
Novel Genetic and Molecular Pathways in Pulmonary Arterial Hypertension Associated with Connective Tissue Disease

 , , ,
, , ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
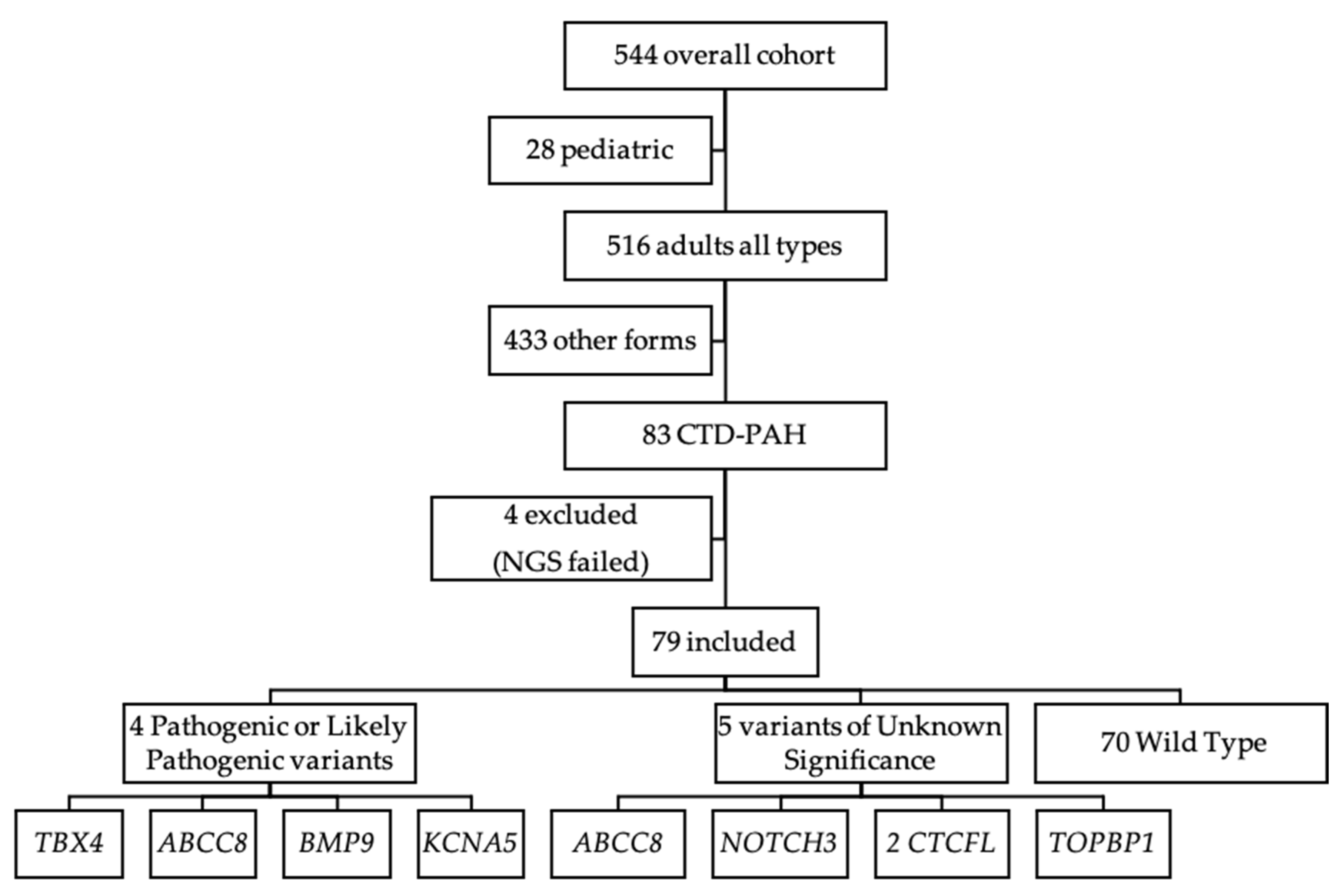
2.1. Study Patients
2.2. Molecular Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rubio-Rivas, M.; Corbella, X.; Guillen-Del-Castillo, A.; Tolosa Vilella, C.; Colunga Arguelles, D.; Argibay, A.; Vargas Hitos, J.A.; Todoli Parra, J.A.; Gonzalez-Echavarri, C.; Ortego-Centeno, N.; et al. Spanish scleroderma risk score (RESCLESCORE) to predict 15-year all-cause mortality in scleroderma patients at the time of diagnosis based on the RESCLE cohort: Derivation and internal validation. Autoimmun. Rev. 2020, 19, 102507. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.K.; Chung, L. Connective tissue disease-associated pulmonary arterial hypertension. Rheum. Dis. Clin. N. Am. 2015, 41, 295–313. [Google Scholar] [CrossRef] [PubMed]
- Morrisroe, K.; Stevens, W.; Sahhar, J.; Rabusa, C.; Nikpour, M.; Proudman, S.; Australian Scleroderma Interest, G. Epidemiology and disease characteristics of systemic sclerosis-related pulmonary arterial hypertension: Results from a real-life screening programme. Arthritis Res. Ther. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Quezada Loaiza, C.A.; Velazquez Martin, M.T.; Jimenez Lopez-Guarch, C.; Ruiz Cano, M.J.; Navas Tejedor, P.; Carreira, P.E.; Flox Camacho, A.; de Pablo Gafas, A.; Delgado Jimenez, J.F.; Gomez Sanchez, M.A.; et al. Trends in Pulmonary Hypertension Over a Period of 30 Years: Experience From a Single Referral Centre. Rev. Esp. Cardiol. 2017, 70, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Subias, P.; Blanco, I.; Lopez-Meseguer, M.; Lopez-Guarch, C.J.; Roman, A.; Morales, P.; Castillo-Palma, M.J.; Segovia, J.; Gomez-Sanchez, M.A.; Barbera, J.A.; et al. Survival in pulmonary hypertension in Spain: Insights from the Spanish registry. Eur. Respir. J. 2012, 40, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; and Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir J. 2019, 53, 1801899. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Pauciulo, M.W.; Welch, C.L.; Lutz, K.A.; Coleman, A.W.; Gonzaga-Jauregui, C.; Wang, J.; Grimes, J.M.; Martin, L.J.; He, H.; et al. Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med. 2019, 11, 69. [Google Scholar] [CrossRef]
- Girerd, B.; Montani, D.; Jais, X.; Eyries, M.; Yaici, A.; Sztrymf, B.; Savale, L.; Parent, F.; Coulet, F.; Godinas, L.; et al. Genetic counselling in a national referral centre for pulmonary hypertension. Eur. Respir. J. 2016, 47, 541–552. [Google Scholar] [CrossRef]
- Liu, D.; Liu, Q.Q.; Guan, L.H.; Jiang, X.; Zhou, D.X.; Beghetti, M.; Qu, J.M.; Jing, Z.C. BMPR2 mutation is a potential predisposing genetic risk factor for congenital heart disease associated pulmonary vascular disease. Int. J. Cardiol. 2016, 211, 132–136. [Google Scholar] [CrossRef]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar]
