
The Emerging Role of HDACs: Pathology and Therapeutic Targets in Diabetes Mellitus

 ,
,  , , ,
, , ,  ,
,  and
and 
Abstract

1. Introduction
2. HDACs: A General Overview
2.1. The HDAC Family: Classes and Inhibitors
2.2. HDACs and DM
3. Role of HDACs in Endocrine Fate of Pancreas
3.1. Role of HDACs in Regulating β Cell Function and Insulin Secretion
3.2. Role of HDACs in Glucose Homeostasis
4. HDACs: The Possible Therapeutic Targets in DM
4.1. Improving β Cell Generation: Via Targeting HDACs
4.2. β. Cell Protection and Promotion of Insulin Secretion: Via Targeting HDACs
4.3. Improving Glucose Homeostasis: Via Targeting HDACs
4.4. Improving Diabetic Complications: Via Targeting HDACs
4.4.1. HDAC-Mediated Therapeutic Options in Diabetic Nephropathy
4.4.2. HDAC-Mediated Therapeutic Options in Diabetic Cardiomyopathy
4.4.3. HDAC-Mediated Therapeutic Options in Diabetic Retinopathy
4.4.4. HDAC-Mediated Therapeutic Options in Diabetic Neuropathy
4.4.5. HDAC-Mediated Therapeutic Options in Diabetic Endothelial Dysfunctions
4.4.6. HDAC-Mediated Therapeutic Options in Other Diabetic Complications
5. Challenges and Future Prospects
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dewanjee, S.; Das, S.; Das, A.K.; Bhattacharjee, N.; Dihingia, A.; Dua, T.K.; Kalita, J.; Manna, P. Molecular mechanism of diabetic neuropathy and its pharmacotherapeutic targets. Eur. J. Pharmacol. 2018, 833, 472–523. [Google Scholar] [CrossRef]
- Rivera-Mancía, S.; Trujillo, J.; Chaverri, J.P. Utility of curcumin for the treatment of diabetes mellitus: Evidence from pre-clinical and clinical studies. J. Nutr. Intermed. Metab. 2018, 14, 29–41. [Google Scholar] [CrossRef]
- Franks, P.W.; Pearson, E.R.; Florez, J.C. Gene-Environment and Gene-Treatment Interactions in Type 2 Diabetes: Progress, pitfalls, and prospects. Diabetes Care 2013, 36, 1413–1421. [Google Scholar] [CrossRef]
- Blanter, M.; Sork, H.; Tuomela, S.; Flodström-Tullberg, M. Genetic and Environmental Interaction in Type 1 Diabetes: A Relationship Between Genetic Risk Alleles and Molecular Traits of Enterovirus Infection? Curr. Diabetes Rep. 2019, 19, 82. [Google Scholar] [CrossRef] [PubMed]
- Böni-Schnetzler, M.; Meier, D.T. Islet inflammation in type 2 diabetes. Semin. Immunopathol. 2019, 41, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Shanak, S.; Saad, B.; Zaid, H. Metabolic and Epigenetic Action Mechanisms of Antidiabetic Medicinal Plants. Evidence-Based Complement. Altern. Med. 2019, 2019, 3583067. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Prim. 2015, 1, 15019. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Stumvoll, M.; Goldstein, B.J.; van Haeften, T.W. Type 2 diabetes: Principles of pathogenesis and therapy. Lancet 2005, 365, 1333–1346. [Google Scholar] [CrossRef]
- Wilkin, T.J. Changing perspectives in diabetes: Their impact on its classification. Diabetologia 2007, 50, 1587–1592. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Jiang, Y. Endogenous Pancreatic β Cell Regeneration: A Potential Strategy for the Recovery of β Cell Deficiency in Diabetes. Front. Endocrinol. 2019, 10, 101. [Google Scholar] [CrossRef]
- Jerram, S.T.; Dang, M.N.; Leslie, R.D. The Role of Epigenetics in Type 1 Diabetes. Curr. Diabetes Rep. 2017, 17, 1–11. [Google Scholar] [CrossRef]
- Ling, C.; Rönn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef]
- Khullar, M.; Cheema, B.S.; Raut, S.K. Emerging Evidence of Epigenetic Modifications in Vascular Complication of Diabetes. Front. Endocrinol. 2017, 8, 237. [Google Scholar] [CrossRef]
- Astratenkova, I.V.; Rogozkin, V.A. The Role of Acetylation/Deacetylation of Histones and Transcription Factors in Regu-lating Metabolism in Skeletal Muscles. Neurosci. Behav. Phys. 2019, 49, 281–288. [Google Scholar] [CrossRef]
- Wang, X.; Wei, X.; Pang, Q.; Yi, F. Histone deacetylases and their inhibitors: Molecular mechanisms and therapeutic im-plications in diabetes mellitus. Acta Pharm. Sin. B 2012, 2, 387–395. [Google Scholar] [CrossRef]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. BioMed Res. Int. 2016, 2016, 8797206. [Google Scholar] [CrossRef] [PubMed]
- Dominy, J.E.; Lee, Y.; Jedrychowski, M.P.; Chim, H.; Jurczak, M.J.; Camporez, J.P.; Ruan, H.-B.; Feldman, J.; Pierce, K.; Mostoslavsky, R.; et al. The Deacetylase Sirt6 Activates the Acetyltransferase GCN5 and Suppresses Hepatic Gluconeogenesis. Mol. Cell 2012, 48, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Bilokapic, S.; Strauss, M.; Halic, M. Structural rearrangements of the histone octamer translocate DNA. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Lawlor, L.; Yang, X.B. Harnessing the HDAC–histone deacetylase enzymes, inhibitors and how these can be utilised in tissue engineering. Int. J. Oral Sci. 2019, 11, 1–11. [Google Scholar] [CrossRef]
- Trisciuoglio, D.; Di Martile, M.; Del Bufalo, D. Emerging Role of Histone Acetyltransferase in Stem Cells and Cancer. Stem Cells Int. 2018, 2018, 8908751. [Google Scholar] [CrossRef] [PubMed]
- Fenley, A.T.; Anandakrishnan, R.; Kidane, Y.H.; Onufriev, A.V. Modulation of nucleosomal DNA accessibility via charge-altering post-translational modifications in histone core. Epigenetics Chromatin 2018, 11, 1–19. [Google Scholar] [CrossRef]
- Sanchez, R.; Meslamani, J.; Zhou, M.-M. The bromodomain: From epigenome reader to druggable target. Biochim. et Biophys. Acta (BBA) Bioenerg. 2014, 1839, 676–685. [Google Scholar] [CrossRef]
- Schütz, L.F.; Park, M.H.; Choudhury, M. HDACs in diabetes: A new era of epigenetic drug. In Nutritional and Therapeutic Interventions for Diabetes and Metabolic Syndrome; Academic Press: Cambridge, MA, USA, 2018; pp. 475–486. [Google Scholar]
- Elmallah, M.I.Y.; Micheau, O. Epigenetic Regulation of TRAIL Signaling: Implication for Cancer Therapy. Cancers 2019, 11, 850. [Google Scholar] [CrossRef]
- Shakespear, M.R.; Halili, M.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhao, N.; Ge, D.; Chen, Y. Next-generation of selective histone deacetylase inhibitors. RSC Adv. 2019, 9, 19571–19583. [Google Scholar] [CrossRef]
- Park, S.-Y.; Kim, G.S.; Hwang, H.-J.; Nam, T.-H.; Park, H.-S.; Song, J.; Jang, T.-H.; Lee, Y.C.; Kim, J.-S. Structural basis of the specific interaction of SMRT corepressor with histone deacetylase 4. Nucleic Acids Res. 2018, 46, 11776–11788. [Google Scholar] [CrossRef]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase modulators provided by Mother Nature. Genes Nutr. 2012, 7, 357–367. [Google Scholar] [CrossRef]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef]
- Faller, Y.D.A.D.V. Transcription Regulation by Class III Histone Deacetylases (HDACs)—Sirtuins. Transl. Oncogenomics 2008, 1, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Yanginlar, C.; Logie, C. HDAC11 is a regulator of diverse immune functions. Biochim. et Biophys. Acta (BBA) Bioenerg. 2018, 1861, 54–59. [Google Scholar] [CrossRef]
- Tang, J.; Yan, H.; Zhuang, S. Histone deacetylases as targets for treatment of multiple diseases. Clin. Sci. 2013, 124, 651–662. [Google Scholar] [CrossRef]
- Bertrand, P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Glaser, K.B. HDAC inhibitors: Clinical update and mechanism-based potential. Biochem. Pharmacol. 2007, 74, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Hu, J.; Jing, H.; Lin, H. Sirtuin inhibitors as anticancer agents. Futur. Med. Chem. 2014, 6, 945–966. [Google Scholar] [CrossRef] [PubMed]
- Zwergel, C.; Stazi, G.; Valente, S.; Mai, A. Histone Deacetylase Inhibitors: Updated Studies in Various Epigenetic-Related Diseases. J. Clin. Epigenetics 2016, 2. [Google Scholar] [CrossRef]
- Chen, Y.; Du, J.; Zhao, Y.T.; Zhang, L.; Lv, G.; Zhuang, S.; Qin, G.; Zhao, T.C. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc. Diabetol. 2015, 14, 99. [Google Scholar] [CrossRef]
- Sharma, S.; Taliyan, R. Histone deacetylase inhibitors: Future therapeutics for insulin resistance and type 2 diabetes. Pharmacol. Res. 2016, 113, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Hadden, M.J.; Advani, A. Histone Deacetylase Inhibitors and Diabetic Kidney Disease. Int. J. Mol. Sci. 2018, 19, 2630. [Google Scholar] [CrossRef]
- Noh, H.; Oh, E.Y.; Seo, J.Y.; Yu, M.R.; Kim, Y.O.; Ha, H.; Lee, H.B. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-β1-induced renal injury. Am. J. Physiol. Physiol. 2009, 297, F729–F739. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.P.; Dahllöf, M.; Lundh, M.; Rasmussen, D.N.; Nielsen, M.D.; Billestrup, N.; Grunnet, L.G.; Mandrup-Poulsen, T. Histone Deacetylase (HDAC) Inhibition as a Novel Treatment for Diabetes Mellitus. Mol. Med. 2011, 17, 378–390. [Google Scholar] [CrossRef]
- Hou, Q.; Hu, K.; Liu, X.; Quan, J.; Liu, Z. HADC regulates the diabetic vascular endothelial dysfunction by targetting MnSOD. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Liao, R.; Yao, Z.; Zhou, W.; Ye, P.; Zheng, X.; Li, X.; Huang, Y.; Chen, S.; Chen, Q. Three single nucleotide variants of the HDAC gene are associated with type 2 diabetes mellitus in a Chinese population: A community-based case–control study. Gene 2014, 533, 427–433. [Google Scholar] [CrossRef]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. New York Acad. Sci. 2018, 1411, 21–35. [Google Scholar] [CrossRef]
- Chen, J.; Wang, N.; Dong, M.; Guo, M.; Zhao, Y.; Zhuo, Z.; Zhang, C.; Chi, X.; Pan, Y.; Jiang, J.; et al. The Metabolic Regulator Histone Deacetylase 9 Contributes to Glucose Homeostasis Abnormality Induced by Hepatitis C Virus Infection. Diabetes 2015, 64, 4088–4098. [Google Scholar] [CrossRef] [PubMed]
- Benerofe, S.A.; Carrillo-Sepulveda, M.A. Downregulation of Histone Deacetylase 9 (HDAC9) is Associated with Vascular Calcification in Diabetic Mice. FASEB J. 2017, 31, 673–679. [Google Scholar]
- Liu, F.; Zong, M.; Wen, X.; Li, X.; Wang, J.; Wang, Y.; Jiang, W.; Li, X.; Guo, Z.; Qi, H. Silencing of Histone Deacetylase 9 Expression in Podocytes Attenuates Kidney Injury in Diabetic Nephropathy. Sci. Rep. 2016, 6, 33676. [Google Scholar] [CrossRef]
- Winkler, R.; Benz, V.; Clemenz, M.; Bloch, M.; Foryst-Ludwig, A.; Wardat, S.; Witte, N.; Trappiel, M.; Namsolleck, P.; Mai, K.; et al. Histone Deacetylase 6 (HDAC6) Is an Essential Modifier of Glucocorticoid-Induced Hepatic Gluconeogenesis. Diabetes 2011, 61, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Wu, Y.; Lei, S.; Zhou, B.; Qiu, Z.; Wang, K.; Xia, Z. Inhibition of HDAC6 Activity Alleviates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats: Potential Role of Peroxiredoxin 1 Acetylation and Redox Regulation. Oxidative Med. Cell. Longev. 2018, 2018, 9494052. [Google Scholar] [CrossRef]
