
SARS-CoV-2 Cellular Entry Is Independent of the ACE2 Cytoplasmic Domain Signaling

 , , , , , and
, , , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Bioinformatic Analysis of ACE2 Sequences
2.3. Generation of Plasmid Constructs
2.4. Production of Recombinant Proteins
2.5. Analysis of ACE2 Transcript by Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.6. SDS-PAGE and Western Blot Analysis
2.7. Immunofluorescence Analysis of wtACE2 and ∆cytACE2 Cell Surface Expression
2.8. Quantification of wtACE2 and ∆cytACE2 Expression by Flow Cytometry
2.9. Coronavirus Spike Protein-Binding Assay on ∆cytACE2
2.10. Production and Validation of Pseudotyped Coronaviruses (CoV PVs)
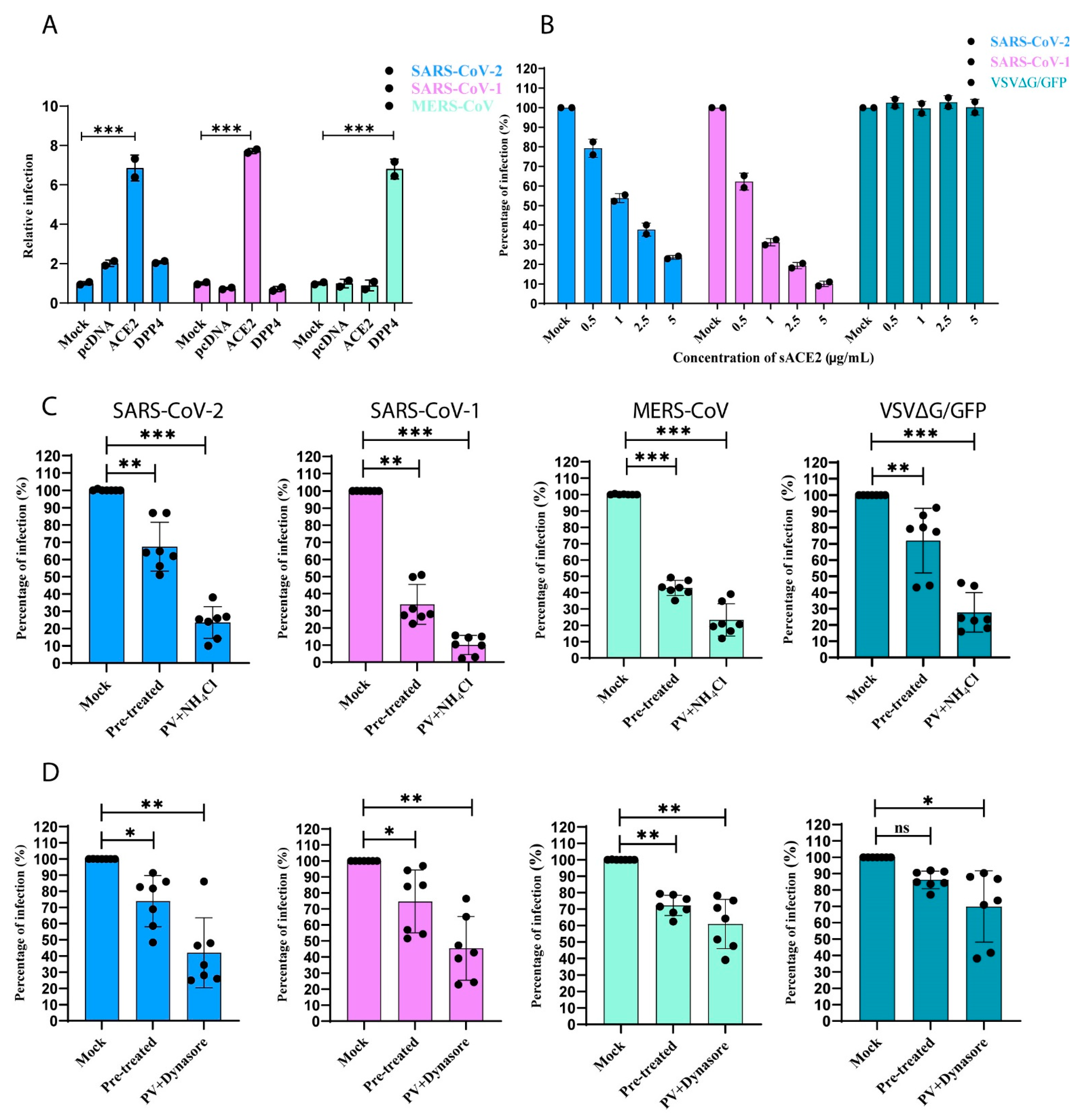
2.11. Soluble ACE2 (sACE2) Blocking Assay
2.12. Effect of Endocytosis Inhibitors on the Entry of CoV PVs
2.13. Colocalization of SARS-CoV-1 and -2 Spike S1-Fc with ACE2 Mutant Receptor
2.14. Receptor-Mediated Internalization Assay of CoV Spike S1-Fc and RBD-Fc Proteins
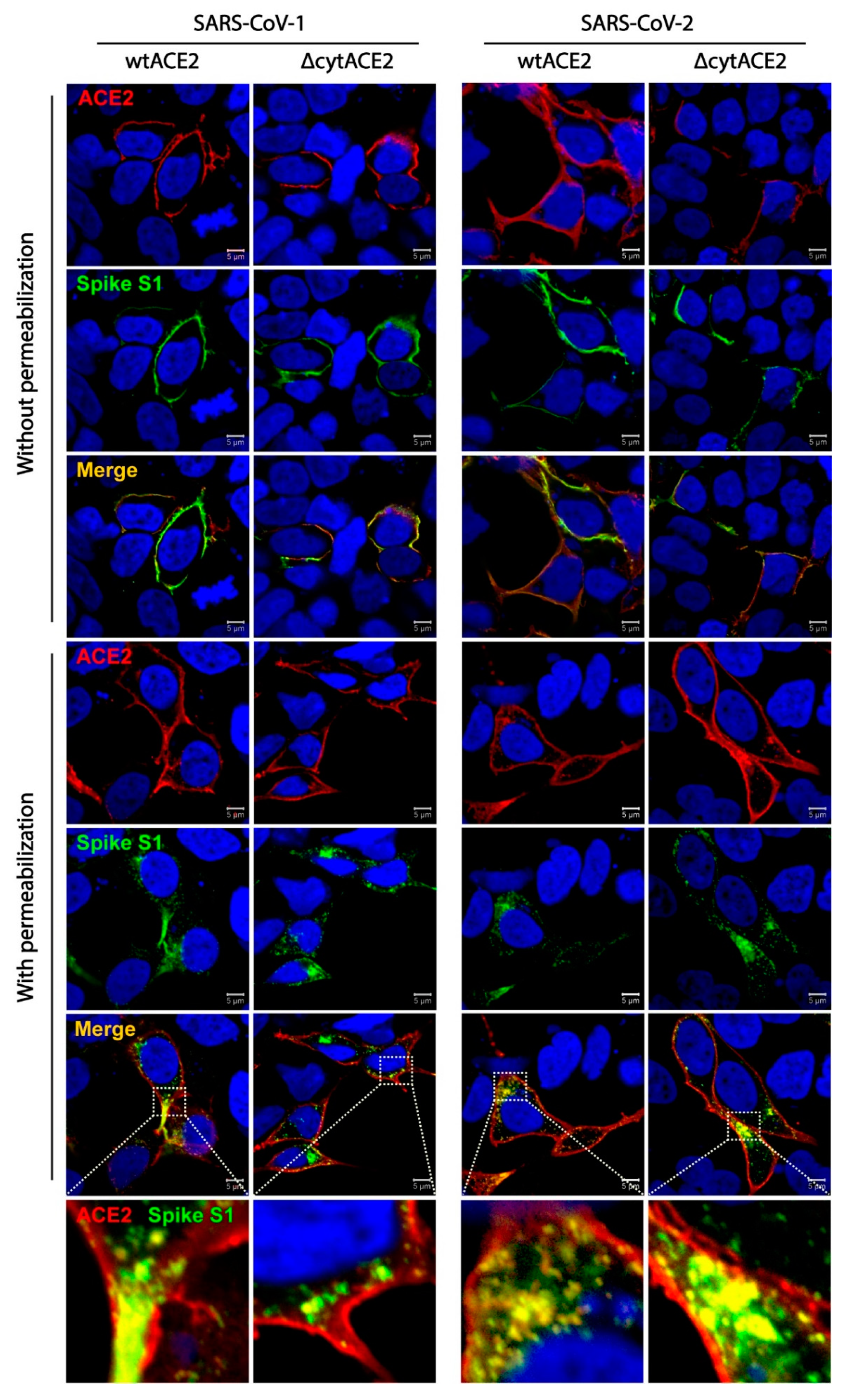
2.15. Colocalization of wtACE2 or ΔcytACE2 with SARS-CoV-1 and SARS-CoV-2 Spike S1-Fc Proteins
2.16. Infectivity of SARS-CoV-1 and -2 PVs in wtACE2- and ∆cytACE2-Expressing Cells
2.17. Statistical Analysis
3. Results
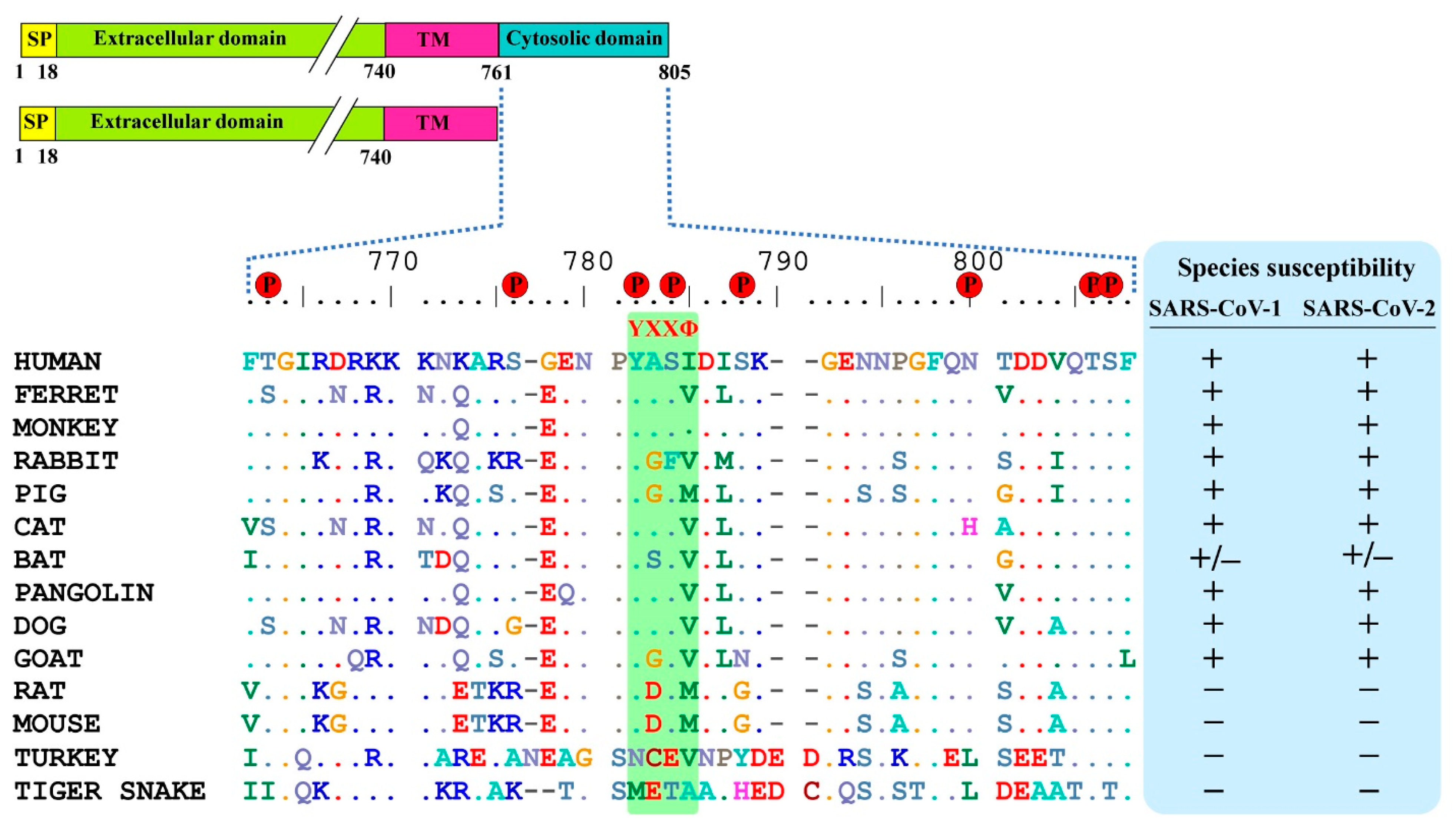
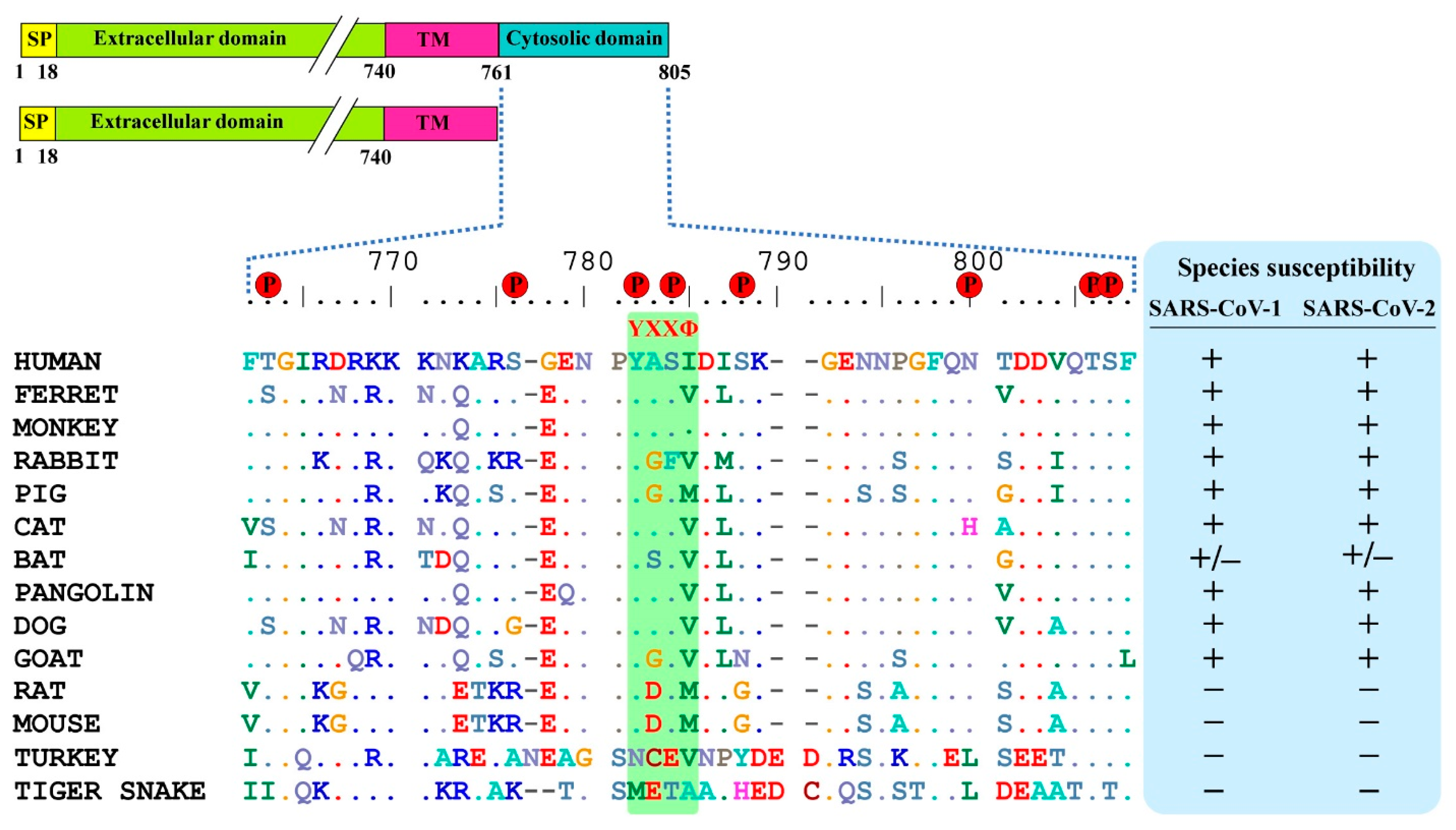
3.1. ACE2 Cytoplasmic Domain Contains Conserved Endocytosis Motif
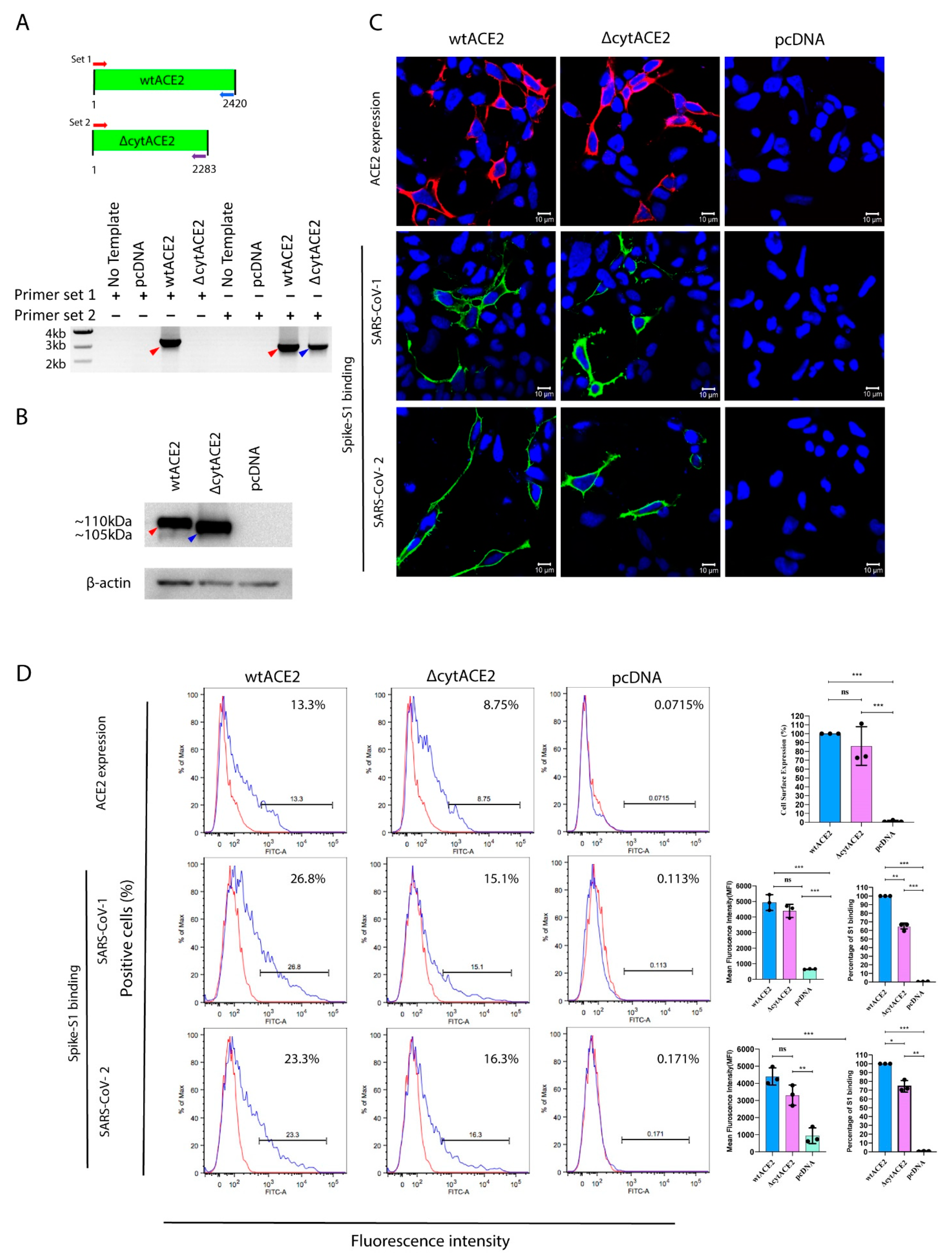
3.2. Surface Expression of Cytoplasmic Deletion Mutant of ACE2 Is Similar to Wildtype ACE2
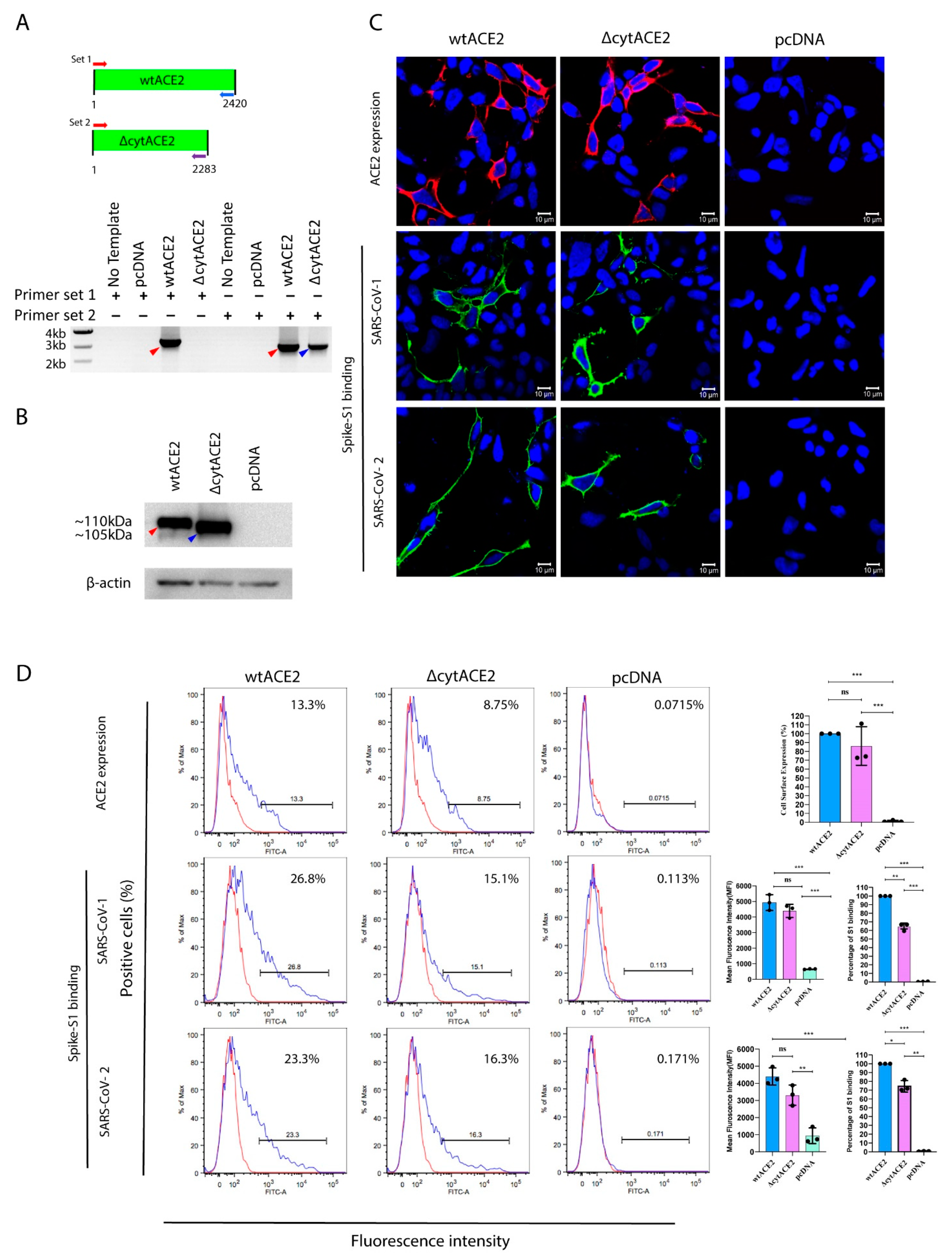
3.3. SARS-CoV-2 Spike S1-Fc Protein Binds on wtACE2 and ΔcytACE2
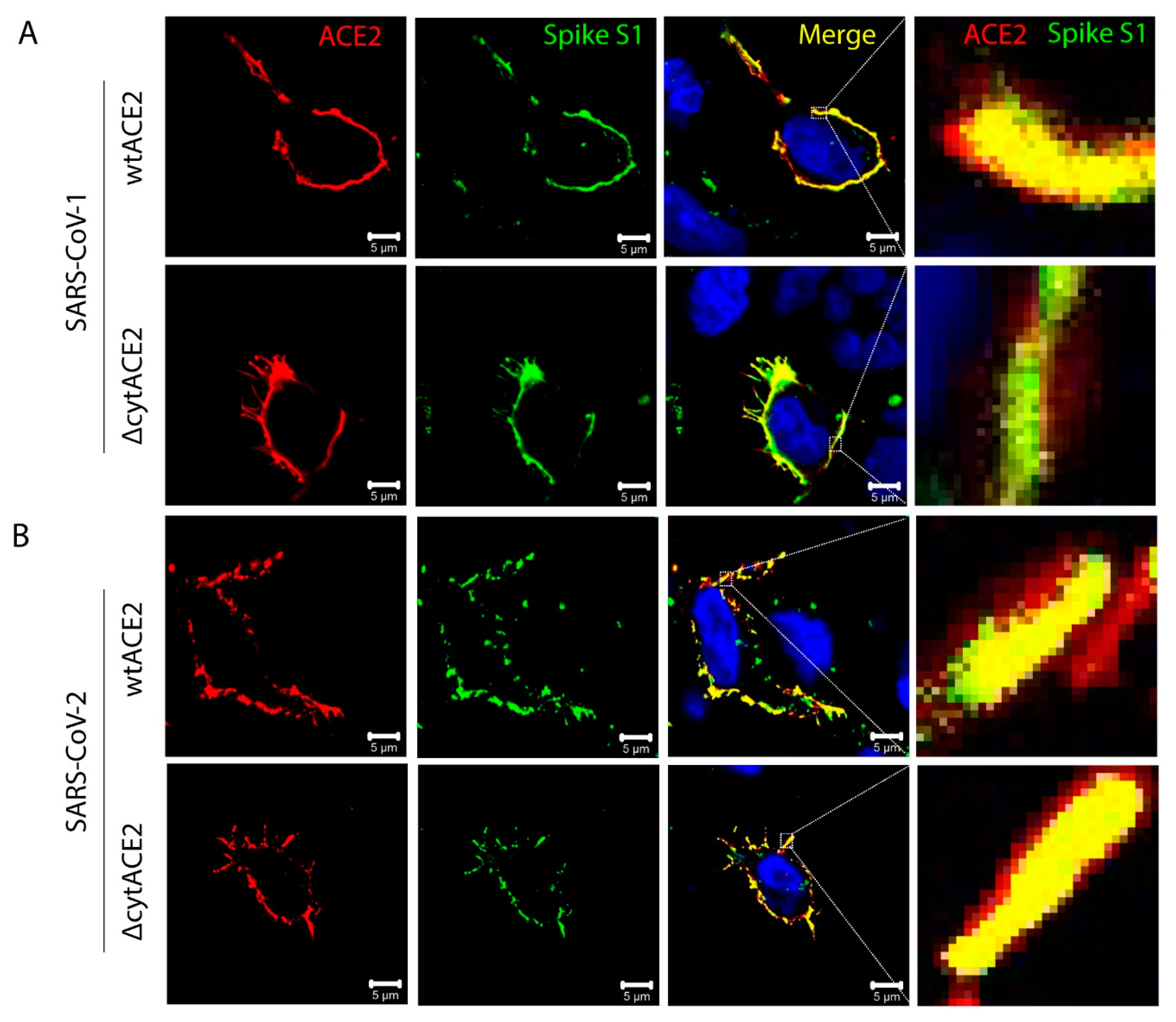
