
The Role of NKT Cells in Glioblastoma

Abstract
1. Introduction
2. The Immune Landscapes of Glioblastoma
2.1. Neuroimmunity
2.2. The GBM Microenvironment
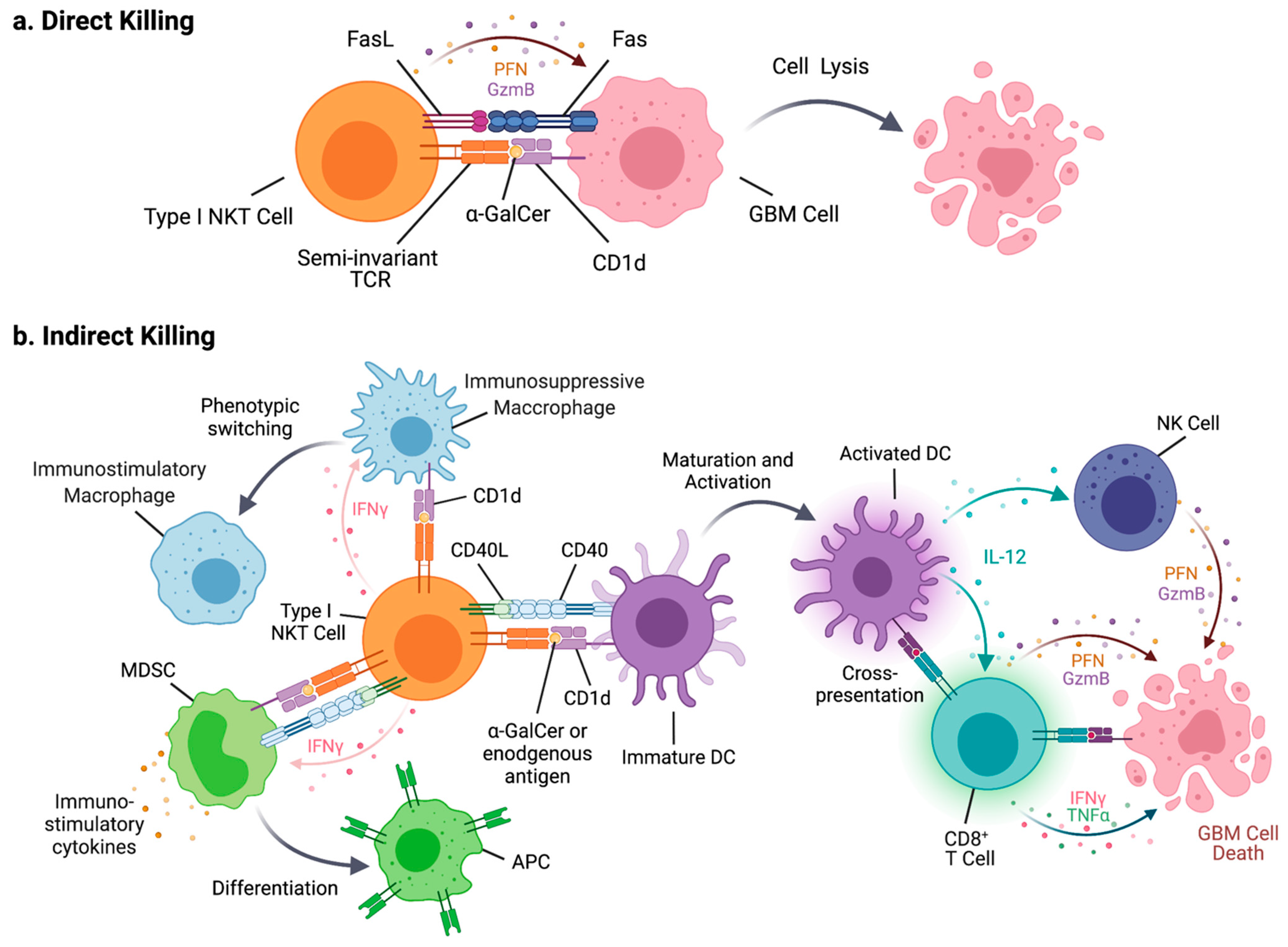
3. NKT Cells
4. NKT Cells in Tumor Immunity
4.1. Type I NKT Cells
4.2. Type II NKT Cells
4.3. Interactions between Two Types of NKT Cells and Tumor Immunity
5. Lipids in Brain Immunology and GBM
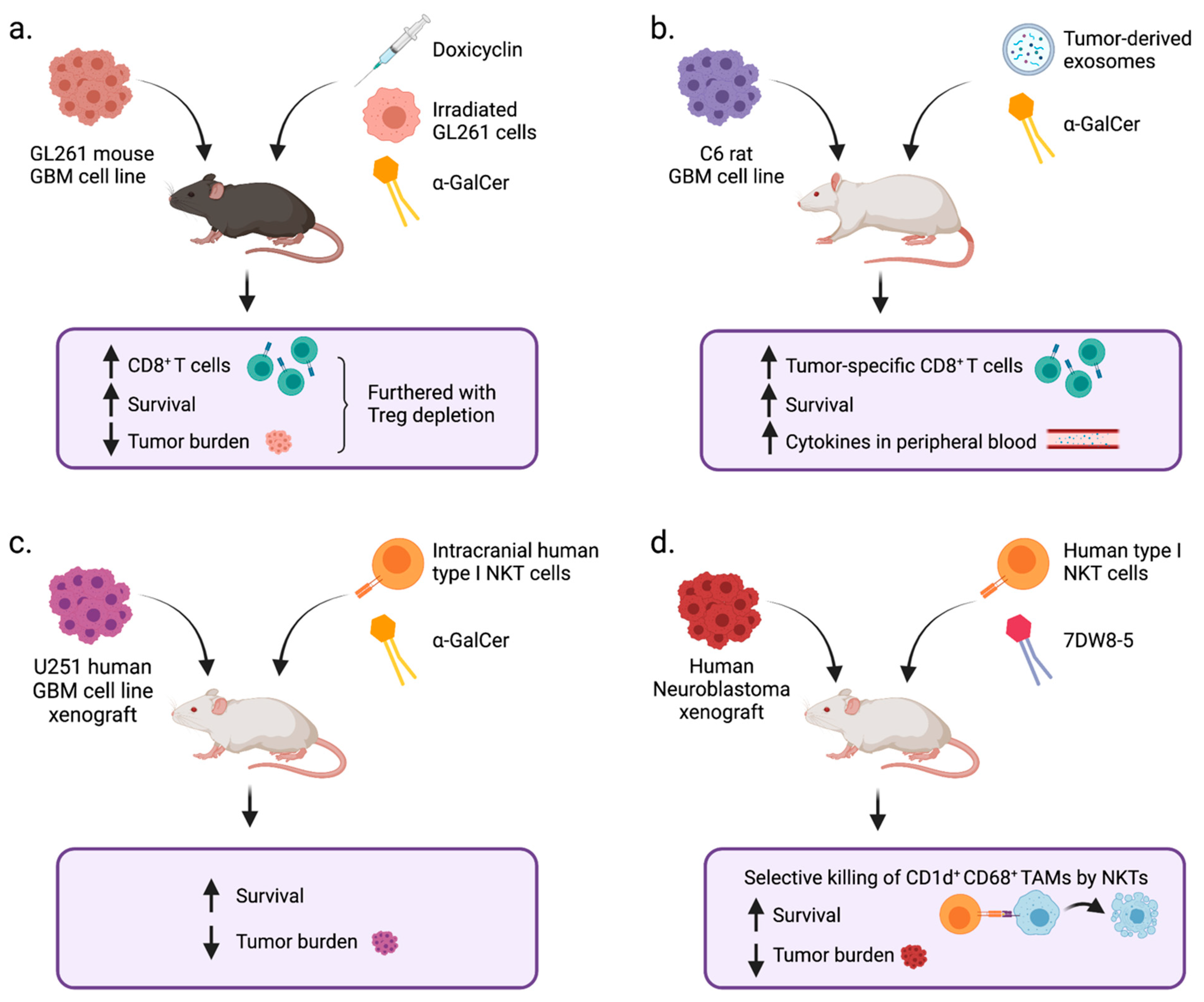
6. NKT Cells in GBM
7. NKT Cells in Multiple Sclerosis
8. Further Questions Regarding NKT Cells in GBM Immunity
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lukas, R.V.; Wainwright, D.A.; Ladomersky, E.; Sachdev, S.; Sonabend, A.M.; Stupp, R. Newly Diagnosed Glioblastoma: A Review on Clinical Management. Oncology 2019, 33, 91–100. [Google Scholar]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Alfonso, J.; Osswald, M.; Monyer, H.; Wick, W.; Winkler, F. Emerging intersections between neuroscience and glioma biology. Nat. Neurosci. 2019, 22, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.M.; Taylor, J.W.; Wiencke, J.K.; Wrensch, M.R. Genetic and molecular epidemiology of adult diffuse glioma. Nat. Rev. Neurol. 2019, 15, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Lyon, J.G.; Mokarram, N.; Saxena, T.; Carroll, S.L.; Bellamkonda, R.V. Engineering challenges for brain tumor immunotherapy. Adv. Drug Deliv. Rev. 2017, 114, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef]
- Frederico, S.C.; Hancock, J.C.; Brettschneider, E.E.S.; Ratnam, N.M.; Gilbert, M.R.; Terabe, M. Making a Cold Tumor Hot: The Role of Vaccines in the Treatment of Glioblastoma. Front. Oncol. 2021, 11, 672508. [Google Scholar] [CrossRef]
- Bendelac, A.; Lantz, O.; Quimby, M.E.; Yewdell, J.W.; Bennink, J.R.; Brutkiewicz, R.R. CD1 recognition by mouse NK1+ T lymphocytes. Science 1995, 268, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Zengeler, K.E.; Lukens, J.R. Innate immunity at the crossroads of healthy brain maturation and neurodevelopmental disorders. Nat. Rev. Immunol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2019, 216, 60–70. [Google Scholar] [CrossRef]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Ratnam, N.M.; Gilbert, M.R.; Giles, A.J. Immunotherapy in CNS cancers: The role of immune cell trafficking. Neuro Oncol. 2019, 21, 37–46. [Google Scholar] [CrossRef]
- Kipnis, J. Multifaceted interactions between adaptive immunity and the central nervous system. Science 2016, 353, 766–771. [Google Scholar] [CrossRef]
- Cronk, J.C.; Kipnis, J. Microglia—The brain’s busy bees. F1000Prime Rep. 2013, 5, 53. [Google Scholar] [CrossRef]
- Kipnis, J.; Filiano, A.J. Neuroimmunology in 2017: The central nervous system: Privileged by immune connections. Nat. Rev. Immunol. 2018, 18, 83–84. [Google Scholar] [CrossRef]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Davies, D.C. Blood-brain barrier breakdown in septic encephalopathy and brain tumours. J. Anat. 2002, 200, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.H.; Weninger, W.; Hunter, C.A. Trafficking of immune cells in the central nervous system. J. Clin. Investig. 2010, 120, 1368–1379. [Google Scholar] [CrossRef]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol. 2008, 67, 1113–1121. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Radjavi, A.; Smirnov, I.; Derecki, N.; Kipnis, J. Dynamics of the meningeal CD4(+) T-cell repertoire are defined by the cervical lymph nodes and facilitate cognitive task performance in mice. Mol. Psychiatry 2014, 19, 531–533. [Google Scholar] [CrossRef]
- Rustenhoven, J.; Drieu, A.; Mamuladze, T.; de Lima, K.A.; Dykstra, T.; Wall, M.; Papadopoulos, Z.; Kanamori, M.; Salvador, A.F.; Baker, W.; et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell 2021, 184, 1000–1016.e27. [Google Scholar] [CrossRef]
- Roth, T.L.; Nayak, D.; Atanasijevic, T.; Koretsky, A.P.; Latour, L.L.; McGavern, D.B. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014, 505, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Deng, Q.; Ma, L.; Li, Q.; Chen, Y.; Liao, Y.; Zhou, F.; Zhang, C.; Shao, L.; Feng, J.; et al. Meningeal lymphatic vessels regulate brain tumor drainage and immunity. Cell Res. 2020, 30, 229–243. [Google Scholar] [CrossRef]
- Song, E.; Mao, T.; Dong, H.; Boisserand, L.S.B.; Antila, S.; Bosenberg, M.; Alitalo, K.; Thomas, J.L.; Iwasaki, A. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 2020, 577, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Kallies, A. T cell responses in the central nervous system. Nat. Rev. Immunol. 2017, 17, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Woodward-Davis, A.; Liu, R.; Hu, Y.; Villadangos, J.; Smyth, G.; Bevan, M.J. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J. Immunol. 2012, 189, 3462–3471. [Google Scholar] [CrossRef]
- Steinbach, K.; Vincenti, I.; Kreutzfeldt, M.; Page, N.; Muschaweckh, A.; Wagner, I.; Drexler, I.; Pinschewer, D.; Korn, T.; Merkler, D. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J. Exp. Med. 2016, 213, 1571–1587. [Google Scholar] [CrossRef]
- Urban, S.L.; Jensen, I.J.; Shan, Q.; Pewe, L.L.; Xue, H.H.; Badovinac, V.P.; Harty, J.T. Peripherally induced brain tissue-resident memory CD8(+) T cells mediate protection against CNS infection. Nat. Immunol. 2020, 21, 938–949. [Google Scholar] [CrossRef]
