Axis. Onco Targets Ther. 2019, 12, 8125–8138. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Graeber, M.B. The Molecular Profile of Microglia under the Influence of Glioma. Neuro Oncol. 2012, 14, 958–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, T.; Jackson, E.; Giamas, G. Breaking through the Glioblastoma Micro-Environment via Extracellular Vesicles. Oncogene 2020, 39, 4477–4490. [Google Scholar] [CrossRef]
- Oushy, S.; Hellwinkel, J.E.; Wang, M.; Nguyen, G.J.; Gunaydin, D.; Harland, T.A.; Anchordoquy, T.J.; Graner, M.W. Glioblastoma Multiforme-Derived Extracellular Vesicles Drive Normal Astrocytes towards a Tumour-Enhancing Phenotype. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160477. [Google Scholar] [CrossRef] [Green Version]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune Evasion Mediated by PD-L1 on Glioblastoma-Derived Extracellular Vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef] [Green Version]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy Resistance of Glioblastoma Stem Cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [Green Version]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma Stem Cells: Signaling, Microenvironment, and Therapy. Stem Cells Int. 2016, 2016, e7849890. [Google Scholar] [CrossRef] [Green Version]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The Role of Glioma Stem Cells in Chemotherapy Resistance and Glioblastoma Multiforme Recurrence. Expert Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef] [Green Version]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem Cell-Associated Heterogeneity in Glioblastoma Results from Intrinsic Tumor Plasticity Shaped by the Microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Hayashi, Y.; Stephens, C.; Georgescu, M.-M. Invasive Glioblastoma Cells Acquire Stemness and Increased Akt Activation. Neoplasia 2010, 12, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated Invasive Potential of Glioblastoma Stem Cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bien-Möller, S.; Balz, E.; Herzog, S.; Plantera, L.; Vogelgesang, S.; Weitmann, K.; Seifert, C.; Fink, M.A.; Marx, S.; Bialke, A.; et al. Association of Glioblastoma Multiforme Stem Cell Characteristics, Differentiation, and Microglia Marker Genes with Patient Survival. Stem Cells Int. 2018, 2018, e9628289. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Miska, J.; Lee-Chang, C.; Rashidi, A.; Panek, W.K.; An, S.; Zannikou, M.; Lopez-Rosas, A.; Han, Y.; Xiao, T.; et al. Therapeutic Targeting of Tumor-Associated Myeloid Cells Synergizes with Radiation Therapy for Glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 23714–23723. [Google Scholar] [CrossRef]
- De Leo, A.; Ugolini, A.; Veglia, F. Myeloid Cells in Glioblastoma Microenvironment. Cells 2020, 10, 18. [Google Scholar] [CrossRef]
- Lu-Emerson, C.; Snuderl, M.; Kirkpatrick, N.D.; Goveia, J.; Davidson, C.; Huang, Y.; Riedemann, L.; Taylor, J.; Ivy, P.; Duda, D.G.; et al. Increase in Tumor-Associated Macrophages after Antiangiogenic Therapy Is Associated with Poor Survival among Patients with Recurrent Glioblastoma. Neuro Oncol. 2013, 15, 1079–1087. [Google Scholar] [CrossRef]
- Zhu, H.; Leiss, L.; Yang, N.; Rygh, C.B.; Mitra, S.S.; Cheshier, S.H.; Weissman, I.L.; Huang, B.; Miletic, H.; Bjerkvig, R.; et al. Surgical Debulking Promotes Recruitment of Macrophages and Triggers Glioblastoma Phagocytosis in Combination with CD47 Blocking Immunotherapy. Oncotarget 2017, 8, 12145–12157. [Google Scholar] [CrossRef] [Green Version]
- Akins, E.A.; Aghi, M.K.; Kumar, S. Incorporating Tumor-Associated Macrophages into Engineered Models of Glioma. iScience 2020, 23, 101770. [Google Scholar] [CrossRef] [PubMed]
- De Vleeschouwer, S.; Bergers, G. Glioblastoma: To Target the Tumor Cell or the Microenvironment? In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; ISBN 978-0-9944381-2-6. [Google Scholar]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, Y.; Guo, N.; Luan, J.; Cheng, J.; Hu, Z.; Jiang, P.; Jin, W.; Gao, X. The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front. Immunol. 2020, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of Myeloid-Derived Suppressor Cells in Cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Bossman, S.A.J.F.H.; ter Laan, M.; Wesseling, P.; Adema, G.J. Elevated Levels of Polymorphonuclear Myeloid-Derived Suppressor Cells in Patients with Glioblastoma Highly Express S100A8/9 and Arginase and Suppress T Cell Function. Neuro Oncol. 2016, 18, 1253–1264. [Google Scholar] [CrossRef]