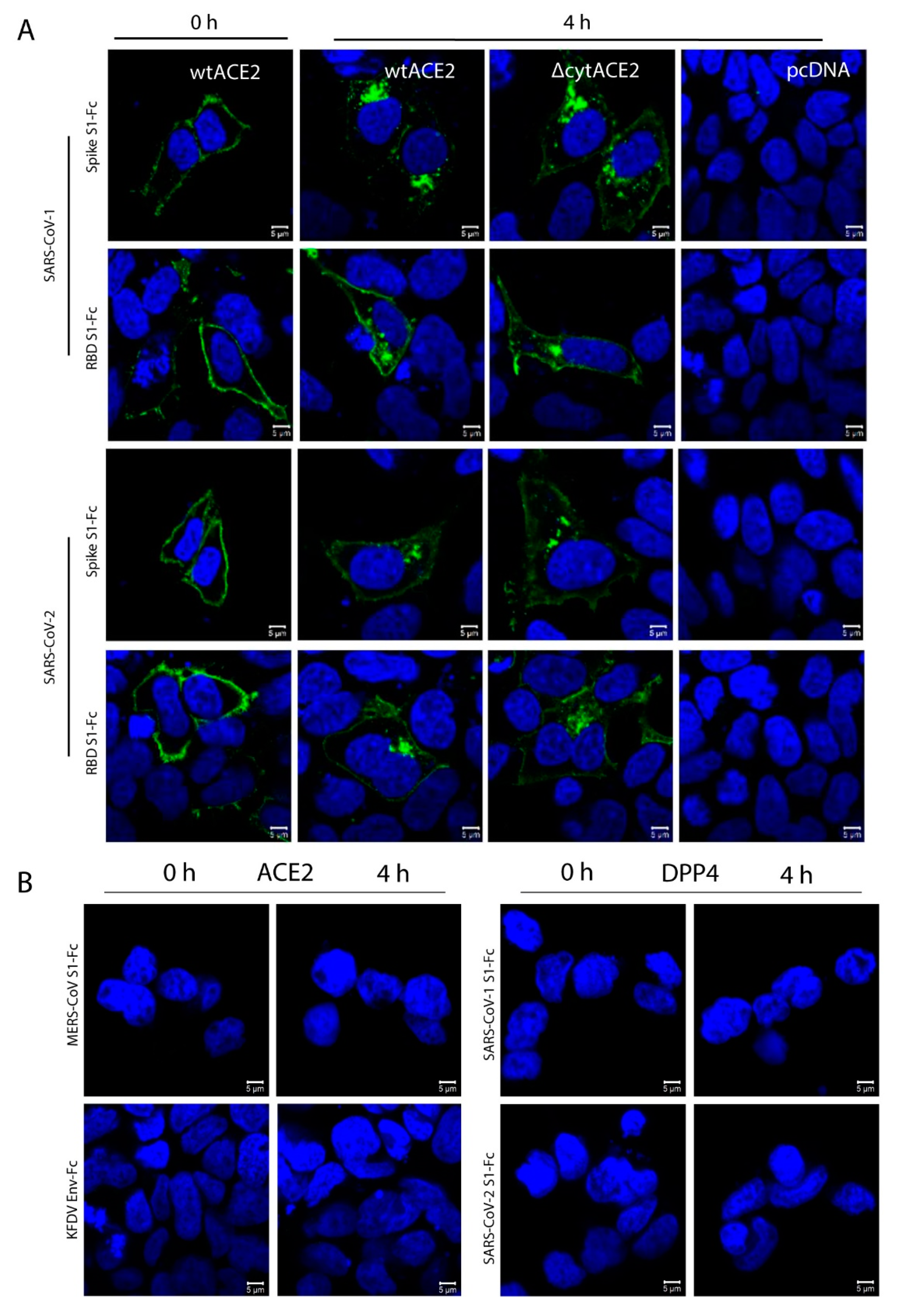
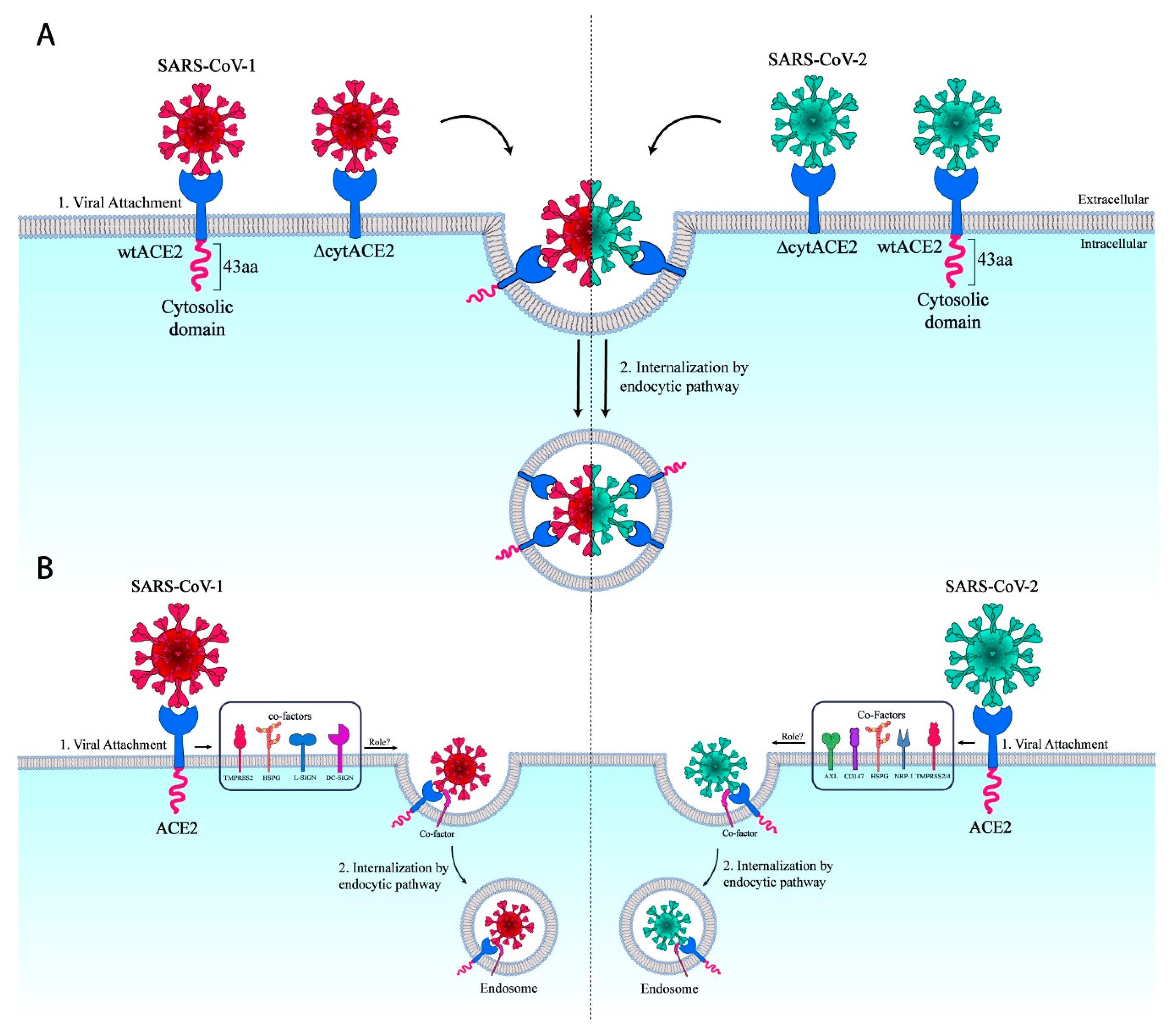
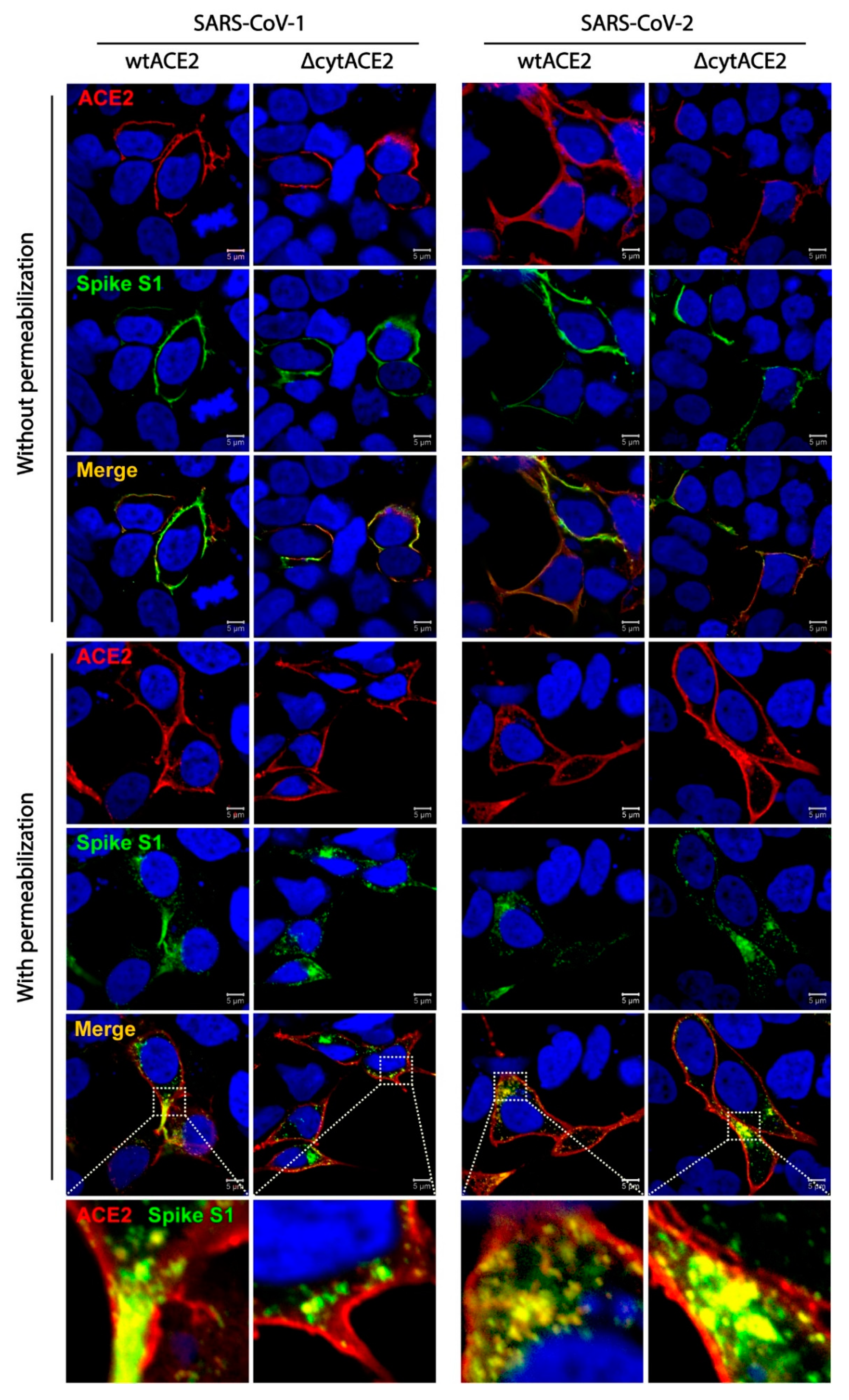
3.4. Cytoplasmic Domain of ACE2 Is Not Necessary for Internalization of Spike S1 or RBD-Fc in HEK293T Cells

3.5. SARS-CoV-1 and -2 Pseudotyped Coronaviruses Enter Cells via pH and Dynamin-Dependent Endocytosis
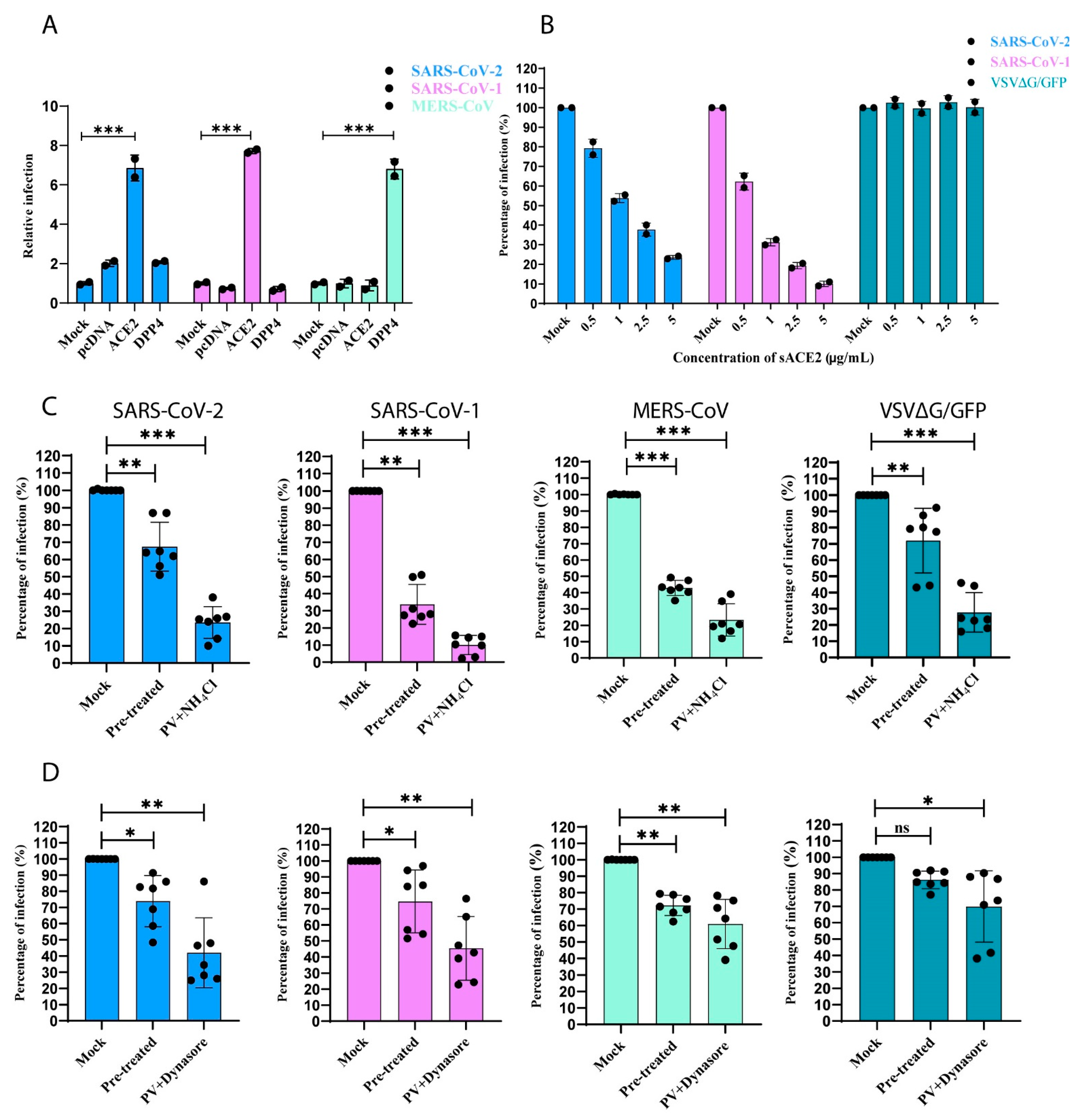
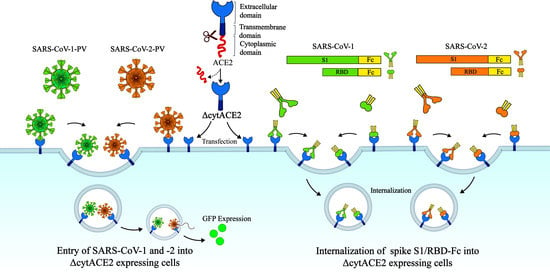
3.6. Removal of Cytoplasmic Domain of ACE2 Does Not Affect SARS-CoV-1 and -2 Entry

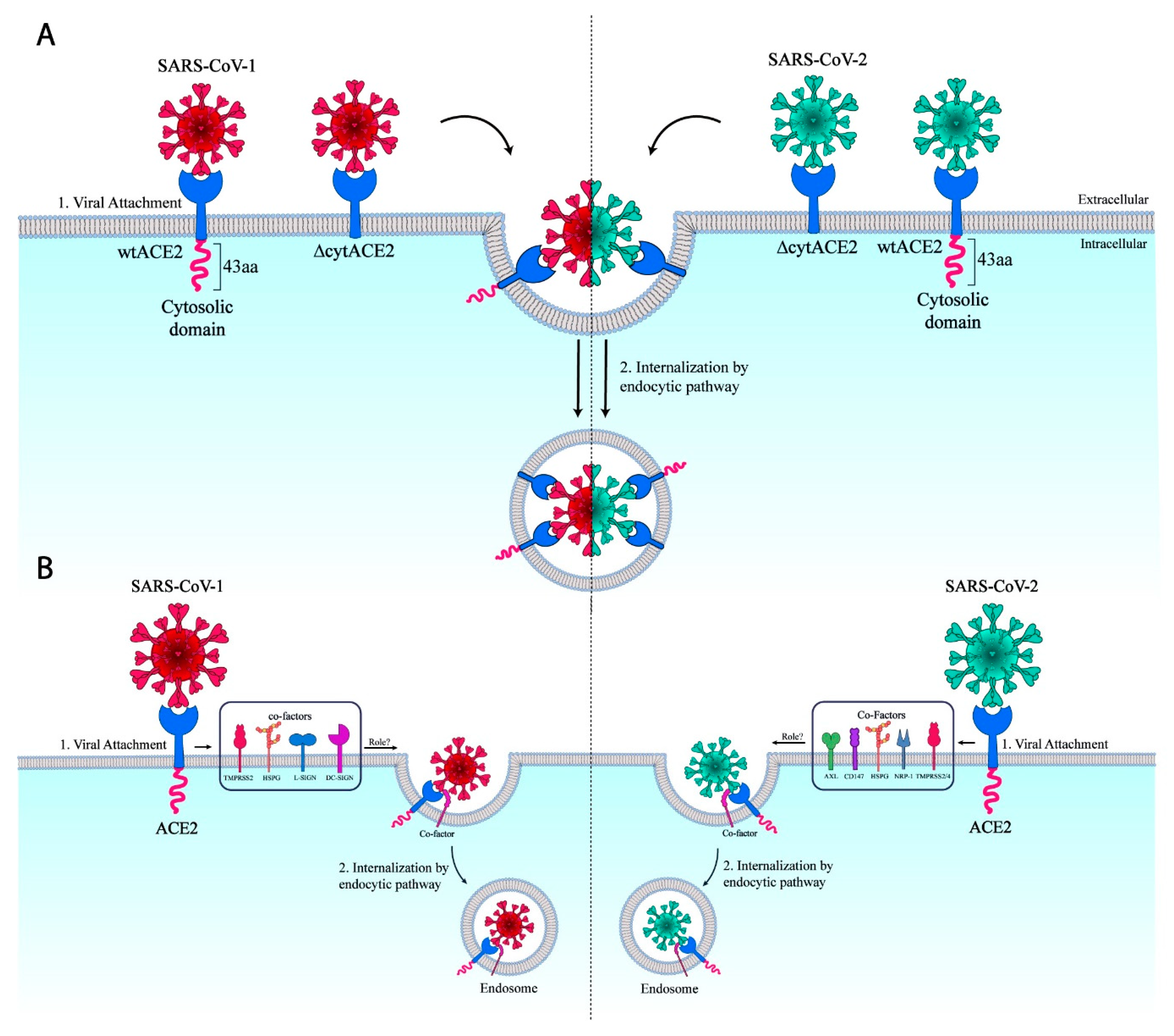
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, X.; Lau, E.H.Y.; Wu, P.; Deng, X.; Wang, J.; Hao, X.; Lau, Y.C.; Wong, J.Y.; Guan, Y.; Tan, X.; et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 2020, 26, 672–675. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Wu, T.; Liu, Q.; Yang, Z. The SARS-CoV-2 outbreak: What we know. Int. J. Infect. Dis. 2020, 94, 44–48. [Google Scholar] [CrossRef]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Peñaranda, S.; Bankamp, B.; Maher, K.; Chen, M.-H.; et al. Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memish, Z.A.; Cotten, M.; Meyer, B.; Watson, S.J.; Alsahafi, A.J.; Al Rabeeah, A.A.; Corman, V.M.; Sieberg, A.; Makhdoom, H.Q.; Assiri, A.; et al. Human Infection with MERS Coronavirus after Exposure to Infected Camels, Saudi Arabia, 2013. Emerg. Infect. Dis. 2014, 20, 1012–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, G.A.; Sacco, O.; Mancino, E.; Cristiani, L.; Midulla, F. Differences and similarities between SARS-CoV and SARS-CoV-2: Spike receptor-binding domain recognition and host cell infection with support of cellular serine proteases. Infection 2020, 48, 665–669. [Google Scholar] [CrossRef] [PubMed]