- Smolders, J.; Heutinck, K.M.; Fransen, N.L.; Remmerswaal, E.B.M.; Hombrink, P.; Ten Berge, I.J.M.; van Lier, R.A.W.; Huitinga, I.; Hamann, J. Tissue-resident memory T cells populate the human brain. Nat. Commun. 2018, 9, 4593. [Google Scholar] [CrossRef]
- Won, W.J.; Deshane, J.S.; Leavenworth, J.W.; Oliva, C.R.; Griguer, C.E. Metabolic and functional reprogramming of myeloid-derived suppressor cells and their therapeutic control in glioblastoma. Cell Stress 2019, 3, 47–65. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef]
- Wu, S.Y.; Watabe, K. The roles of microglia/macrophages in tumor progression of brain cancer and metastatic disease. Front. Biosci. 2017, 22, 1805–1829. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006, 8, 261–279. [Google Scholar] [CrossRef]
- Schartner, J.M.; Hagar, A.R.; Van Handel, M.; Zhang, L.; Nadkarni, N.; Badie, B. Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia 2005, 51, 279–285. [Google Scholar] [CrossRef]
- Wei, J.; Gabrusiewicz, K.; Heimberger, A. The controversial role of microglia in malignant gliomas. Clin. Dev. Immunol. 2013, 2013, 285246. [Google Scholar] [CrossRef]
- Dubinski, D.; Wolfer, J.; Hasselblatt, M.; Schneider-Hohendorf, T.; Bogdahn, U.; Stummer, W.; Wiendl, H.; Grauer, O.M. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol. 2016, 18, 807–818. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Hu, J.; Zhao, Q.; Kong, L.Y.; Wang, J.; Yan, J.; Xia, X.; Jia, Z.; Heimberger, A.B.; Li, S. Regulation of tumor immune suppression and cancer cell survival by CXCL1/2 elevation in glioblastoma multiforme. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Mason, M.; Maurice, C.; McNamara, M.G.; Tieu, M.T.; Lwin, Z.; Millar, B.A.; Menard, C.; Laperriere, N.; Milosevic, M.; Atenafu, E.G.; et al. Neutrophil-lymphocyte ratio dynamics during concurrent chemo-radiotherapy for glioblastoma is an independent predictor for overall survival. J. Neurooncol. 2017, 132, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils promote the malignant glioma phenotype through S100A4. Clin Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef]
- Bertaut, A.; Truntzer, C.; Madkouri, R.; Kaderbhai, C.G.; Derangere, V.; Vincent, J.; Chauffert, B.; Aubriot-Lorton, M.H.; Farah, W.; Mourier, K.L.; et al. Blood baseline neutrophil count predicts bevacizumab efficacy in glioblastoma. Oncotarget 2016, 7, 70948–70958. [Google Scholar] [CrossRef]
- Yee, P.P.; Wei, Y.; Kim, S.Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 2020, 11, 5424. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef]
- Learn, C.A.; Fecci, P.E.; Schmittling, R.J.; Xie, W.; Karikari, I.; Mitchell, D.A.; Archer, G.E.; Wei, Z.; Dressman, H.; Sampson, J.H. Profiling of CD4+, CD8+, and CD4+CD25+CD45RO+FoxP3+ T cells in patients with malignant glioma reveals differential expression of the immunologic transcriptome compared with T cells from healthy volunteers. Clin. Cancer Res. 2006, 12, 7306–7315. [Google Scholar] [CrossRef] [PubMed]
- Fecci, P.E.; Sweeney, A.E.; Grossi, P.M.; Nair, S.K.; Learn, C.A.; Mitchell, D.A.; Cui, X.; Cummings, T.J.; Bigner, D.D.; Gilboa, E.; et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin. Cancer Res. 2006, 12, 4294–4305. [Google Scholar] [CrossRef]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E., 2nd; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.; Ratliff, T.; Huppertz, A.; Ge, Y.; Dictus, C.; Ahmadi, R.; Grau, S.; Hiraoka, N.; Eckstein, V.; Ecker, R.C.; et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin. Cancer Res. 2011, 17, 4296–4308. [Google Scholar] [CrossRef]
- Daubon, T.; Hemadou, A.; Romero Garmendia, I.; Saleh, M. Glioblastoma Immune Landscape and the Potential of New Immunotherapies. Front. Immunol. 2020, 11, 585616. [Google Scholar] [CrossRef]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed]
- Crane, A.T.; Chrostek, M.R.; Krishna, V.D.; Shiao, M.; Toman, N.G.; Pearce, C.M.; Tran, S.K.; Sipe, C.J.; Guo, W.; Voth, J.P.; et al. Zika virus-based immunotherapy enhances long-term survival of rodents with brain tumors through upregulation of memory T-cells. PLoS ONE 2020, 15, e0232858. [Google Scholar] [CrossRef]
- Wintterle, S.; Schreiner, B.; Mitsdoerffer, M.; Schneider, D.; Chen, L.; Meyermann, R.; Weller, M.; Wiendl, H. Expression of the B7-related molecule B7-H1 by glioma cells: A potential mechanism of immune paralysis. Cancer Res. 2003, 63, 7462–7467. [Google Scholar] [PubMed]
- Fabris, D.; Rozman, M.; Sajko, T.; Vukelic, Z. Aberrant ganglioside composition in glioblastoma multiforme and peritumoral tissue: A mass spectrometry characterization. Biochimie 2017, 137, 56–68. [Google Scholar] [CrossRef]
- Godfrey, D.I.; MacDonald, H.R.; Kronenberg, M.; Smyth, M.J.; Van Kaer, L. NKT cells: What’s in a name? Nat. Rev. Immunol. 2004, 4, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Berzofsky, J.A. The role of NKT cells in tumor immunity. Adv. Cancer Res. 2008, 101, 277–348. [Google Scholar]
- Terabe, M.; Berzofsky, J.A. Tissue-Specific Roles of NKT Cells in Tumor Immunity. Front. Immunol. 2018, 9, 1838. [Google Scholar] [CrossRef] [PubMed]