- Wang, L.; Beier, U.H.; Akimova, T.; Dahiya, S.; Han, R.; Samanta, A.; Levine, M.H.; Hancock, W.W. Histone/protein deacetylase inhibitor therapy for enhancement of Foxp3+ T-regulatory cell function posttransplantation. Arab. Archaeol. Epigr. 2018, 18, 1596–1603. [Google Scholar] [CrossRef]
- Gurley, J.M.; Griesel, B.A.; Olson, A.L. Increased Skeletal Muscle GLUT4 Expression in Obese Mice After Voluntary Wheel Running Exercise Is Posttranscriptional. Diabetes 2016, 65, 2911–2919. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, N.; Raychaudhuri, S.; Thamotharan, M.; Devaskar, S.U. Histone Code Modifications Repress Glucose Transporter 4 Expression in the Intrauterine Growth-restricted Offspring. J. Biol. Chem. 2008, 283, 13611–13626. [Google Scholar] [CrossRef]
- Yao, X.; Nguyen, K.H.; Nyomba, B.L.G. Reversal of glucose intolerance in rat offspring exposed to ethanol before birth through reduction of nuclear skeletal muscle HDAC expression by the bile acid TUDCA. Physiol. Rep. 2014, 2. [Google Scholar] [CrossRef]
- Ye, X.; Li, M.; Hou, T.; Gao, T.; Zhu, W.-G.; Yang, Y. Sirtuins in glucose and lipid metabolism. Oncotarget 2016, 8, 1845–1859. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Yang, B.; Jia, X.; Li, M.; Tan, W.; Ma, S.; Shi, X.; Feng, L. Distinctive Roles of Sirtuins on Diabetes, Protective or Detrimental? Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Monno, I.; Koya, D. Sirtuins and Type 2 Diabetes: Role in Inflammation, Oxidative Stress, and Mitochondrial Function. Front. Endocrinol. 2019, 10, 187. [Google Scholar] [CrossRef]
- Tang, W.; Fan, Y. SIRT6 as a potential target for treating insulin resistance. Life Sci. 2019, 231, 116558. [Google Scholar] [CrossRef]
- Kanwal, A.; Dsouza, L.A. Sirtuins and diabetes: Optimizing the sweetness in the blood. Transl. Med. Commun. 2019, 4, 3. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, J.; Hou, X.; Liu, H.; Guo, F.; Zhou, Y.; Zhang, Y.; Qu, Y.; Gu, J.; Zhou, Y.; et al. SIRT1 rs10823108 and FOXO1 rs17446614 responsible for genetic susceptibility to diabetic nephropathy. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Liu, T.; Yang, W.; Pang, S.; Yu, S.; Yan, B. Functional genetic variants within the SIRT2 gene promoter in type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2018, 137, 200–207. [Google Scholar] [CrossRef]
- Shi, J.-X.; Wang, Q.-J.; Li, H.; Huang, Q. SIRT4 overexpression protects against diabetic nephropathy by inhibiting podocyte apoptosis. Exp. Ther. Med. 2017, 13, 342–348. [Google Scholar] [CrossRef]
- Kutil, Z.; Mikešová, J.; Zessin, M.; Meleshin, M.; Nováková, Z.; Alquicer, G.; Kozikowski, A.; Sippl, W.; Bařinka, C.; Schutkowski, M. Continuous Activity Assay for HDAC11 Enabling Reevaluation of HDAC Inhibitors. ACS Omega 2019, 4, 19895–19904. [Google Scholar] [CrossRef]
- Bhaskara, S. Histone deacetylase 11 as a key regulator of metabolism and obesity. EBioMedicine 2018, 35, 27–28. [Google Scholar] [CrossRef]
- Sun, L.; De Evsikova, C.M.; Bian, K.; Achille, A.; Telles, E.; Pei, H.; Seto, E. Programming and Regulation of Metabolic Homeostasis by HDAC11. EBioMedicine 2018, 33, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.-D.; Wan, L.-L.; Duan, M.; Lu, S. HDAC11 deletion reduces fructose-induced cardiac dyslipidemia, apoptosis and inflammation by attenuating oxidative stress injury. Biochem. Biophys. Res. Commun. 2018, 503, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Bonnefond, A.; Froguel, P.; Vaxillaire, M. The emerging genetics of type 2 diabetes. Trends Mol. Med. 2010, 16, 407–416. [Google Scholar] [CrossRef]
- Gray, S.G.; De Meyts, P. Role of histone and transcription factor acetylation in diabetes pathogenesis. Diabetes/Metabolism Res. Rev. 2005, 21, 416–433. [Google Scholar] [CrossRef]
- Da Silva Xavier, G. The Cells of the Islets of Langerhans. J. Clin. Med. 2018, 7, 54. [Google Scholar] [CrossRef]
- Bastidas-Ponce, A.; Scheibner, K.; Lickert, H.; Bakhti, M. Cellular and molecular mechanisms coordinating pancreas development. Development 2017, 144, 2873–2888. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.L.; O’Driscoll, M.; Sheets, T.P.; Hruban, R.H.; Oberholzer, J.; McGarrigle, J.J.; Shamblott, M.J. Neurogenin 3 Expressing Cells in the Human Exocrine Pancreas Have the Capacity for Endocrine Cell Fate. PLoS ONE 2015, 10, e0133862. [Google Scholar] [CrossRef]
- Lorenzo, P.I.; Juárez-Vicente, F.; Cobo-Vuilleumier, N.; García-Domínguez, M.; Gauthier, B.R. The Diabetes-Linked Transcription Factor PAX4: From Gene to Functional Consequences. Genes 2017, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Carrano, A.C.; Sander, M. A systems view of epigenetic networks regulating pancreas development and β-cell function. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Stoffers, D.A.; Nicholls, R.D.; Simmons, R.A. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J. Clin. Investig. 2008, 118, 2316–2324. [Google Scholar] [CrossRef]
- Haumaitre, C.; Lenoir, O.; Scharfmann, R. Histone Deacetylase Inhibitors Modify Pancreatic Cell Fate Determination and Amplify Endocrine Progenitors. Mol. Cell. Biol. 2008, 28, 6373–6383. [Google Scholar] [CrossRef]
- Mosley, A.L.; Özcan, S. The Pancreatic Duodenal Homeobox-1 Protein (Pdx-1) Interacts with Histone Deacetylases Hdac-1 and Hdac-2 on Low Levels of Glucose. J. Biol. Chem. 2004, 279, 54241–54247. [Google Scholar] [CrossRef]
- Plaisance, V.; Rolland, L.; Gmyr, V.; Annicotte, J.-S.; Kerr-Conte, J.; Pattou, F.; Abderrahmani, A. The Class I Histone Deacetylase Inhibitor MS-275 Prevents Pancreatic Beta Cell Death Induced by Palmitate. J. Diabetes Res. 2014, 2014, 195739. [Google Scholar] [CrossRef]
- Lenoir, O.; Flosseau, K.; Ma, F.X.; Blondeau, B.; Mai, A.; Bassel-Duby, R.; Ravassard, P.; Olson, E.N.; Haumaitre, C.; Scharfmann, R. Specific Control of Pancreatic Endocrine β- and δ-Cell Mass by Class IIa Histone Deacetylases HDAC4, HDAC5, and HDAC9. Diabetes 2011, 60, 2861–2871. [Google Scholar] [CrossRef]
- Bramswig, N.C.; Kaestner, K.H. Epigenetics and diabetes treatment: An unrealized promise? Trends Endocrinol. Metab. 2012, 23, 286–291. [Google Scholar] [CrossRef]
- Wu, S.Y.; Liang, J.; Yang, B.C.; Leung, P.S. SIRT1 Activation Promotes β-Cell Regeneration by Activating Endocrine Progenitor Cells via AMPK Signaling-Mediated Fatty Acid Oxidation. Stem Cells 2019, 37, 1416–1428. [Google Scholar] [CrossRef]
- Wang, R.-H.; Xu, X.; Kim, H.-S.; Xiao, Z.; Deng, C.-X. SIRT1 Deacetylates FOXA2 and Is Critical for Pdx1 Transcription and β-Cell Formation. Int. J. Biol. Sci. 2013, 9, 934–946. [Google Scholar] [CrossRef]
- Pacifici, F.; Di Cola, D.; Pastore, D.; Abete, P.; Guadagni, F.; Donadel, G.; Bellia, A.; Esposito, E.; Salimei, C.; Salimei, P.S.; et al. Proposed Tandem Effect of Physical Activity and Sirtuin 1 and 3 Activation in Regulating Glucose Homeostasis. Int. J. Mol. Sci. 2019, 20, 4748. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Fei, X. SIRT5 regulates pancreatic β-cell proliferation and insulin secretion in type 2 diabetes. Exp. Ther. Med. 2018, 16, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Qin, K.; Zhang, N.; Zhang, Z.; Nipper, M.; Zhu, Z.; Leighton, J.; Xu, K.; Musi, N.; Wang, P. SIRT6-mediated transcriptional suppression of Txnip is critical for pancreatic beta cell function and survival in mice. Diabetologia 2018, 61, 906–918. [Google Scholar] [CrossRef]
- Lundh, M.; Christensen, D.; Rasmussen, D.N.; Mascagni, P.; Dinarello, C.A.; Billestrup, N.; Grunnet, L.G.; Mandruppoulsen, T. Lysine deacetylases are produced in pancreatic beta cells and are differentially regulated by proinflammatory cytokines. Diabetologia 2010, 53, 2569–2578. [Google Scholar] [CrossRef]
- Bhattacharjee, N.; Dua, T.K.; Khanra, R.; Joardar, S.; Nandy, A.; Saha, A.; De Feo, V.; Dewanjee, S. Protocatechuic Acid, a Phenolic from Sansevieria roxburghiana Leaves, Suppresses Diabetic Cardiomyopathy via Stimulating Glucose Metabolism, Ameliorating Oxidative Stress, and Inhibiting Inflammation. Front. Pharmacol. 2017, 8, 251. [Google Scholar] [CrossRef]
- Pinney, S.E.; Simmons, R.A. Epigenetic mechanisms in the development of type 2 diabetes. Trends Endocrinol. Metab. 2010, 21, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, Q.; Zhou, Z.; Ikeda, Y. PDX1, Neurogenin-3, and MAFA: Critical transcription regulators for beta cell development and regeneration. Stem Cell Res. Ther. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, J.M.; Walker, E.M.; Stein, R. Impact of Pdx1-associated chromatin modifiers on islet β-cells. Diabetes Obes. Metab. 2016, 18, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Sampley, M.L.; Özcan, S. Regulation of Insulin Gene Transcription by Multiple Histone Acetyltransferases. DNA Cell Biol. 2012, 31, 8–14. [Google Scholar] [CrossRef]
- Kong, Y.; Sharma, R.B.; Nwosu, B.U.; Alonso, L.C. Islet biology, the CDKN2A/B locus and type 2 diabetes risk. Diabetologia 2016, 59, 1579–1593. [Google Scholar] [CrossRef]
- Salas, E.; Rabhi, N.; Froguel, P.; Annicotte, J.-S. Role of Ink4a/Arf Locus in Beta Cell Mass Expansion under Physiological and Pathological Conditions. J. Diabetes Res. 2014, 2014, 873679. [Google Scholar] [CrossRef]
- Remsberg, J.R.; Ediger, B.N.; Ho, W.Y.; Damle, M.; Li, Z.; Teng, C.; Lanzillotta, C.; Stoffers, D.A.; Lazar, M.A. Deletion of histone deacetylase 3 in adult beta cells improves glucose tolerance via increased insulin secretion. Mol. Metab. 2017, 6, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-B.; Gao, L.; Wang, J.; Wang, Y.-G.; Dong, Z.; Zhao, J.; Mi, Q.-S.; Zhou, L. Conditional ablation of HDAC3 in islet beta cells results in glucose intolerance and enhanced susceptibility to STZ-induced diabetes. Oncotarget 2016, 7, 57485–57497. [Google Scholar] [CrossRef]
- Daneshpajooh, M.; Eliasson, L.; Bacos, K.; Ling, C. MC1568 improves insulin secretion in islets from type 2 diabetes patients and rescues β-cell dysfunction caused by Hdac7 upregulation. Acta Diabetol. 2018, 55, 1231–1235. [Google Scholar] [CrossRef]
- Makinistoglu, M.P.; Karsenty, G. The class II histone deacetylase HDAC4 regulates cognitive, metabolic and endocrine functions through its expression in osteoblasts. Mol. Metab. 2015, 4, 64–69. [Google Scholar] [CrossRef]
- McCann, J.; Ellis, M.; McGee, S.L.; Aston-Mourney, K. Class IIa HDACs do not influence beta-cell function under normal or high glucose conditions. Islets 2019, 11, 112–118. [Google Scholar] [CrossRef]