- WHO. Weekly Operational Update on COVID19. Available online: https://www.who.int/publications/m/item/weekly-operational-update-on-covid-19---14-june-2021 (accessed on 14 June 2021).
- Krishnamoorthy, S.; Swain, B.; Verma, R.S.; Gunthe, S.S. SARS-CoV, MERS-CoV, and 2019-nCoV viruses: An overview of origin, evolution, and genetic variations. Virus Disease 2020, 31, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; Zee, R.; van der Haan, C.A.M.; de Rottier, P.J.M. The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Prabakaran, P.; Xiao, X.; Dimitrov, D.S. A model of the ACE2 structure and function as a SARS-CoV receptor. Biochem. Biophys. Res. Commun. 2004, 314, 235–241. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Y.; Pan, Y.; Zhao, Z.J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys. Res. Commun. 2020, 525, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Xu, W.; Zheng, T.; Wu, P.; Xie, S.; Bian, W.; Zhang, C.; et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021, 31, 126–140. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; Van Der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.-Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.-X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.L.; Teng, J.L.L.; Jia, L.; Zhang, C.; Huang, C.; Cai, J.-P.; Zhou, R.; Chan, K.-H.; Zhao, H.; Zhu, L.; et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell 2021, 184, 2212–2228. [Google Scholar] [CrossRef]
- Grove, J.; Marsh, M. The cell biology of receptor-mediated virus entry. J. Cell Biol. 2011, 195, 1071–1082. [Google Scholar] [CrossRef] [Green Version]