- Vidal, K.; Allen, P.M. The effect of endogenous altered peptide ligands on peripheral T-cell responses. Semin. Immunol. 1996, 8, 117–122. [Google Scholar] [CrossRef]
- Terabe, M.; Berzofsky, J.A. The immunoregulatory role of type I and type II NKT cells in cancer and other diseases. Cancer Immunol. Immunother. 2014, 63, 199–213. [Google Scholar] [CrossRef]
- Anderson, B.L.; Teyton, L.; Bendelac, A.; Savage, P.B. Stimulation of natural killer T cells by glycolipids. Molecules 2013, 18, 15662–15688. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.O.; Im, J.S.; Molano, A.; Dutronc, Y.; Illarionov, P.A.; Forestier, C.; Fujiwara, N.; Arias, I.; Miyake, S.; Yamamura, T.; et al. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of alpha-galactosylceramides. Proc. Natl. Acad. Sci. USA 2005, 102, 3383–3388. [Google Scholar] [CrossRef] [PubMed]
- Schmieg, J.; Yang, G.; Franck, R.W.; Tsuji, M. Superior protection against malaria and melanoma metastases by a C-glycoside analogue of the natural killer T cell ligand alpha-Galactosylceramide. J. Exp. Med. 2003, 198, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.N.; Lin, K.H.; Chang, Y.J.; Huang, J.R.; Cheng, J.Y.; Yu, A.L.; Wong, C.H. Avidity of CD1d-ligand-receptor ternary complex contributes to T-helper 1 (Th1) polarization and anticancer efficacy. Proc. Natl. Acad. Sci. USA 2011, 108, 17275–17280. [Google Scholar] [CrossRef] [PubMed]
- Borg, N.A.; Wun, K.S.; Kjer-Nielsen, L.; Wilce, M.C.; Pellicci, D.G.; Koh, R.; Besra, G.S.; Bharadwaj, M.; Godfrey, D.I.; McCluskey, J.; et al. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature 2007, 448, 44–49. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Uldrich, A.P.; McCluskey, J.; Rossjohn, J.; Moody, D.B. The burgeoning family of unconventional T cells. Nat. Immunol. 2015, 16, 1114–1123. [Google Scholar] [CrossRef]
- Matsuda, J.L.; Gapin, L.; Baron, J.L.; Sidobre, S.; Stetson, D.B.; Mohrs, M.; Locksley, R.M.; Kronenberg, M. Mouse V alpha 14i natural killer T cells are resistant to cytokine polarization in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 8395–8400. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Wang, H.; Starrett, G.J.; Phuong, V.; Jameson, S.C.; Hogquist, K.A. Tissue-Specific Distribution of iNKT Cells Impacts Their Cytokine Response. Immunity 2015, 43, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Wingender, G.; Stepniak, D.; Krebs, P.; Lin, L.; McBride, S.; Wei, B.; Braun, J.; Mazmanian, S.K.; Kronenberg, M. Intestinal microbes affect phenotypes and functions of invariant natural killer T cells in mice. Gastroenterology 2012, 143, 418–428. [Google Scholar] [CrossRef]
- Mars, L.T.; Mas, M.; Beaudoin, L.; Bauer, J.; Leite-de-Moraes, M.; Lehuen, A.; Bureau, J.F.; Liblau, R.S. Invariant NKT cells regulate the CD8 T cell response during Theiler’s virus infection. PLoS ONE 2014, 9, e87717. [Google Scholar] [CrossRef]
- Lee, Y.J.; Holzapfel, K.L.; Zhu, J.; Jameson, S.C.; Hogquist, K.A. Steady-state production of IL-4 modulates immunity in mouse strains and is determined by lineage diversity of iNKT cells. Nat. Immunol. 2013, 14, 1146–1154. [Google Scholar] [CrossRef]
- Moreira-Teixeira, L.; Resende, M.; Devergne, O.; Herbeuval, J.P.; Hermine, O.; Schneider, E.; Dy, M.; Cordeiro-da-Silva, A.; Leite-de-Moraes, M.C. Rapamycin combined with TGF-beta converts human invariant NKT cells into suppressive Foxp3+ regulatory cells. J. Immunol. 2012, 188, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Sag, D.; Krause, P.; Hedrick, C.C.; Kronenberg, M.; Wingender, G. IL-10-producing NKT10 cells are a distinct regulatory invariant NKT cell subset. J. Clin. Investig. 2014. [Google Scholar] [CrossRef]
- Lynch, L.; Michelet, X.; Zhang, S.; Brennan, P.J.; Moseman, A.; Lester, C.; Besra, G.; Vomhof-Dekrey, E.E.; Tighe, M.; Koay, H.F.; et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat. Immunol. 2015, 16, 85–95. [Google Scholar] [CrossRef] [PubMed]
- King, I.L.; Fortier, A.; Tighe, M.; Dibble, J.; Watts, G.F.; Veerapen, N.; Haberman, A.M.; Besra, G.S.; Mohrs, M.; Brenner, M.B.; et al. Invariant natural killer T cells direct B cell responses to cognate lipid antigen in an IL-21-dependent manner. Nat. Immunol. 2011, 13, 44–50. [Google Scholar] [CrossRef]
- Chang, P.P.; Barral, P.; Fitch, J.; Pratama, A.; Ma, C.S.; Kallies, A.; Hogan, J.J.; Cerundolo, V.; Tangye, S.G.; Bittman, R.; et al. Identification of Bcl-6-dependent follicular helper NKT cells that provide cognate help for B cell responses. Nat. Immunol. 2012, 13, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Baranek, T.; Lebrigand, K.; de Amat Herbozo, C.; Gonzalez, L.; Bogard, G.; Dietrich, C.; Magnone, V.; Boisseau, C.; Jouan, Y.; Trottein, F.; et al. High Dimensional Single-Cell Analysis Reveals iNKT Cell Developmental Trajectories and Effector Fate Decision. Cell Rep. 2020, 32, 108116. [Google Scholar] [CrossRef]
- Zhou, L.; Adrianto, I.; Wang, J.; Wu, X.; Datta, I.; Mi, Q.S. Single-Cell RNA-Seq Analysis Uncovers Distinct Functional Human NKT Cell Sub-Populations in Peripheral Blood. Front. Cell Dev. Biol. 2020, 8, 384. [Google Scholar] [CrossRef]
- Gumperz, J.E.; Miyake, S.; Yamamura, T.; Brenner, M.B. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J. Exp. Med. 2002, 195, 625–636. [Google Scholar] [CrossRef]