- Gong, M.; Yu, Y.; Liang, L.; Vuralli, D.; Froehler, S.; Kuehnen, P.; Du Bois, P.; Zhang, J.; Cao, A.; Liu, Y.; et al. HDAC4 mutations cause diabetes and induce β-cell FoxO1 nuclear exclusion. Mol. Genet. Genom. Med. 2019, 7, e602. [Google Scholar] [CrossRef]
- Shin, J.-S.; Min, B.-H.; Lim, J.-Y.; Kim, B.-K.; Han, H.-J.; Yoon, K.-H.; Kim, S.-J.; Park, C.-G. Novel Culture Technique Involving an Histone Deacetylase Inhibitor Reduces the Marginal Islet Mass to Correct Streptozotocin-Induced Diabetes. Cell Transplant. 2011, 20, 1321–1332. [Google Scholar] [CrossRef]
- Yamato, E. High dose of histone deacetylase inhibitors affects insulin secretory mechanism of pancreatic beta cell line. Endocr. Regul. 2018, 52, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-Y.; Wang, J.; Ka, S.-O.; Bae, E.J.; Park, B.-H. Insulin secretion impairment in Sirt6 knockout pancreatic β cells is mediated by suppression of the FoxO1-Pdx1-Glut2 pathway. Sci. Rep. 2016, 6, 30321. [Google Scholar] [CrossRef]
- Kanwal, A.; Pillai, V.B.; Samant, S.; Gupta, M.; Gupta, M.P. The nuclear and mitochondrial sirtuins, Sirt6 and Sirt3, regulate each other’s activity and protect the heart from developing obesity-mediated diabetic cardiomyopathy. FASEB J. 2019, 33, 10872–10888. [Google Scholar] [CrossRef]
- Anderson, K.A.; Huynh, F.K.; Fisher-Wellman, K.; Stuart, J.D.; Peterson, B.S.; Douros, J.D.; Wagner, G.R.; Thompson, J.W.; Madsen, A.S.; Green, M.F.; et al. SIRT4 Is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion. Cell Metab. 2017, 25, 838–855.e15. [Google Scholar] [CrossRef]
- Röder, P.V.; Wu, B.; Liu, Y.; Han, W. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 2016, 48, e219. [Google Scholar] [CrossRef]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetologia 2018, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, S.; Chen, J.; Su, Z. Unraveling the Regulation of Hepatic Gluconeogenesis. Front. Endocrinol. 2019, 9, 802. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yu, Y.; Zhang, Z.; Chen, X.; Hu, Z.; Tong, Q.; Chang, J.; Feng, X.-H.; Lin, X. SCP4 Promotes Gluconeogenesis Through FoxO1/3a Dephosphorylation. Diabetes 2018, 67, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhao, J.; Deng, W.; Chen, Y.; Shang, J.; Song, K.; Zhang, L.; Wang, C.; Lu, S.; Yang, X.; et al. Identification of a cellularly active SIRT6 allosteric activator. Nat. Chem. Biol. 2018, 14, 1118–1126. [Google Scholar] [CrossRef]
- Bhattacharjee, N.; Barma, S.; Konwar, N.; Dewanjee, S.; Manna, P. Mechanistic insight of diabetic nephropathy and its pharmacotherapeutic targets: An update. Eur. J. Pharmacol. 2016, 791, 8–24. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef]
- Hossini, A.M.; Quast, A.S.; Plötz, M.; Grauel, K.; Exner, T.; Küchler, J.; Stachelscheid, H.; Eberle, J.; Rabien, A.; Makrantonaki, E.; et al. PI3K/AKT Signaling Pathway Is Essential for Survival of Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0154770. [Google Scholar] [CrossRef]
- Zhang, Z.; Ding, X.; Zhou, Z.; Qiu, Z.; Shi, N.; Zhou, S.; Du, L.; Zhu, X.; Wu, Y.; Yin, X.; et al. Sirtuin 1 alleviates diabetic neuropathic pain by regulating synaptic plasticity of spinal dorsal horn neurons. Pain 2019, 160, 1082–1092. [Google Scholar] [CrossRef]
- Fan, J.; Krautkramer, K.A.; Feldman, J.L.; Denu, J.M. Metabolic Regulation of Histone Post-Translational Modifications. ACS Chem. Biol. 2015, 10, 95–108. [Google Scholar] [CrossRef]
- Kimura, K.; Yamada, T.; Matsumoto, M.; Kido, Y.; Hosooka, T.; Asahara, S.-I.; Matsuda, T.; Ota, T.; Watanabe, H.; Sai, Y.; et al. Endoplasmic Reticulum Stress Inhibits STAT3-Dependent Suppression of Hepatic Gluconeogenesis via Dephosphorylation and Deacetylation. Diabetes 2011, 61, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Zhang, X.; Yi, J.; Huang, J.; He, J.; Tao, Y. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016, 35, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kawada, Y.; Asahara, S.-I.; Sugiura, Y.; Sato, A.; Furubayashi, A.; Kawamura, M.; Bartolome, A.; Terashi-Suzuki, E.; Takai, T.; Kanno, A.; et al. Histone deacetylase regulates insulin signaling via two pathways in pancreatic β cells. PLoS ONE 2017, 12, e0184435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, Z.; Gu, J.; Jiang, S.; Liu, Q.; Zheng, Y.; Freedman, J.H.; Sun, J.; Cai, L. HDAC3 inhibition in diabetic mice may activate Nrf2 preventing diabetes-induced liver damage and FGF21 synthesis and secretion leading to aortic protection. Am. J. Physiol. Metab. 2018, 315, E150–E162. [Google Scholar] [CrossRef]
- Bayley, J.S.; Pedersen, T.H.; Nielsen, O.B. Skeletal muscle dysfunction in the db/db mouse model of type 2 diabetes. Muscle Nerve 2016, 54, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Sathishkumar, C.; Prabu, P.; Balakumar, M.; Lenin, R.; Prabhu, D.; Anjana, R.M.; Mohan, V.; Balasubramanyam, M. Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin. Epigenetics 2016, 8, 1–12. [Google Scholar] [CrossRef]
- Jiang, X.; Ye, X.; Guo, W.; Lu, H.; Gao, Z. Inhibition of HDAC3 promotes ligand-independent PPARγ activation by protein acetylation. J. Mol. Endocrinol. 2014, 53, 191–200. [Google Scholar] [CrossRef]
- Cho, H.M.; Seok, Y.M.; Lee, H.A.; Song, M.; Kim, I. Repression of Transcriptional Activity of Forkhead Box O1 by Histone Deacetylase Inhibitors Ameliorates Hyperglycemia in Type 2 Diabetic Rats. Int. J. Mol. Sci. 2018, 19, 3539. [Google Scholar] [CrossRef]
- Vincent Wai-Sun Wong, M.T.-S.M.; Cheng, A.S.-L. Handbook of Nutrition, Diet, and Epigenetics; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.-D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class IIa Histone Deacetylases Are Hormone-Activated Regulators of FOXO and Mammalian Glucose Homeostasis. Cell 2011, 145, 607–621. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Z.; Wang, N.; Guo, M.; Chi, X.; Pan, Y.; Jiang, J.; Niu, J.; Ksimu, S.; Li, J.Z.; et al. Role of HDAC9-FoxO1 Axis in the Transcriptional Program Associated with Hepatic Gluconeogenesis. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.L. Regulation of GLUT4 and Insulin-Dependent Glucose Flux. ISRN Mol. Biol. 2012, 2012, 856987. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, T.K.; Basford, J.E.; Knoll, E.; Tong, W.S.; Blanco, V.; Blomkalns, A.L.; Rudich, S.; Lentsch, A.B.; Hui, D.Y.; Weintraub, N.L. HDAC9 Knockout Mice Are Protected From Adipose Tissue Dysfunction and Systemic Metabolic Disease During High-Fat Feeding. Diabetes 2013, 63, 176–187. [Google Scholar] [CrossRef]
- Gomes, P.; Outeiro, T.F.; Cavadas, C. Emerging Role of Sirtuin 2 in the Regulation of Mammalian Metabolism. Trends Pharmacol. Sci. 2015, 36, 756–768. [Google Scholar] [CrossRef]
- Ramakrishnan, G.; Davaakhuu, G.; Kaplun, L.; Chung, W.-C.; Rana, A.; Atfi, A.; Miele, L.; Tzivion, G. Sirt2 Deacetylase Is a Novel AKT Binding Partner Critical for AKT Activation by Insulin. J. Biol. Chem. 2014, 289, 6054–6066. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Dey, C.S. SIRT2 negatively regulates insulin resistance in C2C12 skeletal muscle cells. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1372–1378. [Google Scholar] [CrossRef]
- Wang, J.; Wang, K.; Huang, C.; Lin, D.; Zhou, Y.; Wu, Y.; Tian, N.; Fan, P.; Pan, X.; Xu, D.; et al. SIRT3 Activation by Dihydromyricetin Suppresses Chondrocytes Degeneration via Maintaining Mitochondrial Homeostasis. Int. J. Biol. Sci. 2018, 14, 1873–1882. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Chen, L.; Tang, Q.; Zhang, J.; Li, Y.; He, J. The Role of Sirt6 in Obesity and Diabetes. Front. Physiol. 2018, 9, 135. [Google Scholar] [CrossRef]
- Li, S.; Zheng, W. Mammalian Sirtuins SIRT4 and SIRT7. Prog. Mol. Biol. Transl. Sci. 2018, 154, 147–168. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Wu, F.; Qin, S.; Nowsheen, S.; Shan, S.; Zayas, J.; Pei, H.; Lou, Z.; Wang, L. Regulation of Serine-Threonine Kinase Akt Activation by NAD + -Dependent Deacetylase SIRT. Cell Rep. 2017, 18, 1229–1240. [Google Scholar] [CrossRef]
- Bagchi, R.A.; Ferguson, B.S.; Stratton, M.S.; Hu, T.; Cavasin, M.A.; Sun, L.; Lin, Y.-H.; Liu, D.; Londono, P.; Song, K.; et al. HDAC11 suppresses the thermogenic program of adipose tissue via BRD2. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Haumaitre, C.; Lenoir, O.; Scharfmann, R. Directing cell differentiation with small-molecule histone deacetylase inhibitors: The example of promoting pancreatic endocrine cells. Cell Cycle 2009, 8, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD+in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jena, G. Valproic Acid Improves Glucose Homeostasis by Increasing Beta-Cell Proliferation, Function, and Reducing its Apoptosis through HDAC Inhibition in Juvenile Diabetic Rat. J. Biochem. Mol. Toxicol. 2016, 30, 438–446. [Google Scholar] [CrossRef]
- Dirice, E.; Ng, R.; Martinez, R.; Hu, J.; Wagner, F.F.; Holson, E.B.; Wagner, B.K.; Kulkarni, R.N. Isoform-selective inhibitor of histone deacetylase 3 (HDAC3) limits pancreatic islet infiltration and protects female nonobese diabetic mice from diabetes. J. Biol. Chem. 2017, 292, 17598–17608. [Google Scholar] [CrossRef]
- Jiang, J.; Au, M.; Lu, K.; Eshpeter, A.; Korbutt, G.; Fisk, G.; Majumdar, A.S. Generation of Insulin-Producing Islet-Like Clusters from Human Embryonic Stem Cells. STEM CELLS 2007, 25, 1940–1953. [Google Scholar] [CrossRef]
- Ikemoto, T.; Feng, R.; Shimada, M.; Saito, Y.; Iwahashi, S.; Morine, Y.; Imura, S. A New 2-Step Acceleration Protocol Using a Histone Deacetylase Inhibitor to Generate Insulin-Producing Cells From Adipose-Derived Mesenchymal Stem Cells. Pancreas 2018, 47, 477–481. [Google Scholar] [CrossRef]
- El-Serafi, A.; ElSharkawi, I.; Sandeep, D. Investigating the role of the histone deacetylases-inhibitor suberanilohydroxamic acid in the differentiation of stem cells into insulin secreting cells. Hamdan Med. J. 2019, 12, 10–14. [Google Scholar] [CrossRef]
- Ikemoto, T.; Feng, R.; Iwahashi, S.-I.; Yamada, S.; Saito, Y.; Morine, Y.; Imura, S.; Matsuhisa, M.; Shimada, M. In vitro and in vivo effects of insulin-producing cells generated by xeno-antigen free 3D culture with RCP piece. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Helker, C.S.M.; Mullapudi, S.-T.; Mueller, L.M.; Preussner, J.; Tunaru, S.; Skog, O.; Kwon, H.-B.; Kreuder, F.; Lancman, J.J.; Bonnavion, R.; et al. Whole organism small molecule screen identifies novel regulators of pancreatic endocrine development. Development 2019, 146. [Google Scholar] [CrossRef]