- Kliche, J.; Kuss, H.; Ali, M.; Ivarsson, Y. Cytoplasmic short linear motifs in ACE2 and integrin β3 link SARS-CoV-2 host cell receptors to mediators of endocytosis and autophagy. Sci. Signal. 2021, 14, eabf1117. [Google Scholar] [CrossRef]
- Joseph, J.; Karthika, T.; Das, V.A.; Raj, V.S. Epigallocatechin-3-gallate (EGCG): A potential molecule for the development of therapeutics against emerging SARS-CoV-1, MERS-CoV and SARS-CoV-2 coronaviruses. J. Glob. Antimicrob. Resist. 2021, 26, 26–28. [Google Scholar] [CrossRef]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.W.; Müller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.A.; Zaki, A.; Fouchier, R.A.M.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Prokscha, A.; Naim, H.Y.; Müller, M.A.; Drosten, C.; Pöhlmann, S.; Hoffmann, M. Polymorphisms in dipeptidyl peptidase 4 reduce host cell entry of Middle East respiratory syndrome coronavirus. Emerg. Microbes Infect. 2020, 9, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.-L.; Wu, Y.; Cao, J.-L.; Yang, R.; Liu, Y.-X.; Ma, J.; Qiao, X.-Y.; Yao, X.-Y.; Zhang, B.-H.; Zhang, Y.-L.; et al. Robust neutralization assay based on SARS-CoV-2 S-protein-bearing vesicular stomatitis virus (VSV) pseudovirus and ACE2-overexpressing BHK21 cells. Emerg. Microbes Infect. 2020, 9, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Mitra, D.; Sharma, P.; McCandless, M.G.; Stray, S.J.; Bates, J.T.; Marshall, G.D. Effective screening of SARS-CoV-2 neutralizing antibodies in patient serum using lentivirus particles pseudotyped with SARS-CoV-2 spike glycoprotein. Sci. Rep. 2020, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Karthika, T.; Ajay, A.; Das, V.R.A.; Raj, V.S. Green Tea and Spirulina Extracts