- Lee, P.T.; Benlagha, K.; Teyton, L.; Bendelac, A. Distinct functional lineages of human V(alpha)24 natural killer T cells. J. Exp. Med. 2002, 195, 637–641. [Google Scholar] [CrossRef]
- Crowe, N.Y.; Smyth, M.J.; Godfrey, D.I. A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas. J. Exp. Med. 2002, 196, 119–127. [Google Scholar] [CrossRef]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; Cretney, E.; Trapani, J.A.; Taniguchi, M.; Kawano, T.; Pelikan, S.B.; Crowe, N.Y.; Godfrey, D.I. Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med. 2000, 191, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Bellone, M.; Ceccon, M.; Grioni, M.; Jachetti, E.; Calcinotto, A.; Napolitano, A.; Freschi, M.; Casorati, G.; Dellabona, P. iNKT cells control mouse spontaneous carcinoma independently of tumor-specific cytotoxic T cells. PLoS ONE 2010, 5, e8646. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Liu, K.; Smith, C.; Bonito, A.J.; Steinman, R.M. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J. Exp. Med. 2004, 199, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Shimizu, K.; Hemmi, H.; Steinman, R.M. Innate Valpha14(+) natural killer T cells mature dendritic cells, leading to strong adaptive immunity. Immunol. Rev. 2007, 220, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.C.; Shaulov, A.; Wang, R.; Balk, S.P.; Exley, M.A. CD1d ligation on human monocytes directly signals rapid NF-kappaB activation and production of bioactive IL-12. Proc. Natl. Acad. Sci. USA 2005, 102, 11811–11816. [Google Scholar] [CrossRef]
- Hermans, I.F.; Silk, J.D.; Gileadi, U.; Salio, M.; Mathew, B.; Ritter, G.; Schmidt, R.; Harris, A.L.; Old, L.; Cerundolo, V. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J. Immunol. 2003, 171, 5140–5147. [Google Scholar] [CrossRef]
- Shimizu, K.; Goto, A.; Fukui, M.; Taniguchi, M.; Fujii, S. Tumor cells loaded with alpha-galactosylceramide induce innate NKT and NK cell-dependent resistance to tumor implantation in mice. J. Immunol. 2007, 178, 2853–2861. [Google Scholar] [CrossRef]
- Hunn, M.K.; Farrand, K.J.; Broadley, K.W.; Weinkove, R.; Ferguson, P.; Miller, R.J.; Field, C.S.; Petersen, T.; McConnell, M.J.; Hermans, I.F. Vaccination with irradiated tumor cells pulsed with an adjuvant that stimulates NKT cells is an effective treatment for glioma. Clin. Cancer Res. 2012, 18, 6446–6459. [Google Scholar] [CrossRef]
- Paul, S.; Chhatar, S.; Mishra, A.; Lal, G. Natural killer T cell activation increases iNOS(+)CD206(-) M1 macrophage and controls the growth of solid tumor. J. Immunother. Cancer 2019, 7, 208. [Google Scholar] [CrossRef]
- Wang, Y.; Sedimbi, S.K.; Lofbom, L.; Besra, G.S.; Porcelli, S.A.; Cardell, S.L. Promotion or Suppression of Murine Intestinal Polyp Development by iNKT Cell Directed Immunotherapy. Front. Immunol. 2019, 10, 352. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.J.; Lee, J.M.; Kim, Y.J.; Kim, Y.S.; Lee, K.A.; Kang, C.Y. Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of activated NKT cells: An alternative cell-based antitumor vaccine. J. Immunol. 2009, 182, 1818–1828. [Google Scholar] [CrossRef]
- Wu, D.; Shi, Y.; Wang, C.; Chen, H.; Liu, Q.; Liu, J.; Zhang, L.; Wu, Y.; Xia, D. Activated NKT cells facilitated functional switch of myeloid-derived suppressor cells at inflammation sites in fulminant hepatitis mice. Immunobiology 2017, 222, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Gebremeskel, S.; Clattenburg, D.R.; Slauenwhite, D.; Lobert, L.; Johnston, B. Natural killer T cell activation overcomes immunosuppression to enhance clearance of postsurgical breast cancer metastasis in mice. Oncoimmunology 2015, 4, e995562. [Google Scholar] [CrossRef]
- Lee, J.M.; Seo, J.H.; Kim, Y.J.; Kim, Y.S.; Ko, H.J.; Kang, C.Y. The restoration of myeloid-derived suppressor cells as functional antigen-presenting cells by NKT cell help and all-trans-retinoic acid treatment. Int. J. Cancer 2012, 131, 741–751. [Google Scholar] [CrossRef]
- De Santo, C.; Salio, M.; Masri, S.H.; Lee, L.Y.; Dong, T.; Speak, A.O.; Porubsky, S.; Booth, S.; Veerapen, N.; Besra, G.S.; et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J. Clin. Investig. 2008, 118, 4036–4048. [Google Scholar] [CrossRef]
- Horinaka, A.; Sakurai, D.; Ihara, F.; Makita, Y.; Kunii, N.; Motohashi, S.; Nakayama, T.; Okamoto, Y. Invariant NKT cells are resistant to circulating CD15+ myeloid-derived suppressor cells in patients with head and neck cancer. Cancer Sci. 2016, 107, 207–216. [Google Scholar] [CrossRef] [PubMed]
- De Santo, C.; Arscott, R.; Booth, S.; Karydis, I.; Jones, M.; Asher, R.; Salio, M.; Middleton, M.; Cerundolo, V. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat. Immunol. 2010, 11, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Wingender, G.; Hiss, M.; Engel, I.; Peukert, K.; Ley, K.; Haller, H.; Kronenberg, M.; von Vietinghoff, S. Neutrophilic granulocytes modulate invariant NKT cell function in mice and humans. J. Immunol. 2012, 188, 3000–3008. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Berzofsky, J.A. Immunoregulatory T cells in tumor immunity. Curr. Opin. Immunol. 2004, 16, 157–162. [Google Scholar] [CrossRef]