- Gilbert, R.E.; Thai, K.; Advani, S.L.; Cummins, C.L.; Kepecs, D.M.; Schroer, S.A.; Woo, M.; Zhang, Y. SIRT1 activation ameliorates hyperglycaemia by inducing a torpor-like state in an obese mouse model of type 2 diabetes. Diabetologia 2015, 58, 819–827. [Google Scholar] [CrossRef]
- Raghavan, P.R. Metadichol a novel nano lipid emulsion: gpr120 agonist against insulin resistance. J. Diabetes Metab. 2017, 8, 8. [Google Scholar] [CrossRef]
- Xu, Z.; Tong, Q.; Zhang, Z.; Wang, S.; Zheng, Y.; Liu, Q.; Qian, L.-B.; Chen, S.-Y.; Sun, J.; Cai, L. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin. Sci. 2017, 131, 1841–1857. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, H.; Chen, X.; Zou, Y.; Li, J.; Wang, L.; Wu, M.; Zang, J.; Yu, Y.; Zhuang, W.; et al. A small molecule activator of SIRT3 promotes deacetylation and activation of manganese superoxide dismutase. Free. Radic. Biol. Med. 2017, 112, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Kanwal, A.; Fang, Y.H.; Sharp, W.W.; Samant, S.; Arbiser, J.; Gupta, M.P. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget 2017, 8, 34082–34098. [Google Scholar] [CrossRef] [PubMed]
- Feldman, J.L.; Baeza, J.; Denu, J.M. Activation of the Protein Deacetylase SIRT6 by Long-chain Fatty Acids and Widespread Deacylation by Mammalian Sirtuins*. J. Biol. Chem. 2013, 288, 31350–31356. [Google Scholar] [CrossRef]
- Kalbas, D.; Liebscher, S.; Nowak, T.; Meleshin, M.; Pannek, M.; Popp, C.; Alhalabi, Z.; Bordusa, F.; Sippl, W.; Steegborn, C.; et al. Potent and Selective Inhibitors of Human Sirtuin 5. J. Med. Chem. 2018, 61, 2460–2471. [Google Scholar] [CrossRef]
- Cieślak, M.; Wojtczak, A.; Cieślak, M. Role of pro-inflammatory cytokines of pancreatic islets and prospects of elaboration of new methods for the diabetes treatment. Acta Biochim. Pol. 2015, 62, 15–21. [Google Scholar] [CrossRef]
- Yoshimatsu, G.; Kunnathodi, F.; Saravanan, P.B.; Shahbazov, R.; Chang, C.; Darden, C.M.; Zurawski, S.; Boyuk, G.; Kanak, M.A.; Levy, M.F.; et al. Pancreatic β-Cell–Derived IP-10/CXCL10 Isletokine Mediates Early Loss of Graft Function in Islet Cell Transplantation. Diabetes 2017, 66, 2857–2867. [Google Scholar] [CrossRef] [PubMed]
- Larsen, L.; Tonnesen, M.; Ronn, S.G.; Størling, J.; Jørgensen, S.; Mascagni, P.; Dinarello, C.A.; Billestrup, N.; Mandrup-Poulsen, T. Inhibition of histone deacetylases prevents cytokine-induced toxicity in beta cells. Diabetologia 2007, 50, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Susick, L.; Senanayake, T.; Veluthakal, R.; Woster, P.M.; Kowluru, A. A novel histone deacetylase inhibitor prevents IL-1beta induced metabolic dysfunction in pancreatic beta-cells. J. Cell Mol. Med. 2009, 13, 1877–1885. [Google Scholar] [CrossRef]
- Lewis, E.C.; Blaabjerg, L.; Størling, J.; Ronn, S.G.; Mascagni, P.; Dinarello, C.A.; Mandrup-Poulsen, T. The Oral Histone Deacetylase Inhibitor ITF2357 Reduces Cytokines and Protects Islet β Cells In Vivo and In Vitro. Mol. Med. 2011, 17, 369–377. [Google Scholar] [CrossRef]
- Tan, H.W.S.; Sim, A.Y.L.; Huang, S.L.; Leng, Y.; Long, Y.C. HC toxin (a HDAC inhibitor) enhances IRS1–Akt signalling and metabolism in mouse myotubes. J. Mol. Endocrinol. 2015, 55, 197–207. [Google Scholar] [CrossRef]
- Khan, S.; Komarya, S.K.; Jena, G. Phenylbutyrate and β-cell function: Contribution of histone deacetylases and ER stress inhibition. Epigenomics 2017, 9, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jena, G. Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-β1-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem. Toxicol. 2014, 73, 127–139. [Google Scholar] [CrossRef]
- Lundh, M.; Christensen, D.P.; Nielsen, M.D.; Richardson, S.; Dahllöf, M.S.; Skovgaard, T.; Berthelsen, J.; Dinarello, C.A.; Stevenazzi, A.; Mascagni, P.; et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia 2012, 55, 2421–2431. [Google Scholar] [CrossRef]
- Lee, H.-A.; Lee, E.; Do, G.Y.; Moon, E.-K.; Quan, F.-S.; Kim, I. Histone deacetylase inhibitor MGCD0103 protects the pancreas from streptozotocin-induced oxidative stress and β-cell death. Biomed. Pharmacother. 2019, 109, 921–929. [Google Scholar] [CrossRef]
- Chou, D.H.-C.; Holson, E.B.; Wagner, F.F.; Tang, A.J.; Maglathlin, R.L.; Lewis, T.A.; Schreiber, S.L.; Wagner, B.K. Inhibition of Histone Deacetylase 3 Protects Beta Cells from Cytokine-Induced Apoptosis. Chem. Biol. 2012, 19, 669–673. [Google Scholar] [CrossRef]
- Dahllöf, M.S.; Christensen, D.P.; Harving, M.; Wagner, B.K.; Mandrup-Poulsen, T.; Lundh, M. HDAC Inhibitor-Mediated Beta-Cell Protection Against Cytokine-Induced Toxicity Is STAT1 Tyr701 Phosphorylation Independent. J. Interf. Cytokine Res. 2015, 35, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.F.; Lundh, M.; Kaya, T.; McCarren, P.; Zhang, Y.-L.; Chattopadhyay, S.; Gale, J.P.; Galbo, T.; Fisher, S.L.; Meier, B.C.; et al. An Isochemogenic Set of Inhibitors to Define the Therapeutic Potential of Histone Deacetylases in β-Cell Protection. ACS Chem. Biol. 2016, 11, 363–374. [Google Scholar] [CrossRef]
- Pinho, A.V.; Bensellam, M.; Wauters, E.; Rees, M.; Giry-Laterriere, M.; Mawson, A.; Ly, L.Q.; Biankin, A.V.; Wu, J.; Laybutt, D.R.; et al. Pancreas-Specific Sirt1-Deficiency in Mice Compromises Beta-Cell Function without Development of Hyperglycemia. PLoS ONE 2015, 10, e0128012. [Google Scholar] [CrossRef]
- Lee, J.-H.; Song, M.-Y.; Song, E.-K.; Kim, E.-K.; Moon, W.S.; Han, M.-K.; Park, J.-W.; Kwon, K.-B.; Park, B.-H. Overexpression of SIRT1 Protects Pancreatic -Cells Against Cytokine Toxicity by Suppressing the Nuclear Factor- B Signaling Pathway. Diabetes 2008, 58, 344–351. [Google Scholar] [CrossRef]
- Yu, W.-C.; Chen, Y.-L.; Hwang, P.-A.; Chen, T.-H.; Chou, T.-C. Fucoidan ameliorates pancreatic β-cell death and impaired insulin synthesis in streptozotocin-treated β cells and mice via a Sirt-1-dependent manner. Mol. Nutr. Food Res. 2017, 61, 61. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, W.A.; Schaalan, M.F. Antidiabetic efficacy of lactoferrin in type 2 diabetic pediatrics; controlling impact on PPAR-γ, SIRT-1, and TLR4 downstream signaling pathway. Diabetol. Metab. Syndr. 2018, 10, 89. [Google Scholar] [CrossRef]
- Oh, Y.S.; Seo, E.; Park, K.; Jun, H.-S. Compound 19e, a Novel Glucokinase Activator, Protects against Cytokine-Induced Beta-Cell Apoptosis in INS-1 Cells. Front. Pharmacol. 2017, 8, 169. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, L.; Chen, J.F.; Shuai, X.; Xu, Y.; Ding, Y.; Zhang, J.; Yang, W.; Liang, X.; Su, D.; Yan, C. Artesunate protects pancreatic beta cells against cytokine-induced damage via SIRT1 inhibiting NF-κB activation. J. Endocrinol. Investig. 2015, 39, 83–91. [Google Scholar] [CrossRef]
- Caton, P.W.; Richardson, S.; Kieswich, J.; Bugliani, M.; Holland, M.L.; Marchetti, P.; Morgan, N.G.; Yaqoob, M.M.; Holness, M.J.; Sugden, M.C. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia 2013, 56, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Rahnasto-Rilla, M.; Tyni, J.; Huovinen, M.; Jarho, E.; Kulikowicz, T.; Ravichandran, S.; Bohr, V.A.; Ferrucci, L.; Lahtela-Kakkonen, M.; Moaddel, R. Natural polyphenols as sirtuin 6 modulators. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Iachettini, S.; Trisciuoglio, D.; Rotili, D.; Lucidi, A.; Salvati, E.; Zizza, P.; Di Leo, L.; Del Bufalo, D.; Ciriolo, M.R.; Leonetti, C.; et al. Pharmacological activation of SIRT6 triggers lethal autophagy in human cancer cells. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Kim, J.-H.; Lee, J.M.; Kim, J.-H.; Kim, K.R. Fluvastatin activates sirtuin 6 to regulate sterol regulatory element-binding proteins and AMP-activated protein kinase in HepG2 cells. Biochem. Biophys. Res. Commun. 2018, 503, 1415–1421. [Google Scholar] [CrossRef]
- Kumar, S.; Lombard, D.B. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 311–334. [Google Scholar] [CrossRef]
- Wong, C.K.; Wade-Vallance, A.K.; Luciani, D.S.; Brindle, P.K.; Lynn, F.C.; Gibson, W.T. The p300 and CBP Transcriptional Coactivators Are Required for β-Cell and α-Cell Proliferation. Diabetes 2017, 67, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Stiehl, D.P.; Fath, D.M.; Liang, D.; Jiang, Y.; Sang, N. Histone Deacetylase Inhibitors Synergize p300 Autoacetylation that Regulates Its Transactivation Activity and Complex Formation. Cancer Res. 2007, 67, 2256–2264. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-A.; Kang, S.-H.; Kim, M.; Lee, E.; Cho, H.-M.; Moon, E.-K.; Kim, I. Histone deacetylase inhibition ameliorates hypertension and hyperglycemia in a model of Cushing’s syndrome. Am. J. Physiol. Metab. 2018, 314, E39–E52. [Google Scholar] [CrossRef]
- Galmozzi, A.; Mitro, N.; Ferrari, A.; Gers, E.; Gilardi, F.; Godio, C.; Cermenati, G.; Gualerzi, A.; Donetti, E.; Rotili, D.; et al. Inhibition of Class I Histone Deacetylases Unveils a Mitochondrial Signature and Enhances Oxidative Metabolism in Skeletal Muscle and Adipose Tissue. Diabetes 2012, 62, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate Improves Insulin Sensitivity and Increases Energy Expenditure in Mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef]
- Kaiser, C.; James, S.R. Acetylation of insulin receptor substrate-1 is permissive for tyrosine phosphorylation. BMC Biol. 2004, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Muto, N.; Bando, M.; Itoh, Y.; Masaki, A.; Ri, M.; Ota, Y.; Nakagawa, H.; Iida, S.; Shirahige, K.; et al. Design, Synthesis, and Biological Activity of NCC149 Derivatives as Histone Deacetylase 8-Selective Inhibitors. ChemMedChem 2014, 9, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fossati, G.; Marchetti, C.; Modena, D.; Pozzi, P.; Reznikov, L.L.; Moras, M.L.; Azam, T.; Abbate, A.; Mascagni, P.; et al. Specific Inhibition of Histone Deacetylase 8 Reduces Gene Expression and Production of Proinflammatory Cytokines in Vitro and in Vivo. J. Biol. Chem. 2015, 290, 2368–2378. [Google Scholar] [CrossRef]
- Kee, H.J.; Ryu, Y.; Seok, Y.M.; Choi, S.Y.; Sun, S.; Kim, G.R.; Jeong, M.H. Selective inhibition of histone deacetylase 8 improves vascular hypertrophy, relaxation, and inflammation in angiotensin II hypertensive mice. Clin. Hypertens. 2019, 25, 13. [Google Scholar] [CrossRef]
- Karpac, J.; Jasper, H. Metabolic homeostasis: HDACs take center stage. Cell 2011, 145, 497–499. [Google Scholar] [CrossRef]
- Parra, M. Class IIa HDACs—New insights into their functions in physiology and pathology. FEBS J. 2014, 282, 1736–1744. [Google Scholar] [CrossRef]