Inhibit SARS, MERS, and SARS-2 Spike Pseudotyped Virus Entry in Vitro. BioRxiv 2020. [Google Scholar] [CrossRef]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-Dependent Entry of Severe Acute Respiratory Syndrome Coronavirus into Target Cells Expressing ACE2 with the Cytoplasmic Tail Deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulant, S.; Stanifer, M.; Lozach, P.-Y. Dynamics of Virus-Receptor Interactions in Virus Binding, Signaling, and Endocytosis. Viruses 2015, 7, 2794–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izaguirre, G. The Proteolytic Regulation of Virus Cell Entry by Furin and Other Proprotein Convertases. Viruses 2019, 11, 837. [Google Scholar] [CrossRef] [Green Version]
- Gierer, S.; Bertram, S.; Kaup, F.; Wrensch, F.; Heurich, A.; Krämer-Kühl, A.; Welsch, K.; Winkler, M.; Meyer, B.; Drosten, C.; et al. The Spike Protein of the Emerging Betacoronavirus EMC Uses a Novel Coronavirus Receptor for Entry, Can Be Activated by TMPRSS2, and Is Targeted by Neutralizing Antibodies. J. Virol. 2013, 87, 5502–5511. [Google Scholar] [CrossRef] [Green Version]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef]
- Kozik, P.; Francis, R.W.; Seaman, M.N.J.; Robinson, M.S. A Screen for Endocytic Motifs. Traffic 2010, 11, 843–855. [Google Scholar] [CrossRef]
- Busman-Sahay, K.; Drake, L.; Sitaram, A.; Marks, M.; Drake, J.R. Cis and Trans Regulatory Mechanisms Control AP2-Mediated B Cell Receptor Endocytosis via Select Tyrosine-Based Motifs. PLoS ONE 2013, 8, e54938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.-G.; Tang, D.-J.; Hua, Z.; Wang, Z.; An, J. Sunitinib reduces the infection of SARS-CoV, MERS-CoV and SARS-CoV-2 partially by inhibiting AP2M1 phosphorylation. Cell Discov. 2020, 6, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kalia, M.; Jameel, S. Virus entry paradigms. Amino Acids 2009, 41, 1147–1157. [Google Scholar] [CrossRef]