- van der Bruggen, C.E.; Spruijt, O.A.; Nossent, E.J.; Trip, P.; Marcus, J.T.; de Man, F.S.; Jan Bogaard, H.; Vonk Noordegraaf, A. Treatment response in patients with idiopathic pulmonary arterial hypertension and a severely reduced diffusion capacity. Pulm Circ. 2017, 7, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Kawut, S.M.; de Jesus Perez, V.A. New and Emerging Therapies for Pulmonary Arterial Hypertension. Annu. Rev. Med. 2019, 70, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Odler, B.; Foris, V.; Gungl, A.; Muller, V.; Hassoun, P.M.; Kwapiszewska, G.; Olschewski, H.; and Kovacs, G. Biomarkers for Pulmonary Vascular Remodeling in Systemic Sclerosis: A Pathophysiological Approach. Front. Physiol. 2018, 9, 587. [Google Scholar] [CrossRef]
- Wang, Y.; and Kahaleh, B. Epigenetic repression of bone morphogenetic protein receptor II expression in scleroderma. J. Cell Mol. Med. 2013, 17, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Gilbane, A.J.; Derrett-Smith, E.; Trinder, S.L.; Good, R.B.; Pearce, A.; Denton, C.P.; Holmes, A.M. Impaired bone morphogenetic protein receptor II signaling in a transforming growth factor-beta-dependent mouse model of pulmonary hypertension and in systemic sclerosis. Am. J. Respi.r Crit. Care Med. 2015, 191, 665–677. [Google Scholar] [CrossRef]
- Castano, J.A.T.; Hernandez-Gonzalez, I.; Gallego, N.; Perez-Olivares, C.; Ochoa Parra, N.; Arias, P.; Granda, E.; Acebo, G.G.; Lago-Docampo, M.; Palomino-Doza, J.; et al. Customized Massive Parallel Sequencing Panel for Diagnosis of Pulmonary Arterial Hypertension. Genes 2020, 11, 1158. [Google Scholar] [CrossRef]
- Loyd, J.E.; Primm, R.K.; Newman, J.H. Familial primary pulmonary hypertension: Clinical patterns. Am. Rev. Respir. Dis. 1984, 129, 194–197. [Google Scholar] [PubMed]
- Larkin, E.K.; Newman, J.H.; Austin, E.D.; Hemnes, A.R.; Wheeler, L.; Robbins, I.M.; West, J.D.; Phillips III, J.A.; Hamid, R.; Loyd, J.E. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Eichstaedt, C.A.; Song, J.; Benjamin, N.; Harutyunova, S.; Fischer, C.; Grunig, E.; Hinderhofer, K. EIF2AK4 mutation as “second hit” in hereditary pulmonary arterial hypertension. Respir. Res. 2016, 17, 141. [Google Scholar] [CrossRef]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef]
- Thompson, A.A.R.; Lawrie, A. Targeting Vascular Remodeling to Treat Pulmonary Arterial Hypertension. Trends Mol. Med. 2017, 23, 31–45. [Google Scholar] [CrossRef]
- Dorfmuller, P.; Zarka, V.; Durand-Gasselin, I.; Monti, G.; Balabanian, K.; Garcia, G.; Capron, F.; Coulomb-Lhermine, A.; Marfaing-Koka, A.; Simonneau, G.; et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2002, 165, 534–539. [Google Scholar] [CrossRef]