- Raichur, S.; Teh, S.H.; Ohwaki, K.; Gaur, V.; Long, Y.C.; Hargreaves, M.; McGee, S.L.; Kusunoki, J. Histone deacetylase 5 regulates glucose uptake and insulin action in muscle cells. J. Mol. Endocrinol. 2012, 49, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nat. Cell Biol. 2007, 450, 712–716. [Google Scholar] [CrossRef]
- Park, S.-J.; Ahmad, F.; Um, J.-H.; Brown, A.L.; Xu, X.; Kang, H.; Ke, H.; Feng, X.; Ryall, J.; Philp, A.; et al. Specific Sirt1 Activator-mediated Improvement in Glucose Homeostasis Requires Sirt1-Independent Activation of AMPK. EBioMedicine 2017, 18, 128–138. [Google Scholar] [CrossRef]
- Nawaz, A.; Mehmood, A.; Kanatani, Y.; Kado, T.; Igarashi, Y.; Takikawa, A.; Yamamoto, S.; Okabe, K.; Nakagawa, T.; Yagi, K.; et al. Sirt1 activator induces proangiogenic genes in preadipocytes to rescue insulin resistance in diet-induced obese mice. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Dihingia, A.; Ozah, D.; Ghosh, S.; Sarkar, A.; Baruah, P.K.; Kalita, J.; Sil, P.C.; Manna, P. Vitamin K1 inversely correlates with glycemia and insulin resistance in patients with type 2 diabetes (T2D) and positively regulates SIRT1/AMPK pathway of glucose metabolism in liver of T2D mice and hepatocytes cultured in high glucose. J. Nutr. Biochem. 2018, 52, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; An, H.J.; Kim, D.H.; Lee, B.; Lee, H.J.; Ullah, S.; Kim, S.J.; Jeong, H.O.; Moon, K.M.; Lee, E.K.; et al. Novel SIRT1 activator MHY2233 improves glucose tolerance and reduces hepatic lipid accumulation in db/db mice. Bioorganic Med. Chem. Lett. 2018, 28, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Halperin-Sheinfeld, M.; Gertler, A.; Okun, E.; Sredni, B.; Cohen, H.Y. The Tellurium compound, AS101, increases SIRT1 level and activity and prevents type 2 diabetes. Aging 2012, 4, 436–447. [Google Scholar] [CrossRef]
- Choubey, A.; Kar, A.K.; Girdhar, K.; Chattopadhyay, T.; Dogra, S.; Kushwaha, S.; Medhi, B.; Bhansali, A.; Mantri, C.K.; Kolthur-Seetharam, U.; et al. Hyperinsulinemia promotes HMGB1 release leading to inflammation in-duced systemic insulin resistance: An interplay between pancreatic beta-cell and peripheral organs. bioRxiv 2019, 705107. [Google Scholar]
- Lantier, L.; Williams, A.S.; Hughey, C.C.; Bracy, D.P.; James, F.D.; Ansari, M.A.; Gius, D.; Wasserman, D.H. SIRT2 knockout exacerbates insulin resistance in high fat-fed mice. PLoS ONE 2018, 13, e0208634. [Google Scholar] [CrossRef]
- Watanabe, H.; Inaba, Y.; Kimura, K.; Matsumoto, M.; Kaneko, S.; Kasuga, M.; Inoue, H. Sirt2 facilitates hepatic glucose uptake by deacetylating glucokinase regulatory protein. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Zhang, M.; Pan, Y.; Dorfman, R.G.; Yin, Y.; Zhou, Q.; Huang, S.; Liu, J.; Zhao, S. Sirtinol promotes PEPCK1 degradation and inhibits gluconeogenesis by inhibiting deacetylase SIRT2. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Jing, E.; Emanuelli, B.; Hirschey, M.D.; Boucher, J.; Lee, K.Y.; Lombard, D.; Verdin, E.; Kahn, C.R. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2011, 108, 14608–14613. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Cui, Z.-J.; Sun, B.; Han, L.-P.; Li, C.-J.; Chen, L.-M. Celastrol attenuates oxidative stress in the skeletal muscle of diabetic rats by regulating the AMPK-PGC1α-SIRT3 signaling pathway. Int. J. Mol. Med. 2016, 37, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Govindarajulu, M.; Lynd, T.; Briggs, G.; Adamek, D.; Jones, E.; Heiner, J.; Majrashi, M.; Moore, T.; Amin, R.; et al. SIRT3 activator Honokiol attenuates β-Amyloid by modulating amyloidogenic pathway. PLoS ONE 2018, 13, e0190350. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Liu, Q.; Li, Q.; Liu, B.; Yang, Y.; Zhang, N. Berberine Reduces Pyruvate-driven Hepatic Glucose Production by Limiting Mitochondrial Import of Pyruvate through Mitochondrial Pyruvate Carrier 1. EBioMedicine 2018, 34, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.G.; Ramadori, G.; Ioris, R.M.; Galiè, M.; Berglund, E.D.; Coate, K.C.; Fujikawa, T.; Pucciarelli, S.; Moreschini, B.; Amici, A.; et al. Enhanced insulin sensitivity in skeletal muscle and liver by physiological overexpression of SIRT6. Mol. Metab. 2015, 4, 846–856. [Google Scholar] [CrossRef]
- Bae, E.J. Sirtuin 6, a possible therapeutic target for type 2 diabetes. Arch. Pharmacal Res. 2017, 40, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Parenti, M.D.; Grozio, A.; Bauer, I.; Galeno, L.; Damonte, P.; Millo, E.; Sociali, G.; Franceschi, C.; Ballestrero, A.; Bruzzone, S.; et al. Discovery of Novel and Selective SIRT6 Inhibitors. J. Med. Chem. 2013, 57, 4796–4804. [Google Scholar] [CrossRef]
- Sociali, G.; Magnone, M.; Ravera, S.; Damonte, P.; Vigliarolo, T.; Von Holtey, M.; Vellone, V.G.; Millo, E.; Caffa, I.; Cea, M.; et al. Pharmacological Sirt6 inhibition improves glucose tolerance in a type 2 diabetes mouse model. FASEB J. 2017, 31, 3138–3149. [Google Scholar] [CrossRef]
- Khanra, R.; Dewanjee, S.; Dua, T.K.; Sahu, R.; Gangopadhyay, M.; De Feo, V.; Zia-Ul-Haq, M. Abroma augusta L. (Malvaceae) leaf extract attenuates diabetes induced nephropathy and cardiomyopathy via inhibition of oxidative stress and inflammatory response. J. Transl. Med. 2015, 13, 1–14. [Google Scholar] [CrossRef]
- Rehan, M.K. Epigenetics and diabetes mellitus. Egypt. J. Intern. Med. 2016, 28, 39–51. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Zhen, J.; Zhang, C.; Wan, Q.; Liu, G.; Wei, X.; Zhang, Y.; Wang, Z.; Han, H.; et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014, 86, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.L.; De Azevedo, M.J.; Silveiro, S.P.; Canani, L.H.; Caramori, M.L.; Zelmanovitz, T. Diabetic Nephropathy: Diagnosis, Prevention, and Treatment. Diabetes Care 2004, 28, 164–176. [Google Scholar] [CrossRef]
- Dronavalli, S.; Duka, I.; Bakris, G.L. The pathogenesis of diabetic nephropathy. Nat. Clin. Pr. Endocrinol. Metab. 2008, 4, 444–452. [Google Scholar] [CrossRef]
- Advani, A.; Huang, Q.; Thai, K.; Advani, S.L.; White, K.E.; Kelly, D.J.; Yuen, D.A.; Connelly, K.A.; Marsden, P.A.; Gilbert, R.E. Long-Term Administration of the Histone Deacetylase Inhibitor Vorinostat Attenuates Renal Injury in Experimental Diabetes through an Endothelial Nitric Oxide Synthase-Dependent Mechanism. Am. J. Pathol. 2011, 178, 2205–2214. [Google Scholar] [CrossRef]
- Gilbert, R.E.; Huang, Q.; Thai, K.; Advani, S.L.; Lee, K.; Yuen, D.A.; Connelly, K.A.; Advani, A. Histone deacetylase inhibition attenuates diabetes-associated kidney growth: Potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 2011, 79, 1312–1321. [Google Scholar] [CrossRef]
- Dong, W.; Jia, Y.; Liu, X.; Zhang, H.; Li, T.; Huang, W.; Chen, X.; Wang, F.; Sun, W.; Wu, H. Sodium butyrate activates NRF2 to ameliorate diabetic nephropathy possibly via inhibition of HDAC. J. Endocrinol. 2017, 232, 71–83. [Google Scholar] [CrossRef]
- Du, Y.; Tang, G.; Yuan, W. Suppression of HDAC2 by sodium butyrate alleviates apoptosis of kidney cells in db/db mice and HG-induced NRK-52E cells. Int. J. Mol. Med. 2019, 45, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jena, G.; Tikoo, K. Sodium valproate ameliorates diabetes-induced fibrosis and renal damage by the inhibition of histone deacetylases in diabetic rat. Exp. Mol. Pathol. 2015, 98, 230–239. [Google Scholar] [CrossRef]
- Sun, X.-Y.; Qin, H.-J.; Zhang, Z.; Xu, Y.; Yang, X.-C.; Zhao, D.-M.; Li, X.-N.; Sun, L.-K. Valproate attenuates diabetic nephropathy through inhibition of endoplasmic reticulum stress-induced apoptosis. Mol. Med. Rep. 2016, 13, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Zhang, L.; Das, B.; Li, Z.; Liu, B.; Cai, G.; Chen, X.; Chuang, P.Y.; He, J.C.; Lee, K. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int. 2018, 93, 1330–1343. [Google Scholar] [CrossRef]
- Srivastava, S.P.; Li, J.; Kitada, M.; Fujita, H.; Yamada, Y.; Goodwin, J.E.; Kanasaki, K.; Koya, D. SIRT3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Lu, C.; Dai, Q.; Sheng, J.; Xu, M. SIRT3 Facilitates Amniotic Fluid Stem Cells to Repair Diabetic Nephropathy Through Protecting Mitochondrial Homeostasis by Modulation of Mitophagy. Cell. Physiol. Biochem. 2018, 46, 1508–1524. [Google Scholar] [CrossRef]
- Li, G.; Kim, H.-H. 201-LB: The Neuronal Sirt1 Regulate the Metabolic Imbalance in Ischemic Stroke. Diabetes 2019, 68, 201. [Google Scholar] [CrossRef]
- Lewien, P.; Levi, M.; Wang, X.; Myakala, K.; Wang, D.; Luo, Y. 340 Nad+-dependent deacetylase sirt3 activation inhibits diabetic kidney disease. J. Investig. Med. 2018, 66. [Google Scholar] [CrossRef]
- Ma, B.; Zhu, Z.; Zhang, J.; Ren, C.; Zhang, Q. Aucubin alleviates diabetic nephropathy by inhibiting NF-κB activation and inducing SIRT1/SIRT3-FOXO3a signaling pathway in high-fat diet/streptozotocin-induced diabetic mice. J. Funct. Foods 2020, 64, 103702. [Google Scholar] [CrossRef]
- Ji, L.; Chen, Y.; Wang, H.; Zhang, W.; He, L.; Wu, J.; Liu, Y. Overexpression of Sirt6 promotes M2 macrophage transformation, alleviating renal injury in diabetic nephropathy. Int. J. Oncol. 2019, 55, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Hill, M.; Sowers, J.R. Diabetic Cardiomyopathy. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Bagchi, R.A.; Weeks, K.L. Histone deacetylases in cardiovascular and metabolic diseases. J. Mol. Cell. Cardiol. 2019, 130, 151–159. [Google Scholar] [CrossRef]
- Zhang, L.; Du, J.; Yano, N.; Wang, H.; Zhao, Y.T.; Dubielecka, P.M.; Zhuang, S.; Chin, Y.E.; Qin, G.; Zhao, T.C. Sodium Butyrate Protects Against High Fat Diet-Induced Cardiac Dysfunction and Metabolic Disorders in Type II Diabetic Mice. J. Cell. Biochem. 2017, 118, 2395–2408. [Google Scholar] [CrossRef]
- Wu, Y.; Leng, Y.; Meng, Q.; Xue, R.; Zhao, B.; Zhan, L.; Xia, Z. Suppression of Excessive Histone Deacetylases Activity in Diabetic Hearts Attenuates Myocardial Ischemia/Reperfusion Injury via Mitochondria Apoptosis Pathway. J. Diabetes Res. 2017, 2017, 8208065. [Google Scholar] [CrossRef]
- Rabadiya, S.; Bhadada, S.; Dudhrejiya, A.; Vaishnav, D.; Patel, B. Magnesium valproate ameliorates type 1 diabetes and cardiomyopathy in diabetic rats through estrogen receptors. Biomed. Pharmacother. 2018, 97, 919–927. [Google Scholar] [CrossRef]
- Lee, T.-I.; Kao, Y.-H.; Tsai, W.-C.; Chung, C.-C.; Chen, Y.-C.; Chen, Y.-J. HDAC Inhibition Modulates Cardiac PPARs and Fatty Acid Metabolism in Diabetic Cardiomyopathy. PPAR Res. 2016, 2016, 5938740. [Google Scholar] [CrossRef]