- Richard, S.A. Explicating the Pivotal Pathogenic, Diagnostic, and Therapeutic Biomarker Potentials of Myeloid-Derived Suppressor Cells in Glioblastoma. Dis. Markers 2020, 2020, e8844313. [Google Scholar] [CrossRef]
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global Immune Fingerprinting in Glioblastoma Patient Peripheral Blood Reveals Immune-Suppression Signatures Associated with Prognosis. JCI Insight 2018, 3, e122264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raber, P.; Ochoa, A.C.; Rodríguez, P.C. Metabolism of L-Arginine by Myeloid-Derived Suppressor Cells in Cancer: Mechanisms of T Cell Suppression and Therapeutic Perspectives. Immunol. Investig. 2012, 41, 614–634. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression Mediated by Myeloid-Derived Suppressor Cells (MDSCs) during Tumour Progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massara, M.; Persico, P.; Bonavita, O.; Mollica Poeta, V.; Locati, M.; Simonelli, M.; Bonecchi, R. Neutrophils in Gliomas. Front. Immunol. 2017, 8, 1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhang, X.H.-F. Tumor-Associated Neutrophils and Macrophages—Heterogenous but Not Chaotic. Front. Immunol. 2020, 11, 553967. [Google Scholar] [CrossRef]
- Khan, S.; Mittal, S.; McGee, K.; Alfaro-Munoz, K.D.; Majd, N.; Balasubramaniyan, V.; de Groot, J.F. Role of Neutrophils and Myeloid-Derived Suppressor Cells in Glioma Progression and Treatment Resistance. Int. J. Mol. Sci. 2020, 21, 1954. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H. FOXP3 and Its Role in the Immune System. Adv. Exp. Med. Biol. 2009, 665, 17–29. [Google Scholar] [CrossRef]
- Lu, L.; Barbi, J.; Pan, F. The Regulation of Immune Tolerance by FOXP3. Nat. Rev. Immunol. 2017, 17, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Abou-Ghazal, M.; Reina-Ortiz, C.; Yang, D.S.; Sun, W.; Qiao, W.; Hiraoka, N.; Fuller, G.N. Incidence and Prognostic Impact of FoxP3+ Regulatory T Cells in Human Gliomas. Clin. Cancer Res. 2008, 14, 5166–5172. [Google Scholar] [CrossRef] [Green Version]
- Sayour, E.J.; McLendon, P.; McLendon, R.; De Leon, G.; Reynolds, R.; Kresak, J.; Sampson, J.H.; Mitchell, D.A. Increased Proportion of FoxP3+ Regulatory T Cells in Tumor Infiltrating Lymphocytes Is Associated with Tumor Recurrence and Reduced Survival in Patients with Glioblastoma. Cancer Immunol. Immunother. 2015, 64, 419–427. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Richmond, A. Chemokines Modulate Immune Surveillance in Tumorigenesis, Metastasis, and Response to Immunotherapy. Front. Immunol. 2019, 10, 333. [Google Scholar] [CrossRef] [Green Version]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H.G. Suppression of Antitumor Immunity by IL-10 and TGF-β-Producing T Cells Infiltrating the Growing Tumor: Influence of Tumor Environment on the Induction of CD4+ and CD8+ Regulatory T Cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef] [Green Version]
- Humphries, W.; Wei, J.; Sampson, J.H.; Heimberger, A.B. The Role of Tregs in Glioma-Mediated Immunosuppression: Potential Target for Intervention. Neurosurg. Clin. N. Am. 2010, 21, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T Cells in Tumor Microenvironment: New Mechanisms, Potential Therapeutic Strategies and Future Prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Kelly, W.J.; Giles, A.J.; Gilbert, M. T Lymphocyte-Targeted Immune Checkpoint Modulation in Glioma. J. Immunother. Cancer 2020, 8, 379. [Google Scholar] [CrossRef] [PubMed]
- Miska, J.; Rashidi, A.; Chang, A.L.; Muroski, M.E.; Han, Y.; Zhang, L.; Lesniak, M.S. Anti-GITR Therapy Promotes Immunity against Malignant Glioma in a Murine Model. Cancer Immunol. Immunother. 2016, 65, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Wang, D.; Zhang, G.; Guo, X. The Role of PD-1/PD-L1 Axis in Treg Development and Function: Implications for Cancer Immunotherapy. OncoTargets Ther. 2019, 12, 8437–8445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent Glioma Clinical Trial, CheckMate-143: The Game Is Not over Yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; López-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant Nivolumab Modifies the Tumor Immune Microenvironment in Resectable Glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Tremelimumab and Durvalumab in Combination or Alone in Treating Patients with Recurrent Malignant Glioma—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02794883 (accessed on 22 April 2020).