- Nuckols, J.T.; McAuley, A.J.; Huang, Y.-J.S.; Horne, K.M.; Higgs, S.; Davey, R.A.; VanLandingham, D.L. pH-Dependent entry of chikungunya virus fusion into mosquito cells. Virol. J. 2014, 11, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.-Z.; Xu, Q.-Q.; Wu, D.-G.; Ren, H.; Zhao, P.; Lao, W.-G.; Wang, Y.; Tao, Q.-Y.; Qian, X.-J.; Wei, Y.-H.; et al. Japanese Encephalitis Virus Enters Rat Neuroblastoma Cells via a pH-Dependent, Dynamin and Caveola-Mediated Endocytosis Pathway. J. Virol. 2012, 86, 13407–13422. [Google Scholar] [CrossRef] [Green Version]
- Canning, W.M.; Fields, B.N. Ammonium chloride prevents lytic growth of reovirus and helps to establish persistent infection in mouse L cells. Science 1983, 219, 987–988. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Wei, B.L.; Yao, J.; Luo, T.; Garcia, J.V. Inhibition of Endosomal/Lysosomal Degradation Increases the Infectivity of Human Immunodeficiency Virus. J. Virol. 2002, 76, 11440–11446. [Google Scholar] [CrossRef] [Green Version]
- Sloan, R.D.; Kuhl, B.D.; Mesplède, T.; Münch, J.; Donahue, D.A.; Wainberg, M.A. Productive Entry of HIV-1 during Cell-to-Cell Transmission via Dynamin-Dependent Endocytosis. J. Virol. 2013, 87, 8110–8123. [Google Scholar] [CrossRef] [Green Version]
- Schulz, W.; Haj, A.; Schiff, L.A. Reovirus Uses Multiple Endocytic Pathways for Cell Entry. J. Virol. 2012, 86, 12665–12675. [Google Scholar] [CrossRef] [Green Version]