- Cracowski, J.L.; Chabot, F.; Labarere, J.; Faure, P.; Degano, B.; Schwebel, C.; Chaouat, A.; Reynaud-Gaubert, M.; Cracowski, C.; Sitbon, O.; et al. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur. Respir. J. 2014, 43, 915–917. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Oliver, E.; Dorfmuller, P.; Dubois, O.D.; Reed, D.M.; Kirkby, N.S.; Mohamed, N.A.; Perros, F.; Antigny, F.; Fadel, E.; et al. Evidence for the involvement of type I interferon in pulmonary arterial hypertension. Circ. Res. 2014, 114, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Koumakis, E.; Wipff, J.; Dieude, P.; Ruiz, B.; Bouaziz, M.; Revillod, L.; Guedj, M.; Distler, J.H.; Matucci-Cerinic, M.; Humbert, M.; et al. TGFbeta receptor gene variants in systemic sclerosis-related pulmonary arterial hypertension: Results from a multicentre EUSTAR study of European Caucasian patients. Ann. Rheum. Dis. 2012, 71, 1900–1903. [Google Scholar] [CrossRef]
- Dewachter, L.; Adnot, S.; Guignabert, C.; Tu, L.; Marcos, E.; Fadel, E.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Naeije, R.; et al. Bone morphogenetic protein signalling in heritable versus idiopathic pulmonary hypertension. Eur. Respir. J. 2009, 34, 1100–1110. [Google Scholar] [CrossRef]
- Guignabert, C.; Bailly, S.; Humbert, M. Restoring BMPRII functions in pulmonary arterial hypertension: Opportunities, challenges and limitations. Expert Opin. Ther. Targets 2017, 21, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Escribano Subias, P.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N Engl J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Lian, T.Y.; Jiang, X.; Liu, S.F.; Li, S.Q.; Jiang, R.; Wu, W.H.; Ye, J.; Cheng, C.Y.; Du, Y.; et al. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801609. [Google Scholar] [CrossRef]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Graf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Bill, M.; Aldred, M.A.; van de Veerdonk, M.C.; Vonk Noordegraaf, A.; Long-Boyle, J.; Dash, R.; Yang, P.C.; et al. Low-Dose FK506 (Tacrolimus) in End-Stage Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Scott, V.; Del Rosario, P.; Bill, M.; Haddad, F.; Long-Boyle, J.; Hedlin, H.; Zamanian, R.T. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- Kerstjens-Frederikse, W.S.; Bongers, E.M.; Roofthooft, M.T.; Leter, E.M.; Douwes, J.M.; Van Dijk, A.; Vonk-Noordegraaf, A.; Dijk-Bos, K.K.; Hoefsloot, L.H.; Hoendermis, E.S.; et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J. Med. Genet. 2013, 50, 500–506. [Google Scholar] [CrossRef]
- Haarman, M.G.; Kerstjens-Frederikse, W.S.; Vissia-Kazemier, T.R.; Breeman, K.T.N.; Timens, W.; Vos, Y.J.; Roofthooft, M.T.R.; Hillege, H.L.; Berger, R.M.F. The Genetic Epidemiology of Pediatric Pulmonary Arterial Hypertension. J. Pediatr. 2020, 225, 65–73. [Google Scholar] [CrossRef]