- van der Vliet, H.J.; von Blomberg, B.M.; Nishi, N.; Reijm, M.; Voskuyl, A.E.; van Bodegraven, A.A.; Polman, C.H.; Rustemeyer, T.; Lips, P.; van den Eertwegh, A.J.; et al. Circulating V(alpha24+) Vbeta11+ NKT cell numbers are decreased in a wide variety of diseases that are characterized by autoreactive tissue damage. Clin. Immunol. 2001, 100, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Tahir, S.M.; Cheng, O.; Shaulov, A.; Koezuka, Y.; Bubley, G.J.; Wilson, S.B.; Balk, S.P.; Exley, M.A. Loss of IFN-gamma production by invariant NK T cells in advanced cancer. J. Immunol. 2001, 167, 4046–4050. [Google Scholar] [CrossRef] [PubMed]
- Crough, T.; Purdie, D.M.; Okai, M.; Maksoud, A.; Nieda, M.; Nicol, A.J. Modulation of human Valpha24(+)Vbeta11(+) NKT cells by age, malignancy and conventional anticancer therapies. Br. J. Cancer 2004, 91, 1880–1886. [Google Scholar] [CrossRef]
- Yanagisawa, K.; Seino, K.; Ishikawa, Y.; Nozue, M.; Todoroki, T.; Fukao, K. Impaired proliferative response of V alpha 24 NKT cells from cancer patients against alpha-galactosylceramide. J. Immunol. 2002, 168, 6494–6499. [Google Scholar] [CrossRef]
- Giaccone, G.; Punt, C.J.; Ando, Y.; Ruijter, R.; Nishi, N.; Peters, M.; von Blomberg, B.M.; Scheper, R.J.; van der Vliet, H.J.; van den Eertwegh, A.J.; et al. A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin. Cancer Res. 2002, 8, 3702–3709. [Google Scholar]
- Dhodapkar, M.V.; Geller, M.D.; Chang, D.H.; Shimizu, K.; Fujii, S.; Dhodapkar, K.M.; Krasovsky, J. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J. Exp. Med. 2003, 197, 1667–1676. [Google Scholar] [CrossRef]
- van der Vliet, H.J.; Wang, R.; Yue, S.C.; Koon, H.B.; Balk, S.P.; Exley, M.A. Circulating myeloid dendritic cells of advanced cancer patients result in reduced activation and a biased cytokine profile in invariant NKT cells. J. Immunol. 2008, 180, 7287–7293. [Google Scholar] [CrossRef]
- Tachibana, T.; Onodera, H.; Tsuruyama, T.; Mori, A.; Nagayama, S.; Hiai, H.; Imamura, M. Increased intratumor Valpha24-positive natural killer T cells: A prognostic factor for primary colorectal carcinomas. Clin. Cancer Res. 2005, 11, 7322–7327. [Google Scholar] [CrossRef]
- Nieda, M.; Okai, M.; Tazbirkova, A.; Lin, H.; Yamaura, A.; Ide, K.; Abraham, R.; Juji, T.; Macfarlane, D.J.; Nicol, A.J. Therapeutic activation of Valpha24+Vbeta11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood 2004, 103, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Okai, M.; Nieda, M.; Tazbirkova, A.; Horley, D.; Kikuchi, A.; Durrant, S.; Takahashi, T.; Boyd, A.; Abraham, R.; Yagita, H.; et al. Human peripheral blood Valpha24+ Vbeta11+ NKT cells expand following administration of alpha-galactosylceramide-pulsed dendritic cells. Vox Sang 2002, 83, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, E.; Motohashi, S.; Ishikawa, A.; Ito, T.; Uchida, T.; Kaneko, T.; Tanaka, Y.; Horiguchi, S.; Okamoto, Y.; Fujisawa, T.; et al. Dendritic cell maturation by CD11c- T cells and Valpha24+ natural killer T-cell activation by alpha-galactosylceramide. Int. J. Cancer 2005, 117, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, S.; Okamoto, Y.; Yoshino, I.; Nakayama, T. Anti-tumor immune responses induced by iNKT cell-based immunotherapy for lung cancer and head and neck cancer. Clin. Immunol. 2011, 140, 167–176. [Google Scholar] [CrossRef]
- Liu, D.F.; Song, L.P.; Brawley, V.S.; Robison, N.; Wei, J.; Gao, X.H.; Tian, G.W.; Margol, A.; Ahmed, N.; Asgharzadeh, S.; et al. Medulloblastoma expresses CD1d and can be targeted for immunotherapy with NKT cells. Clin. Immunol. 2013, 149, 55–64. [Google Scholar] [CrossRef]
- Yang, W.; Li, H.; Mayhew, E.; Mellon, J.; Chen, P.W.; Niederkorn, J.Y. NKT cell exacerbation of liver metastases arising from melanomas transplanted into either the eyes or spleens of mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3094–3102. [Google Scholar] [CrossRef]
- Kawano, T.; Cui, J.; Koezuka, Y.; Toura, I.; Kaneko, Y.; Sato, H.; Kondo, E.; Harada, M.; Koseki, H.; Nakayama, T.; et al. Natural killer-like nonspecific tumor cell lysis mediated by specific ligand-activated Valpha14 NKT cells. Proc. Natl. Acad. Sci. USA 1998, 95, 5690–5693. [Google Scholar] [CrossRef]
- Crowe, N.Y.; Coquet, J.M.; Berzins, S.P.; Kyparissoudis, K.; Keating, R.; Pellicci, D.G.; Hayakawa, Y.; Godfrey, D.I.; Smyth, M.J. Differential antitumor immunity mediated by NKT cell subsets in vivo. J. Exp. Med. 2005, 202, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Bjordahl, R.L.; Gapin, L.; Marrack, P.; Refaeli, Y. iNKT cells suppress the CD8+ T cell response to a murine Burkitt’s-like B cell lymphoma. PLoS ONE 2012, 7, e42635. [Google Scholar] [CrossRef]
- Wang, Y.; Cardell, S.L. The Yin and Yang of Invariant Natural Killer T Cells in Tumor Immunity-Suppression of Tumor Immunity in the Intestine. Front. Immunol. 2017, 8, 1945. [Google Scholar] [CrossRef]
- Terabe, M.; Matsui, S.; Noben-Trauth, N.; Chen, H.; Watson, C.; Donaldson, D.D.; Carbone, D.P.; Paul, W.E.; Berzofsky, J.A. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat. Immunol. 2000, 1, 515–520. [Google Scholar] [CrossRef]