- Zeltsman, M.; Dozier, J.; McGee, E.; Ngai, D.; Adusumilli, P.S. CAR T-Cell Therapy for Lung Cancer and Malignant Pleural Mesothelioma. Transl. Res. 2017, 187, 1–10. [Google Scholar] [CrossRef]
- Forsberg, E.M.V.; Lindberg, M.F.; Jespersen, H.; Alsén, S.; Bagge, R.O.; Donia, M.; Svane, I.M.; Nilsson, O.; Ny, L.; Nilsson, L.M.; et al. HER2 CAR-T Cells Eradicate Uveal Melanoma and T-Cell Therapy–Resistant Human Melanoma in IL2 Transgenic NOD/SCID IL2 Receptor Knockout Mice. Cancer Res. 2019, 79, 899–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Land, C.A.; Musich, P.R.; Haydar, D.; Krenciute, G.; Xie, Q. Chimeric Antigen Receptor T-Cell Therapy in Glioblastoma: Charging the T Cells to Fight. J. Transl. Med. 2020, 18, 428. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T Cells for Brain Tumors: Lessons Learned and Road Ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A.; et al. Rational Development and Characterization of Humanized Anti-EGFR Variant III Chimeric Antigen Receptor T Cells for Glioblastoma. Sci. Transl. Med. 2015, 7, 275ra22. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Thaci, B.; Crawford, A.C.; Sampath, P. Interleukin-13 Receptor Alpha 2-Targeted Glioblastoma Immunotherapy. Biomed. Res. Int. 2014, 2014, 952128. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T Cells Targeting HER2 and IL13Rα2 Mitigate Tumor Antigen Escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [Green Version]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.-F.; Orange, J.S.; Sumazin, P.; Man, T.-K.; et al. Trivalent CAR T Cells Overcome Interpatient Antigenic Variability in Glioblastoma. Neuro-Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.; Han, S.J.; Sughrue, M.E.; Tihan, T.; Parsa, A.T. Immune Cell Infiltrate Differences in Pilocytic Astrocytoma and Glioblastoma: Evidence of Distinct Immunological Microenvironments That Reflect Tumor Biology. J. Neurosurg. 2011, 115, 505–511. [Google Scholar] [CrossRef]
- Ren, F.; Zhao, Q.; Huang, L.; Zheng, Y.; Li, L.; He, Q.; Zhang, C.; Li, F.; Maimela, N.R.; Sun, Z.; et al. The R132H Mutation in IDH1 Promotes the Recruitment of NK Cells through CX3CL1/CX3CR1 Chemotaxis and Is Correlated with a Better Prognosis in Gliomas. Immunol. Cell Biol. 2019, 97, 457–469. [Google Scholar] [CrossRef]
- Sedgwick, A.J.; Ghazanfari, N.; Constantinescu, P.; Mantamadiotis, T.; Barrow, A.D. The Role of NK Cells and Innate Lymphoid Cells in Brain Cancer. Front. Immunol. 2020, 11, 1549. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Wu, W.; Zhang, H.; Zhang, X.; Zhang, D.; Wang, Q.; Huang, L.; Wang, Y.; Hang, C. High Expression of B7-H6 in Human Glioma Tissues Promotes Tumor Progression. Oncotarget 2017, 8, 37435–37447. [Google Scholar] [CrossRef] [PubMed]
- Burster, T.; Gärtner, F.; Bulach, C.; Zhanapiya, A.; Gihring, A.; Knippschild, U. Regulation of MHC I Molecules in Glioblastoma Cells and the Sensitizing of NK Cells. Pharmaceuticals 2021, 14, 236. [Google Scholar] [CrossRef]
- Martin-Hijano, L.; Sainz, B. The Interactions between Cancer Stem Cells and the Innate Interferon Signaling Pathway. Front. Immunol. 2020, 11, 526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlowska, A.K.; Tseng, H.-C.; Kaur, K.; Topchyan, P.; Inagaki, A.; Bui, V.T.; Kasahara, N.; Cacalano, N.; Jewett, A. Resistance to Cytotoxicity and Sustained Release of Interleukin-6 and Interleukin-8 in the Presence of Decreased Interferon-γ after Differentiation of Glioblastoma by Human Natural Killer Cells. Cancer Immunol. Immunother. 2016, 65, 1085–1097. [Google Scholar] [CrossRef] [Green Version]
- Fares, J.; Fares, M.Y.; Fares, Y. Natural Killer Cells in the Brain Tumor Microenvironment: Defining a New Era in Neuro-Oncology. Surg. Neurol. Int. 2019, 10, 43. [Google Scholar] [CrossRef]