- Hua, C.; Lee, R.; Hussain, K.M.; Chu, J.J.H. Macropinocytosis dependent entry of Chikungunya virus into human muscle cells. PLoS Negl. Trop. Dis. 2019, 13, e0007610. [Google Scholar] [CrossRef]
- Delisser, H.M.; Chilkotowsky, J.; Yan, H.C.; Daise, M.L.; Buck, C.A.; Albelda, S.M. Deletions in the cytoplasmic domain of platelet-endothelial cell adhesion molecule-1 (PECAM-1, CD31) result in changes in ligand binding properties. J. Cell Biol. 1994, 124, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, S.K.; Lukowski, C.M.; Casey, J.R. The cytoplasmic domain is essential for transport function of the integral membrane transport protein SLC4A11. Am. J. Physiol. Cell Physiol. 2016, 310, C161–C174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Guo, F.; Liu, K.; Wang, H.; Rao, S.; Yang, P.; Jiang, C. Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res. 2008, 136, 8–15. [Google Scholar] [CrossRef]
- Lozach, P.-Y.; Burleigh, L.; Staropoli, I.; Navarro-Sanchez, E.; Harriague, J.; Virelizier, J.-L.; Rey, F.A.; Desprès, P.; Arenzana-Seisdedos, F.; Amara, A. Dendritic Cell-specific Intercellular Adhesion Molecule 3-grabbing Non-integrin (DC-SIGN)-mediated Enhancement of Dengue Virus Infection Is Independent of DC-SIGN Internalization Signals. J. Biol. Chem. 2005, 280, 23698–23708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Chen, C.Z.; Swaroop, M.; Xu, M.; Wang, L.; Lee, J.; Wang, A.Q.; Pradhan, M.; Hagen, N.; Chen, L.; et al. Heparan sulfate assists SARS-CoV-2 in cell entry and can be targeted by approved drugs in vitro. Cell Discov. 2020, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, S.; Shen, Y.; Yang, Q. Epidermal growth factor receptor is a co-factor for transmissible gastroenteritis virus entry. Virology 2018, 521, 33–43. [Google Scholar] [CrossRef]