- Terabe, M.; Matsui, S.; Park, J.-M.; Mamura, M.; Noben-Trauth, N.; Donaldson, D.D.; Chen, W.; Wahl, S.M.; Ledbetter, S.; Pratt, B.; et al. Transforming Growth Factor-b production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block Cytotoxic T Lymphocyte-mediated tumor immunosurveillance: Abrogation prevents tumor recurrence. J. Exp. Med. 2003, 198, 1741–1752. [Google Scholar] [CrossRef]
- Fichtner-Feigl, S.; Terabe, M.; Kitani, A.; Young, C.A.; Fuss, I.; Geissler, E.K.; Schlitt, H.J.; Berzofsky, J.A.; Strober, W. Restoration of tumor immunosurveillance via targeting of interleukin-13 receptor-alpha 2. Cancer Res. 2008, 68, 3467–3475. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Terabe, M.; van den Broeke, L.T.; Donaldson, D.D.; Berzofsky, J.A. Unmasking immunosurveillance against a syngeneic colon cancer by elimination of CD4+ NKT regulatory cells and IL-13. Int. J. Cancer 2004, 114, 80–87. [Google Scholar] [CrossRef]
- Ambrosino, E.; Terabe, M.; Halder, R.C.; Peng, J.; Takaku, S.; Miyake, S.; Yamamura, T.; Kumar, V.; Berzofsky, J.A. Cross-regulation between type I and type II NKT cells in regulating tumor immunity: A new immunoregulatory axis. J. Immunol. 2007, 179, 5126–5136. [Google Scholar] [CrossRef] [PubMed]
- Renukaradhya, G.J.; Khan, M.A.; Vieira, M.; Du, W.; Gervay-Hague, J.; Brutkiewicz, R.R. Type I NKT cells protect (and type II NKT cells suppress) the host’s innate antitumor immune response to a B-cell lymphoma. Blood 2008, 111, 5637–5645. [Google Scholar] [CrossRef]
- Chang, D.H.; Deng, H.; Matthews, P.; Krasovsky, J.; Ragupathi, G.; Spisek, R.; Mazumder, A.; Vesole, D.H.; Jagannath, S.; Dhodapkar, M.V. Inflammation-associated lysophospholipids as ligands for CD1d-restricted T cells in human cancer. Blood 2008, 112, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Berzofsky, J.A. NKT cells in immunoregulation of tumor immunity: A new immunoregulatory axis. Trends Immunol. 2007, 28, 491–496. [Google Scholar] [CrossRef]
- Halder, R.C.; Aguilera, C.; Maricic, I.; Kumar, V. Type II NKT cell-mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J. Clin. Investig. 2007, 117, 2302–2312. [Google Scholar] [CrossRef]
- Izhak, L.; Ambrosino, E.; Kato, S.; Parish, S.T.; O’Konek, J.J.; Weber, H.; Xia, Z.; Venzon, D.; Berzofsky, J.A.; Terabe, M. Delicate balance among three types of T cells in concurrent regulation of tumor immunity. Cancer Res. 2013, 73, 1514–1523. [Google Scholar] [CrossRef]
- Izhak, L.; Berzofsky, J.A.; Terabe, M. Balance is a key for happiness. OncoImmunology 2013, 2, e24211. [Google Scholar] [CrossRef]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 26. [Google Scholar] [CrossRef]
- Sedic, M.; Grbcic, P.; Pavelic, S.K. Bioactive Sphingolipids as Biomarkers Predictive of Disease Severity and Treatment Response in Cancer: Current Status and Translational Challenges. Anticancer Res. 2019, 39, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, O.; Iwamori, M.; Matsutani, M.; Takakura, K. Ganglioside GD3 shedding by human gliomas. Acta Neurochir. 1991, 109, 34–36. [Google Scholar] [CrossRef]
- Ariga, T.; Suetake, K.; Nakane, M.; Kubota, M.; Usuki, S.; Kawashima, I.; Yu, R.K. Glycosphingolipid antigens in neural tumor cell lines and anti-glycosphingolipid antibodies in sera of patients with neural tumors. Neurosignals 2008, 16, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, T.; Zhang, P.; Ohkawa, Y.; Momota, H.; Wakabayashi, T.; Ohmi, Y.; Bhuiyan, R.H.; Furukawa, K.; Furukawa, K. Enhancement of malignant properties of human glioma cells by ganglioside GD3/GD2. Int. J. Oncol. 2018, 52, 1255–1266. [Google Scholar] [CrossRef]
- Ohkawa, Y.; Momota, H.; Kato, A.; Hashimoto, N.; Tsuda, Y.; Kotani, N.; Honke, K.; Suzumura, A.; Furukawa, K.; Ohmi, Y.; et al. Ganglioside GD3 Enhances Invasiveness of Gliomas by Forming a Complex with Platelet-derived Growth Factor Receptor alpha and Yes Kinase. J. Biol. Chem. 2015, 290, 16043–16058. [Google Scholar] [CrossRef]
- Yeh, S.C.; Wang, P.Y.; Lou, Y.W.; Khoo, K.H.; Hsiao, M.; Hsu, T.L.; Wong, C.H. Glycolipid GD3 and GD3 synthase are key drivers for glioblastoma stem cells and tumorigenicity. Proc. Natl. Acad. Sci. USA 2016, 113, 5592–5597. [Google Scholar] [CrossRef]
- Giussani, P.; Bassi, R.; Anelli, V.; Brioschi, L.; De Zen, F.; Riccitelli, E.; Caroli, M.; Campanella, R.; Gaini, S.M.; Viani, P.; et al. Glucosylceramide synthase protects glioblastoma cells against autophagic and apoptotic death induced by temozolomide and Paclitaxel. Cancer Investig. 2012, 30, 27–37. [Google Scholar] [CrossRef]
- Sa, G.; Das, T.; Moon, C.; Hilston, C.M.; Rayman, P.A.; Rini, B.I.; Tannenbaum, C.S.; Finke, J.H. GD3, an overexpressed tumor-derived ganglioside, mediates the apoptosis of activated but not resting T cells. Cancer Res. 2009, 69, 3095–3104. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Fox, L.; Tian, R.; Bardet, W.; Skaley, M.; Mojsilovic, D.; Gumperz, J.; Hildebrand, W. Determination of cellular lipids bound to human CD1d molecules. PLoS ONE 2009, 4, e5325. [Google Scholar] [CrossRef]
- Dhodapkar, K.M.; Cirignano, B.; Chamian, F.; Zagzag, D.; Miller, D.C.; Finlay, J.L.; Steinman, R.M. Invariant natural killer T cells are preserved in patients with glioma and exhibit antitumor lytic activity following dendritic cell-mediated expansion. Int. J. Cancer 2004, 109, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Waziri, A.; Killory, B.; Ogden, A.T., 3rd; Canoll, P.; Anderson, R.C.; Kent, S.C.; Anderson, D.E.; Bruce, J.N. Preferential in situ CD4+CD56+ T cell activation and expansion within human glioblastoma. J. Immunol. 2008, 180, 7673–7680. [Google Scholar] [CrossRef]