- Hernandez-Gonzalez, I.; Tenorio, J.; Palomino-Doza, J.; Martinez Menaca, A.; Morales Ruiz, R.; Lago-Docampo, M.; Valverde Gomez, M.; Gomez Roman, J.; Enguita Valls, A.B.; Perez-Olivares, C.; et al. Clinical heterogeneity of Pulmonary Arterial Hypertension associated with variants in TBX4. PLoS ONE 2020, 15, e0232216. [Google Scholar] [CrossRef]
- Le Ribeuz, H.; Capuano, V.; Girerd, B.; Humbert, M.; Montani, D.; Antigny, F. Implication of Potassium Channels in the Pathophysiology of Pulmonary Arterial Hypertension. Biomolecules 2020, 10, 1261. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Trégouët, D.A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef]
- Lago-Docampo, M.; Tenorio, J.; Hernandez-Gonzalez, I.; Perez-Olivares, C.; Escribano-Subias, P.; Pousada, G.; Baloira, A.; Arenas, M.; Lapunzina, P.; Valverde, D. Characterization of rare ABCC8 variants identified in Spanish pulmonary arterial hypertension patients. Sci Rep. 2020, 10, 15135. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, M.S.; Ma, L.; Zhu, N.; Qi, H.; McClenaghan, C.; Gonzaga-Jauregui, C.; Dewey, F.E.; Overton, J.D.; Reid, J.G.; Shuldiner, A.R.; et al. Loss-of-Function ABCC8 Mutations in Pulmonary Arterial Hypertension. Circ. Genom Precis Med. 2018, 11, e002087. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Female | 69 (87.3%) |
| Age (years) | 55.6 ± 1.9 |
| Right Heart Catheterism | |
| RAP (mmHg) | 8.6 ± 0.6 |
| mPAP (mmHg) | 42 ± 1.4 |
| PCWP (mmHg) | 9.8 ± 0.4 |
| CO (l/min) | 4.3 ± 0.1 |
| CI (l/min/m2) | 2.6 ± 0.1 |
| PVR (UW) | 8.6 ± 0.5 |
| SvO2 (%) | 65.8 ± 1.5 |
| Pulmonary Function Test | |
| FEV1 (% predicted) | 80.5 ± 2.2 |
| FVC (% predicted) | 81.7 ± 2.2 |
| TLC (% predicted) | 88.5 ± 2.6 |
| DLCO (% predicted) | 47.5 ± 2 |
| 6MWT (m) | 340 ± 16 |
| Exercise O2 Sat (%) | 87 ± 1.3 |
| Functional Class | |
| I | 2 (2.5%) |
| II | 27 (34.2%) |
| III | 43 (54.4%) |
| IV | 7 (8.9%) |
| Data are median mean ± SD, or n (%) | |
| Patient 1 | Patient 2 | Patient 4 | Patient 5 | |
|---|---|---|---|---|
| Gene | TBX4 | ABCC8 | GDF2 | KCNA5 |
| Age PAH diagnosis | 58 | 26 | 25 | 70 |
| Age CTD diagnosis | 56 | 28 | 22 | NA |
| Gender | F | F | F | F |
| CTD | Mixed CTD | SSc | SLE | SSc |
| CTD manifestations | Arthritis | Raynaud, digital ulcers | Discoid lupus, enteritis, serositis, poliarthritis | NA |
| CTD serology | NA | ANA, ACA | ANA, anti-DNA, anti-Sm | NA |
| CTD treatment | None | Corticosteoids, AZA | Corticosteoids, MTX, cyclophosmamide, hydroxychloroquine | None |
| mPAP (mmHg) | 37 | 71 | 30 | 45 |
| PCWP (mmHg) | 4 | 3 | 6 | 8 |
| CI (l/min/m2) | 2.25 | 4.2 | 4.6 | |
| PVR (WU) | 12.6 | 10.2 | 3.75 | 4.5 |
| FEV1 (% predicted) | 83 | 102 | 86 | 75 |
| TLC or FVC (% predicted) | 109 | 87 | 103 | 81 |
| DLCO (% predicted) | 61 | 74 | ||
| 6MWT (m) | 389 | 463 | NA | 180 |
| FC | II | III | II | III |
| Final status | Alive | Alive | Alive | Death |
| Follow-up (years) | 6 | 18 | 3.5 | 8.5 |