- Borau, M.S.; Stertz, S. Entry of influenza A virus into host cells—Recent progress and remaining challenges. Curr. Opin. Virol. 2021, 48, 23–29. [Google Scholar] [CrossRef]
- Ji, C.-M.; Wang, B.; Zhou, J.; Huang, Y.-W. Aminopeptidase-N-independent entry of porcine epidemic diarrhea virus into Vero or porcine small intestine epithelial cells. Virology 2018, 517, 16–23. [Google Scholar] [CrossRef]
- Madavaraju, K.; Koganti, R.; Volety, I.; Yadavalli, T.; Shukla, D. Herpes Simplex Virus Cell Entry Mechanisms: An Update. Front. Cell. Infect. Microbiol. 2021, 10, 1–18. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karthika, T.; Joseph, J.; Das, V.R.A.; Nair, N.; Charulekha, P.; Roji, M.D.; Raj, V.S. SARS-CoV-2 Cellular Entry Is Independent of the ACE2 Cytoplasmic Domain Signaling. Cells 2021, 10, 1814. https://doi.org/10.3390/cells10071814
Karthika T, Joseph J, Das VRA, Nair N, Charulekha P, Roji MD, Raj VS. SARS-CoV-2 Cellular Entry Is Independent of the ACE2 Cytoplasmic Domain Signaling. Cells. 2021; 10(7):1814. https://doi.org/10.3390/cells10071814
Chicago/Turabian StyleKarthika, Thankamani, Jeswin Joseph, V. R. Akshay Das, Niranjana Nair, Packirisamy Charulekha, Melvin Daniel Roji, and V. Stalin Raj. 2021. "SARS-CoV-2 Cellular Entry Is Independent of the ACE2 Cytoplasmic Domain Signaling" Cells 10, no. 7: 1814. https://doi.org/10.3390/cells10071814
APA StyleKarthika, T., Joseph, J., Das, V. R. A., Nair, N., Charulekha, P., Roji, M. D., & Raj, V. S. (2021). SARS-CoV-2 Cellular Entry Is Independent of the ACE2 Cytoplasmic Domain Signaling. Cells, 10(7), 1814. https://doi.org/10.3390/cells10071814