- Schmidt-Wolf, I.G.; Negrin, R.S.; Kiem, H.P.; Blume, K.G.; Weissman, I.L. Use of a SCID Mouse/Human Lymphoma Model to Evaluate Cytokine-Induced Killer Cells with Potent Antitumor Cell Activity. J. Exp. Med. 1991, 174, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.-S.; Nam, D.-H.; Kang, S.-H.; Lee, J.W.; Chang, J.-H.; Kim, J.-H.; Lim, Y.-J.; Koh, Y.-C.; Chung, Y.-G.; Kim, J.-M.; et al. Phase III Randomized Trial of Autologous Cytokine-Induced Killer Cell Immunotherapy for Newly Diagnosed Glioblastoma in Korea. Oncotarget 2016, 8, 7003–7013. [Google Scholar] [CrossRef] [Green Version]
- Mildner, A.; Jung, S. Development and Function of Dendritic Cell Subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [Green Version]
- D’Agostino, P.M.; Gottfried-Blackmore, A.; Anandasabapathy, N.; Bulloch, K. Brain Dendritic Cells: Biology and Pathology. Acta Neuropathol. 2012, 124, 599–614. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.J.; Ornelles, D.A.; Mitchell, L.M.; Brzoza-Lewis, K.L.; Hiltbold, E.M. IL-12 Produced by Dendritic Cells Augments CD8+ T Cell Activation through the Production of the Chemokines CCL1 and CCL17. J. Immunol. 2008, 181, 8576–8584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef] [Green Version]
- Liau, L.M.; Prins, R.M.; Kiertscher, S.M.; Odesa, S.K.; Kremen, T.J.; Giovannone, A.J.; Lin, J.-W.; Chute, D.J.; Mischel, P.S.; Cloughesy, T.F.; et al. Dendritic Cell Vaccination in Glioblastoma Patients Induces Systemic and Intracranial T-Cell Responses Modulated by the Local Central Nervous System Tumor Microenvironment. Clin. Cancer Res. 2005, 11, 5515–5525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First Results on Survival from a Large Phase 3 Clinical Trial of an Autologous Dendritic Cell Vaccine in Newly Diagnosed Glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, R.; Homma, J.; Yajima, N.; Tsuchiya, N.; Sano, M.; Kobayashi, T.; Yoshida, S.; Abe, T.; Narita, M.; Takahashi, M.; et al. Clinical Evaluation of Dendritic Cell Vaccination for Patients with Recurrent Glioma: Results of a Clinical Phase I/II Trial. Clin. Cancer Res. 2005, 11, 4160–4167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, G.J.; Demissie, E.; Pillai, S. B Lymphocytes and Cancer: A Love-Hate Relationship. Trends Cancer 2016, 2, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, M.C.A.; Nelson, B.H. Prognostic Significance of Tumor-Infiltrating B Cells and Plasma Cells in Human Cancer. Clin. Cancer Res. 2018, 24, 6125–6135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Chang, C.; Bodogai, M.; Moritoh, K.; Olkhanud, P.B.; Chan, A.C.; Croft, M.; Mattison, J.A.; Holst, P.J.; Gress, R.E.; Ferrucci, L.; et al. Accumulation of 4-1BBL+ B Cells in the Elderly Induces the Generation of Granzyme-B+ CD8+ T Cells with Potential Antitumor Activity. Blood 2014, 124, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Lee-Chang, C.; Miska, J.; Hou, D.; Rashidi, A.; Zhang, P.; Burga, R.A.; Jusué-Torres, I.; Xiao, T.; Arrieta, V.A.; Zhang, D.Y.; et al. Activation of 4-1BBL+ B Cells with CD40 Agonism and IFNγ Elicits Potent Immunity against Glioblastoma. J. Exp. Med. 2020, 218, e20200913. [Google Scholar] [CrossRef] [PubMed]