- Lee, T.-W.; Bai, K.; Chen, Y.; Chung, C.; Tsai, W.; Tsao, S.; Kao, Y. Histone deacetylase inhibition of cardiac autophagy in rats on a high-fat diet with low-dose streptozotocin-induced type 2 diabetes mellitus. Mol. Med. Rep. 2017, 17, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Bocchi, L.; Motta, B.M.; Savi, M.; Vilella, R.; Meraviglia, V.; Rizzi, F.; Galati, S.; Buschini, A.; Lazzaretti, M.; Pramstaller, P.P.; et al. The Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid (SAHA) Restores Cardiomyocyte Contractility in a Rat Model of Early Diabetes. Int. J. Mol. Sci. 2019, 20, 1873. [Google Scholar] [CrossRef]
- Tate, M.; Willis, A.M.; Deo, M.; De Blasio, M.J.; Prakoso, D.; Kiriazis, H.; Du, X.-J.; Qian, H.-W.; McGee, S.; Gregorevic, P.; et al. P2841Cardiac-selective targeting of histone deacetylase 4 to limit experimental diabetic cardiomyopathy. Eur. Hear. J. 2018, 39, 2841. [Google Scholar] [CrossRef]
- Kronlage, M.; Dewenter, M.; Grosso, J.; Fleming, T.; Oehl, U.; Lehmann, L.H.; Falcão-Pires, I.; Leite-Moreira, A.F.; Volk, N.; Gröne, H.-J.; et al. O-GlcNAcylation of Histone Deacetylase 4 Protects the Diabetic Heart From Failure. Circulation 2019, 140, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, M.; Matta, M.J.; Sunderesan, N.R.; Gupta, M.P.; Periasamy, M. Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am. J. Physiol. Circ. Physiol. 2010, 298, H833–H843. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, Q.; Sun, Y.; Xing, Y.; Wang, Y.; Lu, X.; Bai, W.; Liu, X.; Zhao, Y. Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J. Cell. Mol. Med. 2014, 18, 1599–1611. [Google Scholar] [CrossRef]
- Ma, S.; Feng, J.; Zhang, R.; Chen, J.; Han, D.; Li, X.; Yang, B.; Li, X.; Fan, M.; Li, C.; et al. SIRT1 Activation by Resveratrol Alleviates Cardiac Dysfunction via Mitochondrial Regulation in Diabetic Cardiomyopathy Mice. Oxidative Med. Cell. Longev. 2017, 2017, 4602715. [Google Scholar] [CrossRef]
- Fang, W.-J.; Wang, C.-J.; He, Y.; Zhou, Y.-L.; Peng, X.-D.; Liu, S.-K. Resveratrol alleviates diabetic cardiomyopathy in rats by improving mitochondrial function through PGC-1α deacetylation. Acta Pharmacol. Sin. 2017, 39, 59–73. [Google Scholar] [CrossRef]
- Bagul, P.K.; Katare, P.B.; Bugga, P.; Dinda, A.K.; Banerjee, S.K. SIRT-3 Modulation by Resveratrol Improves Mitochondrial Oxidative Phosphorylation in Diabetic Heart through Deacetylation of TFAM. Cells 2018, 7, 235. [Google Scholar] [CrossRef] [PubMed]
- Bagul, P.K.; Deepthi, N.; Sultana, R.; Banerjee, S.K. Resveratrol ameliorates cardiac oxidative stress in diabetes through deacetylation of NFkB-p65 and histone 3. J. Nutr. Biochem. 2015, 26, 1298–1307. [Google Scholar] [CrossRef]
- Chen, T.; Li, J.; Liu, J.; Li, N.; Wang, S.; Liu, H.; Zeng, M.; Zhang, Y.; Bu, P. Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF-β/Smad3 pathway. Am. J. Physiol. Circ. Physiol. 2015, 308, H424–H434. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Gao, B.; Li, N.; Wang, J.; Qiu, C.; Zhang, G.; Liu, M.; Zhang, R.; Li, C.; Ji, G.; et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Jiang, C.; Zhang, M.; Jin, J.; Ge, S.; Wang, X. Phloretin protects against cardiac damage and remodeling via restoring SIRT1 and anti-inflammatory effects in the streptozotocin-induced diabetic mouse model. Aging 2019, 11, 2822–2835. [Google Scholar] [CrossRef]
- Rizk, S.M.; El-Maraghy, S.A.; Nassar, N.N. A Novel Role for SIRT-1 in L-Arginine Protection against STZ Induced Myocardial Fibrosis in Rats. PLoS ONE 2014, 9, e114560. [Google Scholar] [CrossRef]
- Zeng, H.; He, X.; Hou, X.; Li, L.; Chen, J.-X. Apelin gene therapy increases myocardial vascular density and ameliorates diabetic cardiomyopathy via upregulation of sirtuin 3. Am. J. Physiol. Circ. Physiol. 2014, 306, H585–H597. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, S.; Cheng, Z.; Xiong, Z.; Lv, J.; Yang, Z.; Li, T.; Jiang, S.; Gu, J.; Sun, D.; et al. Polydatin ameliorates diabetic cardiomyopathy via Sirt3 activation. Biochem. Biophys. Res. Commun. 2017, 493, 1280–1287. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Liu, X.; Ryu, D.; Nelson, O.D.; Stupinski, J.A.; Li, Z.; Chen, W.; Zhang, S.; Weiss, R.S.; Locasale, J.W.; et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. USA 2016, 113, 4320–4325. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhan, L.; Zhou, Q.-Y.; Zhang, L.-L.; Chen, X.-M.; Hu, X.-M.; Yuan, X.-C. SIRT2 regulates microtubule stabilization in diabetic cardiomyopathy. Eur. J. Pharmacol. 2015, 764, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, D.; Liu, Y.; Crosson, C.E.; Ablonczy, Z. Histone Deacetylase Inhibition Restores Retinal Pigment Epithelium Function in Hyperglycemia. PLoS ONE 2016, 11, e0162596. [Google Scholar] [CrossRef]
- Zhong, Q.; Kowluru, R.A. Role of histone acetylation in the development of diabetic retinopathy and the metabolic memory phenomenon. J. Cell. Biochem. 2010, 110, 1306–1313. [Google Scholar] [CrossRef]
- Kadiyala, C.S.R.; Zheng, L.; Du, Y.; Yohannes, E.; Kao, H.-Y.; Miyagi, M.; Kern, T.S. Acetylation of Retinal Histones in Diabetes Increases Inflammatory Proteins. J. Biol. Chem. 2012, 287, 25869–25880. [Google Scholar] [CrossRef]
- Abouhish, H.A.; Thounaojam, M.C.; Jadeja, R.N.; Martin, P.M.; Khriza, M.M.; Bartoli, M. Activation of Histone Deacetylase 6 (HDAC6) in the diabetic retina and in retinal endothelial cells exposed to glucidic stress, promotes oxidative stress through suppression of the thioredoxin system. FASEB J. 2019, 33, lb337. [Google Scholar] [CrossRef]
- Yuan, H.; Li, H.; Yu, P.; Fan, Q.; Zhang, X.; Huang, W.; Shen, J.; Cui, Y.; Zhou, W. Involvement of HDAC6 in ischaemia and reperfusion-induced rat retinal injury. BMC Ophthalmol. 2018, 18, 300. [Google Scholar] [CrossRef]
- Cai, X.; Li, J.; Wang, M.; She, M.; Tang, Y.; Li, J.; Li, H.; Hui, H. GLP-1 Treatment Improves Diabetic Retinopathy by Alleviating Autophagy through GLP-1R-ERK1/2-HDAC6 Signaling Pathway. Int. J. Med. Sci. 2017, 14, 1203–1212. [Google Scholar] [CrossRef]
- Xie, M.; Tian, J.; Luo, Y.; Wei, L.; Lin, S.; Tang, S. Effects of 5-aza-2’-deoxycytidine and trichostatin A on high glucose- and interleukin-1beta-induced secretory mediators from human retinal endothelial cells and retinal pigment epithelial cells. Mol. Vis. 2014, 20, 1411–1421. [Google Scholar] [PubMed]
- Maghbooli, Z.; Emamgholipour, S.; Aliakbar, S.; Amini, M.; Gorgani-Firuzjaee, S.; Hossein-Nezhad, A. Differential expressions of SIRT1, SIRT3, and SIRT4 in peripheral blood mononuclear cells from patients with type 2 diabetic retinopathy. Arch. Physiol. Biochem. 2020, 126, 1–6. [Google Scholar] [CrossRef]
- Mortuza, R.; Feng, B.; Chakrabarti, S. SIRT 1 reduction causes renal and retinal injury in diabetes through endothelin 1 and transforming growth factor β1. J. Cell. Mol. Med. 2015, 19, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Santos, J.M.; Zhong, Q. Sirt1, a Negative Regulator of Matrix Metalloproteinase-9 in Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2014, 55, 5653–6560. [Google Scholar] [CrossRef] [PubMed]
- Karbasforooshan, H.; Karimi, G. The role of SIRT1 in diabetic retinopathy. Biomed. Pharmacother. 2018, 97, 190–194. [Google Scholar] [CrossRef]
- Hammer, S.S.; Vieira, C.P.; McFarland, D.; Sandler, M.; Levitsky, Y.; Dorweiler, T.F.; Lydic, T.A.; Asare-Bediako, B.; Adu-Agyeiwaah, Y.; Sielski, M.S.; et al. Fasting and fasting-mimicking treatment activate SIRT1/LXRα and alleviate diabetes-induced systemic and microvascular dysfunction. Diabetologia 2021, 1–16. [Google Scholar] [CrossRef]
- Li, J.; Yu, S.; Ying, J.; Shi, T.; Wang, P. Resveratrol Prevents ROS-Induced Apoptosis in High Glucose-Treated Retinal Capillary Endothelial Cells via the Activation of AMPK/Sirt1/PGC-1α Pathway. Oxidative Med. Cell. Longev. 2017, 2017, 7584691. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Patel, P.; Steinle, J.J. PKA regulates HMGB1 through activation of IGFBP-3 and SIRT1 in human retinal endothelial cells cultured in high glucose. Inflamm. Res. 2018, 67, 1013–1019. [Google Scholar] [CrossRef]
- Chen, N.; Jiang, K.; Yan, G.G. Effect of fenofibrate on diabetic retinopathy in rats via SIRT1/NF-kappaB signaling pathway. Eur. Rev. Med. Pharmacol Sci. 2019, 23, 8630–8636. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zheng, Z.; Gu, Q.; Chen, X.; Liu, X.; Xu, X. Deacetylation of MnSOD by PARP-regulated SIRT3 protects retinal capillary endothelial cells from hyperglycemia-induced damage. Biochem. Biophys. Res. Commun. 2016, 472, 425–431. [Google Scholar] [CrossRef]
- Lin, J.B.; Lin, J.B.; Chen, H.C.; Chen, T.; Apte, R.S. Combined SIRT3 and SIRT5 deletion is associated with inner retinal dysfunction in a mouse model of type 1 diabetes. Sci. Rep. 2019, 9, 3799. [Google Scholar] [CrossRef] [PubMed]
- Zorrilla-Zubilete, M.A.; Yeste, A.; Quintana, F.J.; Toiber, D.; Mostoslavsky, R.; Silberman, D.M. Epigenetic control of early neurodegenerative events in diabetic retinopathy by the histone deacetylase SIRT6. J. Neurochem. 2018, 144, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Golonzhka, O.; Jarpe, M.B. HDAC Inhibitors for the Treatment of Diabetic Peripheral Neuropathy. US Patent US10040769B2, 7 August 2018. [Google Scholar]
- Du, W.; Wang, N.; Li, F.; Jia, K.; An, J.; Liu, Y.; Wang, Y.; Zhu, L.; Zhao, S.; Hao, J. STAT3 phosphorylation mediates high glucose—impaired cell autophagy in an HDAC1-dependent and -independent manner in Schwann cells of diabetic peripheral neuropathy. FASEB J. 2019, 33, 8008–8021. [Google Scholar] [CrossRef]
- Zhou, C.-H.; Zhang, M.-X.; Zhou, S.-S.; Li, H.; Gao, J.; Du, L.; Yin, X.-X. SIRT1 attenuates neuropathic pain by epigenetic regulation of mGluR1/5 expressions in type 2 diabetic rats. Pain 2017, 158, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, K.; Muragundla, A.; Demarest, T.G.; Choi, J.; Sagi, A.R.; Najimi, N.; Kumar, P.; Singh, A.; Ho, C.-Y.; Fiskum, G.; et al. mGluR2/3 activation of the SIRT1 axis preserves mitochondrial function in diabetic neuropathy. Ann. Clin. Transl. Neurol. 2017, 4, 844–858. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Salimian, M.; Konduru, S.R.; Choi, J.; Kumar, P.; Long, A.; Klimova, N.; Ho, C.-Y.; Kristian, T.; Russell, J.W. Overexpression of Sirtuin 1 protein in neurons prevents and reverses experimental diabetic neuropathy. Brain 2019, 142, 3737–3752. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Shang, Y.; Zhang, R.; Gao, X.; Zeng, Q. A SIRT1 agonist reduces cognitive decline in type 2 diabetic rats through antioxidative and anti-inflammatory mechanisms. Mol. Med. Rep. 2018, 19, 1040–1048. [Google Scholar] [CrossRef]