| Patient | Gene | Genomic Coordinate (hg19) | cDNA and Protein Location | Exon/Intron | Mutation Type | Population Frequency † | Pathogenicity Predictors ‡ | ACMG Prediction § | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | TBX4 | chr17:59560351dup | NM_018488.2:c.1112dupC:p. (Pro372Serfs*14) | 8 | frameshift | 0 | 3/3 | P | PMID: 32348326 |
| 2 | ABCC8 | chr11:17432062C>T | NM_000352.4:c.2694+1G>A | IVS21 | splicing | 0.000003979 | 2/2 | LP | Lago-Docampo et al. |
| 3 | ABCC8 | chr11:17491762G>A | NM_000352.6:c.298G>A p. (Glu100Lys) | 3 | missense | 0.00007162 | 2/9 | VUS | Lago-Docampo et al. |
| 4 | GDF2 | chr10:48414226C>T | NM_016204.3:c.642G>A:p. (Trp214*) | 2 | nonsense | 0 | 3/3 | P | Tenorio et al. |
| 5 | KCNA5 | chr12:5154998del | NM_002234.3:c.1685delC (p.Phe563fs*21) | 1 | frameshift | 0 | 2/2 | P | Tenorio et al. |
| 6 | NOTCH3 | chr19:15278219C>T | NM_000435.2:c.5203G>A:p. (Glu1735Lys) | 29 | missense | 0.00000409 | 8/9 | VUS | Tenorio et al. |
| 7 | CTCFL | chr20:56078510G>A | NM_001269041.1:c.1822G>A (p.Glu608Lys) | 9 | missense | 0 | 1/9 | VUS | This study |
| 8 | CTCFL | chr20:56093935A>C | NM_001269041.1:c.938A>G (p.Tyr313Cys) | 4 | missense | 0 | 4/9 | VUS | This study |
| 9 | TOPBP1 | chr3:133371445T>G | NM_007027.3:c.951T>G (p.Ile317Met) | 8 | missense | 0.0000299 | 4/9 | VUS | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernandez-Gonzalez, I.; Tenorio-Castano, J.; Ochoa-Parra, N.; Gallego, N.; Pérez-Olivares, C.; Lago-Docampo, M.; Palomino Doza, J.; Valverde, D.; Lapunzina, P.; Escribano-Subias, P. Novel Genetic and Molecular Pathways in Pulmonary Arterial Hypertension Associated with Connective Tissue Disease. Cells 2021, 10, 1488. https://doi.org/10.3390/cells10061488
Hernandez-Gonzalez I, Tenorio-Castano J, Ochoa-Parra N, Gallego N, Pérez-Olivares C, Lago-Docampo M, Palomino Doza J, Valverde D, Lapunzina P, Escribano-Subias P. Novel Genetic and Molecular Pathways in Pulmonary Arterial Hypertension Associated with Connective Tissue Disease. Cells. 2021; 10(6):1488. https://doi.org/10.3390/cells10061488
Chicago/Turabian StyleHernandez-Gonzalez, Ignacio, Jair Tenorio-Castano, Nuria Ochoa-Parra, Natalia Gallego, Carmen Pérez-Olivares, Mauro Lago-Docampo, Julian Palomino Doza, Diana Valverde, Pablo Lapunzina, and Pilar Escribano-Subias. 2021. "Novel Genetic and Molecular Pathways in Pulmonary Arterial Hypertension Associated with Connective Tissue Disease" Cells 10, no. 6: 1488. https://doi.org/10.3390/cells10061488
APA StyleHernandez-Gonzalez, I., Tenorio-Castano, J., Ochoa-Parra, N., Gallego, N., Pérez-Olivares, C., Lago-Docampo, M., Palomino Doza, J., Valverde, D., Lapunzina, P., & Escribano-Subias, P. (2021). Novel Genetic and Molecular Pathways in Pulmonary Arterial Hypertension Associated with Connective Tissue Disease. Cells, 10(6), 1488. https://doi.org/10.3390/cells10061488