- Lee-Chang, C.; Rashidi, A.; Miska, J.; Zhang, P.; Pituch, K.C.; Hou, D.; Xiao, T.; Fischietti, M.; Kang, S.J.; Appin, C.L.; et al. Myeloid-Derived Suppressive Cells Promote B Cell–Mediated Immunosuppression via Transfer of PD-L1 in Glioblastoma. Cancer Immunol. Res. 2019, 7, 1928–1943. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, L.; Ye, M.; Kang, C.; You, H. Treatment Progress of Immune Checkpoint Blockade Therapy for Glioblastoma. Front. Immunol. 2020, 11, 3080. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus Investigator-Choice Chemotherapy for Ipilimumab-Refractory Melanoma (KEYNOTE-002): A Randomised, Controlled, Phase 2 Trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Carter, T.; Shaw, H.; Cohn-Brown, D.; Chester, K.; Mulholland, P. Ipilimumab and Bevacizumab in Glioblastoma. Clin. Oncol. 2016, 28, 622–626. [Google Scholar] [CrossRef] [Green Version]
- Kong, Z.; Wang, Y.; Ma, W. Vaccination in the Immunotherapy of Glioblastoma. Hum. Vaccines Immunother. 2017, 14, 255–268. [Google Scholar] [CrossRef]
- Choi, B.D.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; McLendon, R.E.; Bigner, D.D.; Sampson, J.H. EGFRvIII-Targeted Vaccination Therapy of Malignant Glioma. Brain Pathol. 2009, 19, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic Escape after Prolonged Progression-Free Survival with Epidermal Growth Factor Receptor Variant III Peptide Vaccination in Patients with Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Fadul, C.E.; Fisher, J.L.; Hampton, T.H.; Lallana, E.C.; Li, Z.; Gui, J.; Szczepiorkowski, Z.M.; Tosteson, T.D.; Rhodes, C.H.; Wishart, H.A.; et al. Immune Response in Patients With Newly Diagnosed Glioblastoma Multiforme Treated With Intranodal Autologous Tumor Lysate-Dendritic Cell Vaccination After Radiation Chemotherapy. J. Immunother. 2011, 34, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A Phase II, Multicenter Trial of Rindopepimut (CDX-110) in Newly Diagnosed Glioblastoma: The ACT III Study. Neuro Oncol. 2015, 17, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, L.; Brown, C.; Badie, B. Chimeric Antigen Receptor T-Cell Therapy: Updates in Glioblastoma Treatment. Neurosurgery 2021, 88, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhu, X.; Qian, Y.; Yuan, X.; Ding, Y.; Hu, D.; He, X.; Wu, Y. Chimeric Antigen Receptor T-Cell Therapy in Glioblastoma: Current and Future. Front. Immunol. 2020, 11, 2837. [Google Scholar] [CrossRef]
- Sanders, S.; Debinski, W. Challenges to Successful Implementation of the Immune Checkpoint Inhibitors for Treatment of Glioblastoma. Int. J. Mol. Sci. 2020, 21, 2759. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [Green Version]
- Hira, V.V.V.; Aderetti, D.A.; van Noorden, C.J.F. Glioma Stem Cell Niches in Human Glioblastoma Are Periarteriolar. J. Histochem. Cytochem. 2018, 66, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magaña-Maldonado, R.; Chávez-Cortez, E.G.; Olascoaga-Arellano, N.K.; López-Mejía, M.; Maldonado-Leal, F.M.; Sotelo, J.; Pineda, B. Immunological Evasion in Glioblastoma. BioMed Res. Int. 2016, 2016, e7487313. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.F.; Cui, J.W. The Role of Tumor-Infiltrating B Cells in Tumor Immunity. J. Oncol. 2019, 2019, 2592419. [Google Scholar] [CrossRef] [PubMed]
- Matarredona, E.R.; Pastor, A.M. Extracellular Vesicle-Mediated Communication between the Glioblastoma and Its Microenvironment. Cells 2019, 9, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. https://doi.org/10.3390/cells10092257
Dapash M, Hou D, Castro B, Lee-Chang C, Lesniak MS. The Interplay between Glioblastoma and Its Microenvironment. Cells. 2021; 10(9):2257. https://doi.org/10.3390/cells10092257
Chicago/Turabian StyleDapash, Mark, David Hou, Brandyn Castro, Catalina Lee-Chang, and Maciej S. Lesniak. 2021. "The Interplay between Glioblastoma and Its Microenvironment" Cells 10, no. 9: 2257. https://doi.org/10.3390/cells10092257
APA StyleDapash, M., Hou, D., Castro, B., Lee-Chang, C., & Lesniak, M. S. (2021). The Interplay between Glioblastoma and Its Microenvironment. Cells, 10(9), 2257. https://doi.org/10.3390/cells10092257