- Hara, A.; Koyama-Nasu, R.; Takami, M.; Toyoda, T.; Aoki, T.; Ihara, F.; Kobayashi, M.; Hirono, S.; Matsutani, T.; Nakayama, T.; et al. CD1d expression in glioblastoma is a promising target for NKT cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2021, 70, 1239–1254. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Wu, W.; Wei, X.; Li, Y.; Ren, G.; Fan, W. Activation of glioma cells generates immune tolerant NKT cells. J. Biol. Chem. 2014, 289, 34595–34600. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, L.; Peng, Y.; Yu, S.; Liu, J.; Wu, L.; Zhang, L.; Wu, Q.; Chang, X.; Yu, X.; et al. Dendritic cells loaded with tumor derived exosomes for cancer immunotherapy. Oncotarget 2018, 9, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L.; Wu, L.; Parekh, V.V. Natural killer T cells in multiple sclerosis and its animal model, experimental autoimmune encephalomyelitis. Immunology 2015, 146, 1–10. [Google Scholar] [CrossRef]
- Podbielska, M.; O’Keeffe, J.; Hogan, E.L. Autoimmunity in multiple sclerosis: Role of sphingolipids, invariant NKT cells and other immune elements in control of inflammation and neurodegeneration. J. Neurol. Sci. 2018, 385, 198–214. [Google Scholar] [CrossRef]
- Bianchini, E.; De Biasi, S.; Simone, A.M.; Ferraro, D.; Sola, P.; Cossarizza, A.; Pinti, M. Invariant natural killer T cells and mucosal-associated invariant T cells in multiple sclerosis. Immunol. Lett. 2017, 183, 1–7. [Google Scholar] [CrossRef]
- Araki, M.; Kondo, T.; Gumperz, J.E.; Brenner, M.B.; Miyake, S.; Yamamura, T. Th2 bias of CD4+ NKT cells derived from multiple sclerosis in remission. Int. Immunol. 2003, 15, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Demoulins, T.; Gachelin, G.; Bequet, D.; Dormont, D. A biased Valpha24+ T-cell repertoire leads to circulating NKT-cell defects in a multiple sclerosis patient at the onset of his disease. Immunol. Lett. 2003, 90, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Illes, Z.; Kondo, T.; Newcombe, J.; Oka, N.; Tabira, T.; Yamamura, T. Differential expression of NK T cell V alpha 24J alpha Q invariant TCR chain in the lesions of multiple sclerosis and chronic inflammatory demyelinating polyneuropathy. J. Immunol. 2000, 164, 4375–4381. [Google Scholar] [CrossRef] [PubMed]
- O’Keeffe, J.; Gately, C.M.; Counihan, T.; Hennessy, M.; Leahy, T.; Moran, A.P.; Hogan, E.L. T-cells expressing natural killer (NK) receptors are altered in multiple sclerosis and responses to alpha-galactosylceramide are impaired. J. Neurol. Sci. 2008, 275, 22–28. [Google Scholar] [CrossRef]
- Gausling, R.; Trollmo, C.; Hafler, D.A. Decreases in interleukin-4 secretion by invariant CD4(-)CD8(-)V alpha 24J alpha Q T cells in peripheral blood of patientswith relapsing-remitting multiple sclerosis. Clin. Immunol. 2001, 98, 11–17. [Google Scholar] [CrossRef]
- De Biasi, S.; Simone, A.M.; Nasi, M.; Bianchini, E.; Ferraro, D.; Vitetta, F.; Gibellini, L.; Pinti, M.; Del Giovane, C.; Sola, P.; et al. iNKT Cells in Secondary Progressive Multiple Sclerosis Patients Display Pro-inflammatory Profiles. Front. Immunol. 2016, 7, 555. [Google Scholar] [CrossRef] [PubMed]
- Jahng, A.; Maricic, I.; Aguilera, C.; Cardell, S.; Halder, R.C.; Kumar, V. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J. Exp. Med. 2004, 199, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Maricic, I.; Halder, R.; Bischof, F.; Kumar, V. Dendritic cells and anergic type I NKT cells play a crucial role in sulfatide-mediated immune regulation in experimental autoimmune encephalomyelitis. J. Immunol. 2014, 193, 1035–1046. [Google Scholar] [CrossRef]
- Jahng, A.W.; Maricic, I.; Pedersen, B.; Burdin, N.; Naidenko, O.; Kronenberg, M.; Koezuka, Y.; Kumar, V. Activation of natural killer T cells potentiates or prevents experimental autoimmune encephalomyelitis. J. Exp. Med. 2001, 194, 1789–1799. [Google Scholar] [CrossRef]
- Furlan, R.; Bergami, A.; Cantarella, D.; Brambilla, E.; Taniguchi, M.; Dellabona, P.; Casorati, G.; Martino, G. Activation of invariant NKT cells by alphaGalCer administration protects mice from MOG35-55-induced EAE: Critical roles for administration route and IFN-gamma. Eur. J. Immunol. 2003, 33, 1830–1838. [Google Scholar] [CrossRef]
- Singh, A.K.; Wilson, M.T.; Hong, S.; Olivares-Villagomez, D.; Du, C.; Stanic, A.K.; Joyce, S.; Sriram, S.; Koezuka, Y.; Van Kaer, L. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J. Exp. Med. 2001, 194, 1801–1811. [Google Scholar] [CrossRef]
- Teige, A.; Teige, I.; Lavasani, S.; Bockermann, R.; Mondoc, E.; Holmdahl, R.; Issazadeh-Navikas, S. CD1-dependent regulation of chronic central nervous system inflammation in experimental autoimmune encephalomyelitis. J. Immunol. 2004, 172, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Denney, L.; Kok, W.L.; Cole, S.L.; Sanderson, S.; McMichael, A.J.; Ho, L.P. Activation of invariant NKT cells in early phase of experimental autoimmune encephalomyelitis results in differentiation of Ly6Chi inflammatory monocyte to M2 macrophages and improved outcome. J. Immunol. 2012, 189, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Mars, L.T.; Laloux, V.; Goude, K.; Desbois, S.; Saoudi, A.; Van Kaer, L.; Lassmann, H.; Herbelin, A.; Lehuen, A.; Liblau, R.S. Cutting edge: V alpha 14-J alpha 281 NKT cells naturally regulate experimental autoimmune encephalomyelitis in nonobese diabetic mice. J. Immunol. 2002, 168, 6007–6011. [Google Scholar] [CrossRef]