- Yerra, V.G.; Kalvala, A.K.; Kumar, A. Isoliquiritigenin reduces oxidative damage and alleviates mitochondrial impairment by SIRT1 activation in experimental diabetic neuropathy. J. Nutr. Biochem. 2017, 47, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Negi, G.; Sharma, S. Neuroprotection by Resveratrol in Diabetic Neuropathy: Concepts & Mechanisms. Curr. Med. Chem. 2013, 20, 4640–4645. [Google Scholar] [CrossRef]
- Shi, X.; Pi, L.; Zhou, S.; Li, X.; Min, F.; Wang, S.; Liu, Z.; Wu, J. Activation of Sirtuin 1 Attenuates High Glucose-Induced Neuronal Apoptosis by Deacetylating p53. Front. Endocrinol. 2018, 9, 274. [Google Scholar] [CrossRef]
- Li, H.-Y.; Wang, X.-C.; Xu, Y.-M.; Luo, N.-C.; Luo, S.; Hao, X.-Y.; Cheng, S.-Y.; Fang, J.-S.; Wang, Q.; Zhang, S.-J.; et al. Berberine Improves Diabetic Encephalopathy Through the SIRT1/ER Stress Pathway indb/dbMice. Rejuvenation Res. 2018, 21, 200–209. [Google Scholar] [CrossRef]
- Guzyk, M.M.; Tykhonenko, T.M.; Dyakun, K.O.; Yanitska, L.V.; Pryvrotska, I.B.; Kuchmerovska, T.M. Altered sirtuins 1 and 2 expression in the brain of rats induced by experimental diabetes and the ways of its correction. Ukr. Biochem. J. 2019, 91, 21–29. [Google Scholar] [CrossRef]
- Schartner, E.; Saleh, A.; Da Silva, R.V.; Chowdhury, S.R.; Smith, D.; Fernyhough, P. SIRT2 is Required for Axon Regen-eration in Adult Sensory Neurons and High Glucose Concentration Reduces its Expression in Diabetic Neuropathy. Can. J. Diabetes 2014, 38, S62. [Google Scholar] [CrossRef]
- Schartner, E.; Sabbir, M.G.; Saleh, A.; Silva, R.V.; Chowdhury, S.R.; Smith, D.R.; Fernyhough, P. High glucose concentration suppresses a SIRT2 regulated pathway that enhances neurite outgrowth in cultured adult sensory neurons. Exp. Neurol. 2018, 309, 134–147. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, L.; Yang, X.; Huang, H.; Huang, Z.; Shi, L.; Zhang, H.; Du, G. Salvianolic Acid A Protects the Peripheral Nerve Function in Diabetic Rats through Regulation of the AMPK-PGC1α-Sirt3 Axis. Molecules 2012, 17, 11216–11228. [Google Scholar] [CrossRef]
- Magnifico, S.; Saias, L.; Deleglise, B.; Duplus, E.; Kilinc, D.; Miquel, M.; Viovy, J.-L.; Brugg, B.; Peyrin, J. NAD + acts on mitochondrial SirT3 to prevent axonal caspase activation and axonal degeneration. FASEB J. 2013, 27, 4712–4722. [Google Scholar] [CrossRef]
- Kolluru, G.; Bir, S.C.; Kevil, C.G. Endothelial Dysfunction and Diabetes: Effects on Angiogenesis, Vascular Remodeling, and Wound Healing. Int. J. Vasc. Med. 2012, 2012, 918267. [Google Scholar] [CrossRef]
- Jin, G.; Bausch, D.; Knightly, T.; Liu, Z.; Li, Y.; Liu, B.; Lu, J.; Chong, W.; Velmahos, G.C.; Alam, H.B. Histone deacetylase inhibitors enhance endothelial cell sprouting angiogenesis in vitro. Surgery 2011, 150, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Manea, S.-A.; Antonescu, M.-L.; Fenyo, I.M.; Raicu, M.; Simionescu, M.; Manea, A. Epigenetic regulation of vascular NADPH oxidase expression and reactive oxygen species production by histone deacetylase-dependent mechanisms in experimental diabetes. Redox Biol. 2018, 16, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jiang, Z.; Zhang, H.; Liang, W.; Huang, W.; Zhang, H.; Li, Y.; Wang, Z.; Wang, J.; Jia, Y.; et al. Sodium butyrate attenuates diabetes-induced aortic endothelial dysfunction via P300-mediated transcriptional activation of Nrf2. Free. Radic. Biol. Med. 2018, 124, 454–465. [Google Scholar] [CrossRef]
- Zhou, S.; Chen, H.-Z.; Wan, Y.-Z.; Zhang, Q.-J.; Wei, Y.-S.; Huang, S.; Liu, J.-J.; Lu, Y.-B.; Zhang, Z.-Q.; Yang, R.-F.; et al. Repression of P66Shc Expression by SIRT1 Contributes to the Prevention of Hyperglycemia-Induced Endothelial Dysfunction. Circ. Res. 2011, 109, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kim, Y.-R.; Vikram, A.; Naqvi, A.; Li, Q.; Kassan, M.; Kumar, V.; Bachschmid, M.M.; Jacobs, J.S.; Kumar, A.; et al. Sirtuin1-regulated lysine acetylation of p66Shc governs diabetes-induced vascular oxidative stress and endothelial dysfunction. Proc. Natl. Acad. Sci. USA 2017, 114, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wan, Y.; Zhou, S.; Lu, Y.; Zhang, Z.; Zhang, R.; Chen, F.; Hao, D.; Zhao, X.; Guo, Z.; et al. Endothelium-specific SIRT1 overexpression inhibits hyperglycemia-induced upregulation of vascular cell senescence. Sci. China Life Sci. 2012, 55, 467–473. [Google Scholar] [CrossRef]
- Wang, W.; Shang, C.; Zhang, W.; Jin, Z.; Yao, F.; He, Y.; Wang, B.; Li, Y.; Zhang, J.; Lin, R. Hydroxytyrosol NO regulates oxidative stress and NO production through SIRT1 in diabetic mice and vascular endothelial cells. Phytomedicine 2019, 52, 206–215. [Google Scholar] [CrossRef]
- Mortuza, R.; Chen, S.; Feng, B.; Sen, S.; Chakrabarti, S. High Glucose Induced Alteration of SIRTs in Endothelial Cells Causes Rapid Aging in a p300 and FOXO Regulated Pathway. PLoS ONE 2013, 8, e54514. [Google Scholar] [CrossRef]
- Zhao, H.; Ma, T.; Fan, B.; Yang, L.; Han, C.; Luo, J.; Kong, L. Protective effect of trans-?-viniferin against high glucose-induced oxidative stress in human umbilical vein endothelial cells through the SIRT1 pathway. Free. Radic. Res. 2015, 50, 1–16. [Google Scholar] [CrossRef]
- Qin, R.; Zhang, L.; Lin, D.; Xiao, F.; Guo, L. Sirt1 inhibits HG-induced endothelial injury: Role of Mff-based mitochondrial fission and F-actin homeostasis-mediated cellular migration. Int. J. Mol. Med. 2019, 44, 89–102. [Google Scholar] [CrossRef]
- Orimo, M.; Minamino, T.; Miyauchi, H.; Tateno, K.; Okada, S.; Moriya, J.; Komuro, I. Protective Role of SIRT1 in Diabetic Vascular Dysfunction. Arter. Thromb. Vasc. Biol. 2009, 29, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, L.; Zhou, C.; Lin, N.; Liu, A. Sirt 1 activator inhibits the AGE-induced apoptosis and p53 acetylation in human vascular endothelial cells. J. Toxicol. Sci. 2015, 40, 615–624. [Google Scholar] [CrossRef]
- Wu, H.; Wu, J.; Zhou, S.; Huang, W.; Li, Y.; Zhang, H.; Wang, J.; Jia, Y. SRT2104 attenuates diabetes-induced aortic endothelial dysfunction via inhibition of P53. J. Endocrinol. 2018, 237, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Zhao, A.; Li, J. SIRT1 activation inhibits hyperglycemia-induced apoptosis by reducing oxidative stress and mitochondrial dysfunction in human endothelial cells. Mol. Med. Rep. 2017, 16, 3331–3338. [Google Scholar] [CrossRef] [PubMed]
- El Assar, M.; Angulo, J.; Rodríguez-Mañas, L. Diabetes and ageing-induced vascular inflammation. J. Physiol. 2016, 594, 2125–2146. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Shi, M.; Li, L.; Liu, J.; Chen, B.; Chen, Y.; An, X.; Liu, S.; Luo, R.; Long, D.; et al. Mesenchymal stem cell-conditioned media ameliorate diabetic endothelial dysfunction by improving mitochondrial bioenergetics via the Sirt1/AMPK/PGC-1α pathway. Clin. Sci. 2016, 130, 2181–2198. [Google Scholar] [CrossRef]
- Yuen, D.A.; Zhang, Y.; Thai, K.; Spring, C.; Chan, L.; Guo, X.; Advani, A.; Sivak, J.M.; Gilbert, R.E. Angiogenic Dysfunction in Bone Marrow-Derived Early Outgrowth Cells from Diabetic Animals Is Attenuated by SIRT1 Activation. Stem Cells Transl. Med. 2012, 1, 921–926. [Google Scholar] [CrossRef]
- Liu, R.; Liu, H.; Ha, Y.; Tilton, R.G.; Zhang, W. Oxidative Stress Induces Endothelial Cell Senescence via Downregulation of Sirt6. BioMed Res. Int. 2014, 2014, 1–13. [Google Scholar] [CrossRef]
- Shen, J.; Ma, W.; Liu, Y. Deacetylase SIRT6 deaccelerates endothelial senescence. Cardiovasc. Res. 2012, 97, 391–392. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Li, W.; Zhang, J.; Che, Y.; Cui, X.; Sun, B.; Zhao, G. Loss of histone deacetylase 2 inhibits oxidative stress induced by high glucose via the HO-1/SIRT1 pathway in endothelial progenitor cells. Gene 2018, 678, 1–7. [Google Scholar] [CrossRef]
- Bashmakov, Y.K.; Petyaev, I.M. Old Drug Acquires New Target: Metformin and Sirt1. J. Diabetes Metab. 2011, 2, 1000107. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, G.; Tang, H.; Dong, L.; Gao, C.; Yang, X.; Peng, Y.; Xu, Y. Influence of SIRT1 polymorphisms for diabetic foot susceptibility and severity. Medicine 2018, 97, e11455. [Google Scholar] [CrossRef]
- Davis, F.M.; Kimball, A.; Boniakowski, A.; Gallagher, K. Dysfunctional Wound Healing in Diabetic Foot Ulcers: New Crossroads. Curr. Diabetes Rep. 2018, 18, 2. [Google Scholar] [CrossRef]
- Alrdahe, S.; Al Sadoun, H.; Torbica, T.; McKenzie, E.A.; Bowling, F.L.; Boulton, A.J.M.; Mace, K.A. Dysregulation of macrophage development and phenotype in diabetic human macrophages can be rescued by Hoxa3 protein transduction. PLoS ONE 2019, 14, e0223980. [Google Scholar] [CrossRef]
- Yin, J.; Fushin, X.Y. Role of Histone Acetylation in Corneal Epithelial Wound Healing and Diabetic Keratopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5307. [Google Scholar]
- Keshava, R.; Gope, R. Sodium Butyrate Plus EGF and PDGF-BB Aids Cutaneous Wound Healing in Diabetic Mice. Adv. Biol. 2015, 2015, 527231. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, X.; Shi, D.; Chen, P.; Yu, Y.; Yang, L.; Xie, L. Overexpression of SIRT1 Promotes High Glucose–Attenuated Corneal Epithelial Wound Healing via p53 Regulation of the IGFBP3/IGF-1R/AKT Pathway. Investig. Opthalmol. Vis. Sci. 2013, 54, 3806–3814. [Google Scholar] [CrossRef] [PubMed]
- Boniakowski, A.E.; Kimball, A.; Joshi, A.; Kunkel, S.; Gallagher, K. PC200 A Deacetylase Enzyme, Sirtuin 3, Plays a Major Role in Macrophage Inflammation and Wound Healing. J. Vasc. Surg. 2017, 65, 193S–194S. [Google Scholar] [CrossRef][Green Version]
- Thandavarayan, R.A.; Garikipati, V.N.S.; Joladarashi, D.; Babu, S.S.; Jeyabal, P.; Verma, S.K.; Mackie, A.R.; Khan, M.; Arumugam, S.; Watanabe, K.; et al. Sirtuin-6 deficiency exacerbates diabetes-induced impairment of wound healing. Exp. Dermatol. 2015, 24, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Spallotta, F.; Cencioni, C.; Straino, S.; Nanni, S.; Rosati, J.; Artuso, S.; Manni, I.; Colussi, C.; Piaggio, G.; Martelli, F.; et al. A Nitric Oxide-dependent Cross-talk between Class I and III Histone Deacetylases Accelerates Skin Repair*. J. Biol. Chem. 2013, 288, 11004–11012. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Sun, J.; Chen, G.; Niu, C.; Wang, Y.; Zhao, C.; Huang, H.; Huang, S.; Liang, Y.; Shen, Y.; et al. Resveratrol Promotes Diabetic Wound Healing via SIRT1-FOXO1-c-Myc Signaling Pathway-Mediated Angiogenesis. Front. Pharmacol. 2019, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Chen, X.; Su, X.; Su, X.; Hou, C. A novel dressing seeded with embryonic artery CD133+ cells and loaded with the Sirt1 agonist SRT1720 accelerates the healing of diabetic ischemic ulcers. Exp. Ther. Med. 2018, 15, 5243–5250. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, G.; Han, F.; Wang, K.; Bai, X.; Jia, Y.; Li, Z.; Cai, W.; Zhang, W.; Su, L.; et al. SIRT1 activation promotes angiogenesis in diabetic wounds by protecting endothelial cells against oxidative stress. Arch. Biochem. Biophys. 2019, 661, 117–124. [Google Scholar] [CrossRef]