- Miyamoto, K.; Miyake, S.; Yamamura, T. A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing TH2 bias of natural killer T cells. Nature 2001, 413, 531–534. [Google Scholar] [CrossRef]
- Pal, E.; Tabira, T.; Kawano, T.; Taniguchi, M.; Miyake, S.; Yamamura, T. Costimulation-dependent modulation of experimental autoimmune encephalomyelitis by ligand stimulation of V alpha 14 NK T cells. J. Immunol. 2001, 166, 662–668. [Google Scholar] [CrossRef]
- Parekh, V.V.; Wu, L.; Olivares-Villagomez, D.; Wilson, K.T.; Van Kaer, L. Activated invariant NKT cells control central nervous system autoimmunity in a mechanism that involves myeloid-derived suppressor cells. J. Immunol. 2013, 190, 1948–1960. [Google Scholar] [CrossRef]
- Kojo, S.; Seino, K.; Harada, M.; Watarai, H.; Wakao, H.; Uchida, T.; Nakayama, T.; Taniguchi, M. Induction of regulatory properties in dendritic cells by Valpha14 NKT cells. J. Immunol. 2005, 175, 3648–3655. [Google Scholar] [CrossRef] [PubMed]
- Pernber, Z.; Richter, K.; Mansson, J.E.; Nygren, H. Sulfatide with different fatty acids has unique distributions in cerebellum as imaged by time-of-flight secondary ion mass spectrometry (TOF-SIMS). Biochim. Biophys. Acta 2007, 1771, 202–209. [Google Scholar] [CrossRef]
- Blomqvist, M.; Rhost, S.; Teneberg, S.; Lofbom, L.; Osterbye, T.; Brigl, M.; Mansson, J.E.; Cardell, S.L. Multiple tissue-specific isoforms of sulfatide activate CD1d-restricted type II NKT cells. Eur. J. Immunol. 2009, 39, 1726–1735. [Google Scholar] [CrossRef] [PubMed]
- Zajonc, D.M.; Maricic, I.; Wu, D.; Halder, R.; Roy, K.; Wong, C.H.; Kumar, V.; Wilson, I.A. Structural basis for CD1d presentation of a sulfatide derived from myelin and its implications for autoimmunity. J. Exp. Med. 2005, 202, 1517–1526. [Google Scholar] [CrossRef]
- Zajonc, D.M.; Wilson, I.A. Architecture of CD1 proteins. Curr. Top. Microbiol. Immunol. 2007, 314, 27–50. [Google Scholar]
- Robinson, S.A.; Yau, J.; Terabe, M.; Berzofsky, J.A.; Painter, G.F.; Compton, B.J.; Larsen, D.S. Synthetic preparation and immunological evaluation of beta-mannosylceramide and related N-acyl analogues. Org. Biomol. Chem. 2020, 18, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.J.; Hsuan, Y.C.; Lai, A.C.; Han, Y.C.; Hou, D.R. Synthesis of alpha-C-Galactosylceramide via Diastereoselective Aziridination: The New Immunostimulant 4’-epi-C-Glycoside of KRN7000. Org. Lett. 2016, 18, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Fox, L.M.; Cox, D.G.; Lockridge, J.L.; Wang, X.; Chen, X.; Scharf, L.; Trott, D.L.; Ndonye, R.M.; Veerapen, N.; Besra, G.S.; et al. Recognition of lyso-phospholipids by human natural killer T lymphocytes. PLoS Biol. 2009, 7, e1000228. [Google Scholar] [CrossRef] [PubMed]
- Melum, E.; Jiang, X.; Baker, K.D.; Macedo, M.F.; Fritsch, J.; Dowds, C.M.; Wang, J.; Pharo, A.; Kaser, A.; Tan, C.; et al. Control of CD1d-restricted antigen presentation and inflammation by sphingomyelin. Nat. Immunol. 2019, 20, 1644–1655. [Google Scholar] [CrossRef]
- Webb, T.J.; Li, X.; Giuntoli, R.L., 2nd; Lopez, P.H.; Heuser, C.; Schnaar, R.L.; Tsuji, M.; Kurts, C.; Oelke, M.; Schneck, J.P. Molecular identification of GD3 as a suppressor of the innate immune response in ovarian cancer. Cancer Res. 2012, 72, 3744–3752. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.Y.; Segal, N.H.; Sidobre, S.; Kronenberg, M.; Chapman, P.B. Cross-presentation of disialoganglioside GD3 to natural killer T cells. J. Exp. Med. 2003, 198, 173–181. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Type I NKT | Type II NKT | |
|---|---|---|
| Antigen-presenting molecule in immunological synapse | CD1d | CD1d |
| TCRα | Vα14 Jα18 (mice) | Diverse |
| Vα24 Jα18 (humans) | ||
| TCRβ | Vβ2, 7, 8 (mice) | Diverse |
| Vβ11 (humans) | ||
| Functional subsets | NKT1, NKT2, NKT17 | Not Known |
| NKT10, NKTFH, NKTreg | ||
| Representative antigen recognized | α-GalCer | Sulfatide |
| Role in cancer (with exceptions) | Enhance immunity | Suppress immunity |
| Major mechanism of action | Production of IFN-γ, induction of IL-12 production by DCs and CD40–CD40L interaction | Production of IL-13 to induce TGF-β production by myeloid cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brettschneider, E.E.S.; Terabe, M. The Role of NKT Cells in Glioblastoma. Cells 2021, 10, 1641. https://doi.org/10.3390/cells10071641
Brettschneider EES, Terabe M. The Role of NKT Cells in Glioblastoma. Cells. 2021; 10(7):1641. https://doi.org/10.3390/cells10071641
Chicago/Turabian StyleBrettschneider, Emily E. S., and Masaki Terabe. 2021. "The Role of NKT Cells in Glioblastoma" Cells 10, no. 7: 1641. https://doi.org/10.3390/cells10071641
APA StyleBrettschneider, E. E. S., & Terabe, M. (2021). The Role of NKT Cells in Glioblastoma. Cells, 10(7), 1641. https://doi.org/10.3390/cells10071641