- Lucas, T.; Schäfer, F.; Müller, P.; Eming, S.A.; Heckel, A.; Dimmeler, S. Light-inducible antimiR-92a as a therapeutic strategy to promote skin repair in healing-impaired diabetic mice. Nat. Commun. 2017, 8, 15162. [Google Scholar] [CrossRef]
- Manea, S.-A.; Vlad, M.-L.; Fenyo, I.M.; Lazar, A.-G.; Raicu, M.; Muresian, H.; Simionescu, M.; Manea, A. Pharmacological inhibition of histone deacetylase reduces NADPH oxidase expression, oxidative stress and the progression of atherosclerotic lesions in hypercholesterolemic apolipoprotein E-deficient mice; potential implications for human atherosclerosis. Redox Biol. 2020, 28, 101338. [Google Scholar] [CrossRef]
- Cao, Q.; Rong, S.; Repa, J.J.; Clair, R.S.; Parks, J.S.; Mishra, N. Histone Deacetylase 9 Represses Cholesterol Efflux and Alternatively Activated Macrophages in Atherosclerosis Development. Arter. Thromb. Vasc. Biol. 2014, 34, 1871–1879. [Google Scholar] [CrossRef]
- Malhotra, P.; Muthusamy, S.; Gavin, D.P.; Luo, A.Y.; Saksena, S.; Gill, R.K.; Dudeja, P.K.; Alrefai, W.A. Mo1760 Histone Deacetylase Inhibition Decreases the Expression of Intestinal Cholesterol Transporter Niemann Pick-C1-Like 1 (NPC1L1). Gastroenterology 2014, 146, 653. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection. Antioxid. Redox Signal. 2018, 28, 711–732. [Google Scholar] [CrossRef]
- Li, Y.; Ni, J.; Guo, R.; Li, W. In Patients with Coronary Artery Disease and Type 2 Diabetes, SIRT1 Expression in Circulating Mononuclear Cells Is Associated with Levels of Inflammatory Cytokines but Not with Coronary Lesions. Biomed. Res. Int. 2016, 2016, 8734827. [Google Scholar] [CrossRef]
- Escribano-Lopez, I.; de Maranon, A.M.; Iannantuoni, F.; Lopez-Domenech, S.; Abad-Jimenez, Z.; Diaz, P.; Sola, E.; Apostolova, N.; Rocha, M.; Victor, V.M. The Mitochondrial Antioxidant SS-31 Modulates Oxidative Stress, Endoplasmic Reticulum Stress, and Autophagy in Type 2 Diabetes. J. Clin. Med. 2019, 8, 1322. [Google Scholar] [CrossRef]
- Bartoli-Leonard, F.; Wilkinson, F.L.; Schiro, A.; Inglott, F.S.; Alexander, M.Y.; Weston, R. Suppression of SIRT1 in Diabetic Conditions Induces Osteogenic Differentiation of Human Vascular Smooth Muscle Cells via RUNX2 Signalling. Sci. Rep. 2019, 9, 878. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, J.; Huang, X.; Li, Z.; Liu, P. Deletion of sirtuin 6 accelerates endothelial dysfunction and atherosclerosis in apolipoprotein E-deficient mice. Transl. Res. 2016, 172, 18–29.e12. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, M.L.; Rizzo, M.R.; Barbieri, M.; Paolisso, P.; D’Onofrio, N.; Giovane, A.; Siniscalchi, M.; Minicucci, F.; Sardu, C.; D’Andrea, D.; et al. Sirtuin 6 expression and inflammatory activity in diabetic atherosclerotic plaques: Effects of incretin treatment. Diabetes 2015, 64, 1395–1406. [Google Scholar] [CrossRef]
- Fessler, E.B.; Chibane, F.L.; Wang, Z.; Chuang, D.M. Potential roles of HDAC inhibitors in mitigating ischemia-induced brain damage and facilitating endogenous regeneration and recovery. Curr. Pharm. Des. 2013, 19, 5105–5120. [Google Scholar] [CrossRef]
- Zhao, B.; Yuan, Q.; Hou, J.B.; Xia, Z.Y.; Zhan, L.Y.; Li, M.; Jiang, M.; Gao, W.W.; Liu, L. Inhibition of HDAC3 Ameliorates Cerebral Ischemia Reperfusion Injury in Diabetic Mice In Vivo and In Vitro. J. Diabetes Res. 2019, 2019, 8520856. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, J.; Shaik, N.F.; Yi, B.; Wei, X.; Yang, X.F.; Naik, U.P.; Summer, R.; Yan, G.; Xu, X.; et al. The histone deacetylase inhibitor tubacin mitigates endothelial dysfunction by up-regulating the expression of endothelial nitric oxide synthase. J. Biol. Chem. 2019, 294, 19565–19576. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Jackson, C.W.; Khoury, N.; Escobar, I.; Perez-Pinzon, M.A. Brain SIRT1 Mediates Metabolic Homeostasis and Neuroprotection. Front. Endocrinol. 2018, 9, 702. [Google Scholar] [CrossRef]
- Chen, L.; Cao, J.; Cao, D.; Wang, M.; Xiang, H.; Yang, Y.; Ying, T.; Cong, H. Protective effect of dexmedetomidine against diabetic hyperglycemia-exacerbated cerebral ischemia/reperfusion injury: An in vivo and in vitro study. Life Sci. 2019, 235, 116553. [Google Scholar] [CrossRef]
- Fusco, R.; Scuto, M.; Cordaro, M.; D’Amico, R.; Gugliandolo, E.; Siracusa, R.; Peritore, A.F.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; et al. N-Palmitoylethanolamide-Oxazoline Protects against Middle Cerebral Artery Occlusion Injury in Diabetic Rats by Regulating the SIRT1 Pathway. Int. J. Mol. Sci. 2019, 20, 4845. [Google Scholar] [CrossRef]
- Zheng, J.; Shi, L.; Liang, F.; Xu, W.; Li, T.; Gao, L.; Sun, Z.; Yu, J.; Zhang, J. Sirt3 Ameliorates Oxidative Stress and Mitochondrial Dysfunction After Intracerebral Hemorrhage in Diabetic Rats. Front. Neurosci. 2018, 12, 414. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
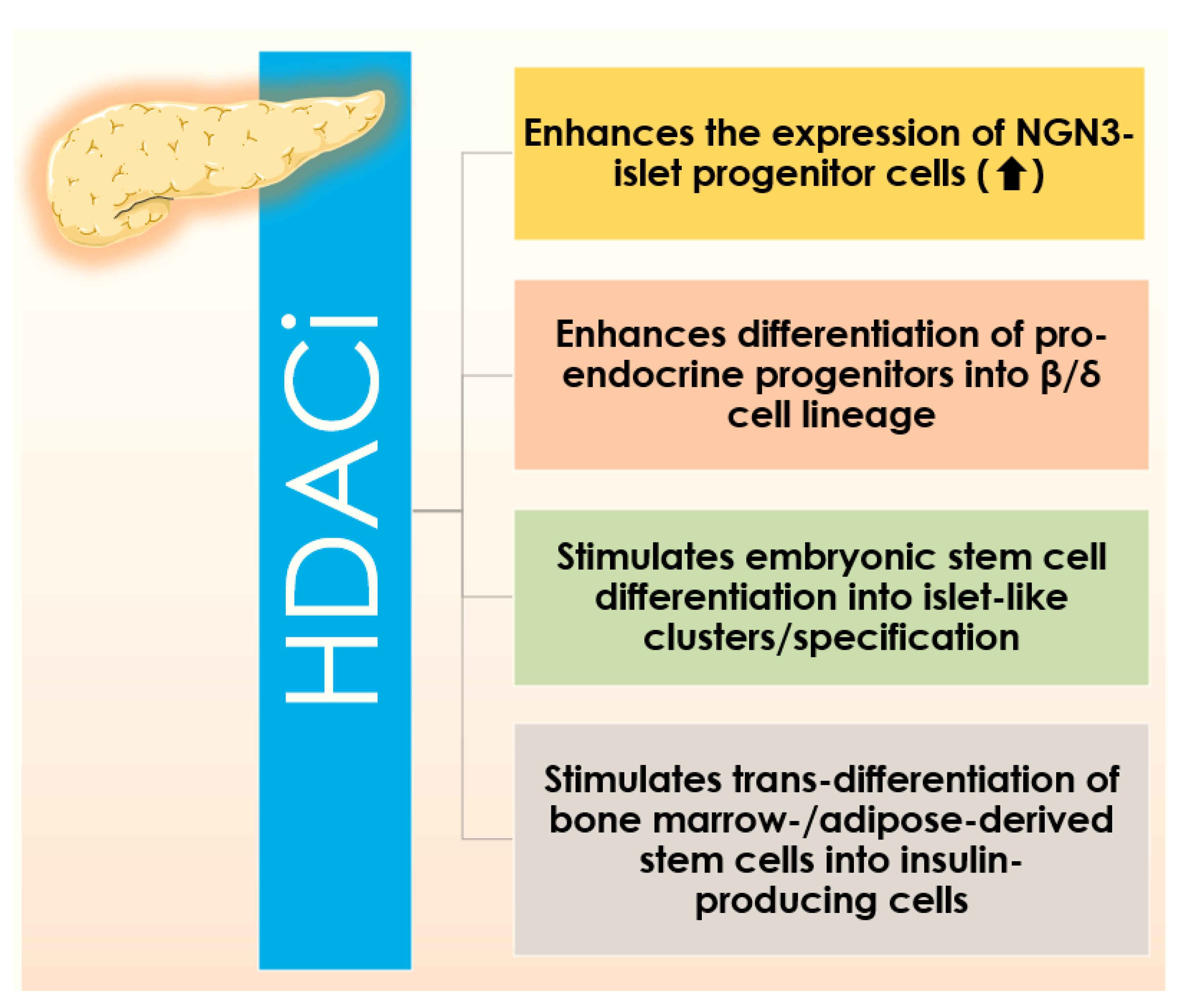{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classes | Members | Localizations | Expressions | Functions | Specific Function in Glucose Metabolism and Diabetes |
|---|---|---|---|---|---|
| Class I (Zn2+-dependent) | HDAC1, 2, 3, 8 | Nucleus of all types of tissues. | Ubiquitous | Promotes proliferation, IFN signalling, and HIF1α function. Contrary, it represses MTA1 & NF-κB functions. | HDAC1: Acts as repressor for the transcription of GLUT4 HDAC2: Binds to IRS-1 leading to reduced acetylation and decreased Insulin receptor mediated tyrosine phosphorylation of IRS-1 HDAC3: Stimulates Glucose production by FOXO transcription factor. HDAC8: Upregulated in obese mice models and promotes Insulin resistance. |
| Class II (Zn2+-dependent) | HDAC4, 5, 6, 7, 9, 10 | Nucleus and cytoplasm of brain, heart, skeletal muscle, pancreas, liver, kidney, and placenta. | Specific | Promotes HIF1α function and HSP 90 function (chaperone). It represses II5 promoter and inhibits Foxp3 and Treg function | HDAC4 & HDAC5: Repress transcription of GLUT4, involved in recruitment of class I HDAC3 to liver and stimulation of glucose production. HDAC6: Stimulates glucocorticoid-stimulated glucose production and contributes to hyperglycemia and glucose intolerance. HDAC7: Levels of HDAC7 are elevated in islets of T2D patients, elevated levels of HDAC7 lead to impairment in Insulin secretion and increased β-cell apoptosis in vitro HDAC9: Suppression of HDAC9 expression leads to improvement in Insulin sensitivity and glucose tolerance |
| Class III / SIRTUINS (NAD+-dependent) | SIRT1-7 | Mitochondria, nucleus, and cytoplasm. Brain, and in all the oncogenic tissues. | Ubiquitous | Promotes immune function across various types synapses by NAD+-dependent mechanism through SIRT1. | SIRT2 and SIRT4: SIRT2 impair insulin signaling and may contribute to pathogenesis of T2D |
| Class IV (Zn2+-dependent) | HDAC11 | Cytoplasm and nucleus of heart, brain, kidney, and skeletal muscle. | Ubiquitous | Inhibits II5 promoter activity in APCs. | NA |
| Specificity | Classification | Candidate HDACi Compounds/Drugs | HDAC Class Selectivity |
|---|---|---|---|
| Pan-HDACis | Hydroxamates | Vorinostat (SAHA) | class I, II and IV |
| Belinostat(PXD-101) | class I and II | ||
| Panobinostat (LBH-589) | class I, II and IV | ||
| Trichostatin A (TSA) | class I and II | ||
| Quisinostat (JNJ-16241199) | class I and II | ||
| WW437 | HDAC 2 and 4 | ||
| Benzamides | Entinostat (MS-275) | class I | |
| Tacedinaline (CI-994) | class I | ||
| Mocetinostat (MG-0103) | class I and IV | ||
| Aliphatic fatty acids | Pivaloyloxmethyl butyrate (AN-9) | class I and IIa | |
| Sodium Butyrate (NaB) | class I and IIa | ||
| Sodium Phenylbutyrate (4-PB) | class I and IIa | ||
| Valproate (valproic acid) | class I and IIa | ||
| Electrophilic ketones | trapoxins(TPX) | class I | |
| a-ketoamides | NA | ||
| heterocyclic ketones | NA | ||
| SIRT inhibitors | cambinol | SIRT1 and 2 | |
| EX-527 | SIRT1 and 2 | ||
| sirtinol | SIRT1 and 2 | ||
| nicotinamide | class III | ||
| Cyclic inhibitors/peptides | Romidepsin (depsipeptide, FK228) | class I | |
| Other compounds | Diallyl Trisulfide (DATS) | NA | |
| Class-specific HDACis | Benzamides | Ricolinosta(ACY-1215) | HDAC 6 |
| tubacin | HDAC 6 | ||
| Hydroxamate derivatives | Azelaic Bishydroxamic Acid (ABHA) | HDAC 3 | |
| CBHA (m-carboxycinnamic acid bis-hydroxamide) | HDAC 3 | ||
| SIRTs inhibitors | SEN196 | SIRT1 | |
| COMPOUND 6J | SIRT2 | ||
| JGB1741 | SIRT1 | ||
| N/A | I-7ab | HDAC 3 | |
| RGFP966 | HDAC 3 | ||
| PCI34051 | HDAC 8 | ||
| C149 | HDAC 8 | ||
| Polyketides | Depudecin | HDAC 1 | |
| Bromodomain HDACis | BET inhibitors | JQ1 | BRD4 |
| I-BET | BET | ||
| BY27 | BD2 | ||
| Hybrid HDACis | Chimerics | CUDC907 | HDAC /PI3K |
| CUDC101 | EGFR/Her-2/HDAC 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dewanjee, S.; Vallamkondu, J.; Kalra, R.S.; Chakraborty, P.; Gangopadhyay, M.; Sahu, R.; Medala, V.; John, A.; Reddy, P.H.; De Feo, V.; et al. The Emerging Role of HDACs: Pathology and Therapeutic Targets in Diabetes Mellitus. Cells 2021, 10, 1340. https://doi.org/10.3390/cells10061340
Dewanjee S, Vallamkondu J, Kalra RS, Chakraborty P, Gangopadhyay M, Sahu R, Medala V, John A, Reddy PH, De Feo V, et al. The Emerging Role of HDACs: Pathology and Therapeutic Targets in Diabetes Mellitus. Cells. 2021; 10(6):1340. https://doi.org/10.3390/cells10061340
Chicago/Turabian StyleDewanjee, Saikat, Jayalakshmi Vallamkondu, Rajkumar Singh Kalra, Pratik Chakraborty, Moumita Gangopadhyay, Ranabir Sahu, Vijaykrishna Medala, Albin John, P. Hemachandra Reddy, Vincenzo De Feo, and et al. 2021. "The Emerging Role of HDACs: Pathology and Therapeutic Targets in Diabetes Mellitus" Cells 10, no. 6: 1340. https://doi.org/10.3390/cells10061340
APA StyleDewanjee, S., Vallamkondu, J., Kalra, R. S., Chakraborty, P., Gangopadhyay, M., Sahu, R., Medala, V., John, A., Reddy, P. H., De Feo, V., & Kandimalla, R. (2021). The Emerging Role of HDACs: Pathology and Therapeutic Targets in Diabetes Mellitus. Cells, 10(6), 1340. https://doi.org/10.3390/cells10061340