
The Role of Heparan Sulfate and Neuropilin 2 in VEGFA Signaling in Human Endothelial Tip Cells and Non-Tip Cells during Angiogenesis In Vitro

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Flow Cytometric Analysis
2.3. Apoptosis Assay
2.4. Spheroid-Based In Vitro Angiogenesis Assay
2.5. SiRNA Knockdown
2.6. VLDL Isolation and Labeling
2.7. VLDL Uptake
2.8. Immunohistochemistry and Quantification of Protein Levels
2.9. RNA Isolation and Quantitative PCR
2.10. Western Blots
2.11. Statistics and Data Correction
3. Results
3.1. Role of SULF2 in VEGFA165 and VEGFA121 Stimulation of HUVECs
3.2. NRP2 Is a VEGFR2 Co-Receptor Involved in VEGFA-Induced Sprouting, but Not in Tip Cell Formation
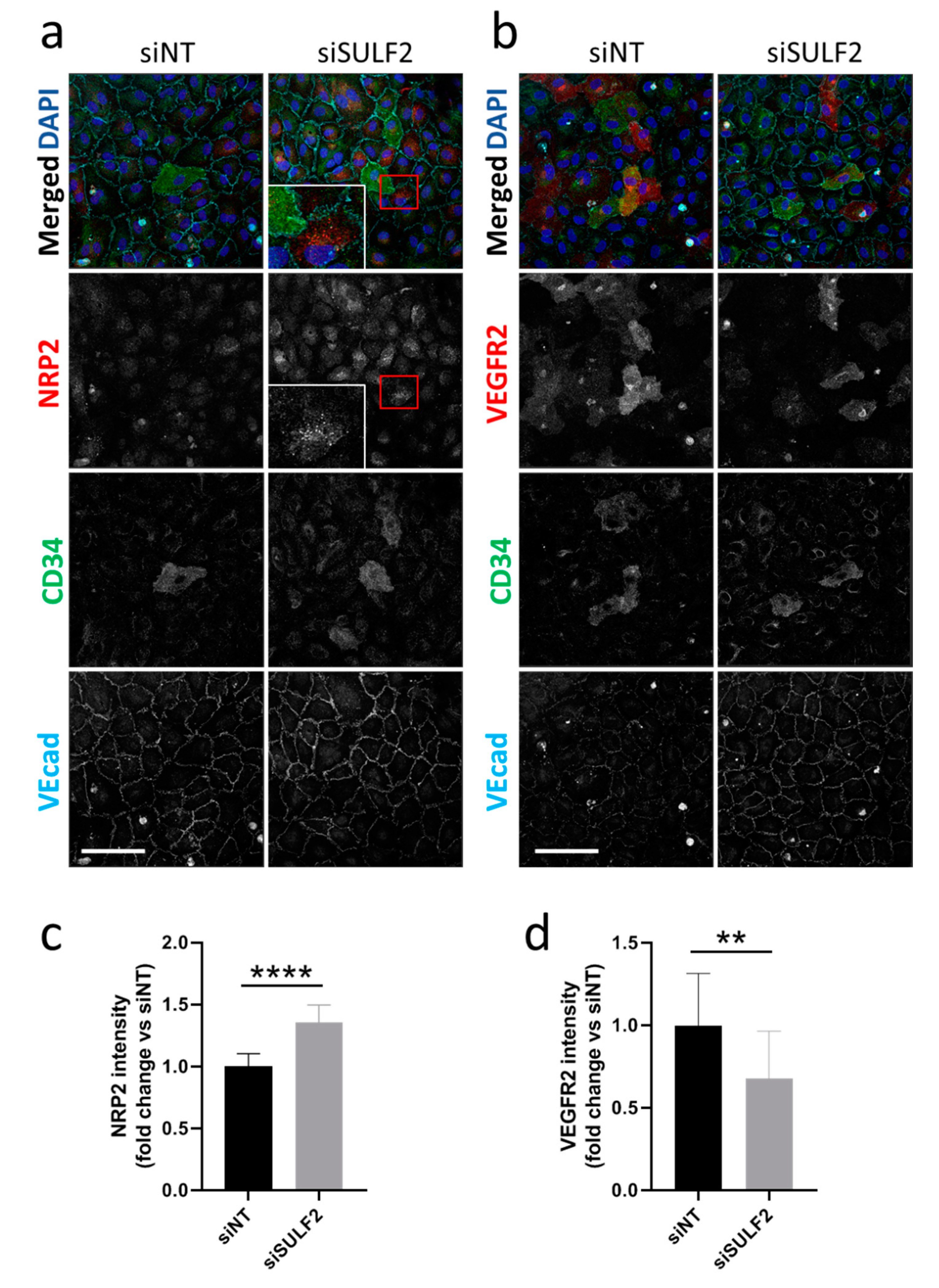
3.3. Role of SULF2 in NRP2 and VEGFR2 Protein Expression in Tip Cells and Non-Tip Cells
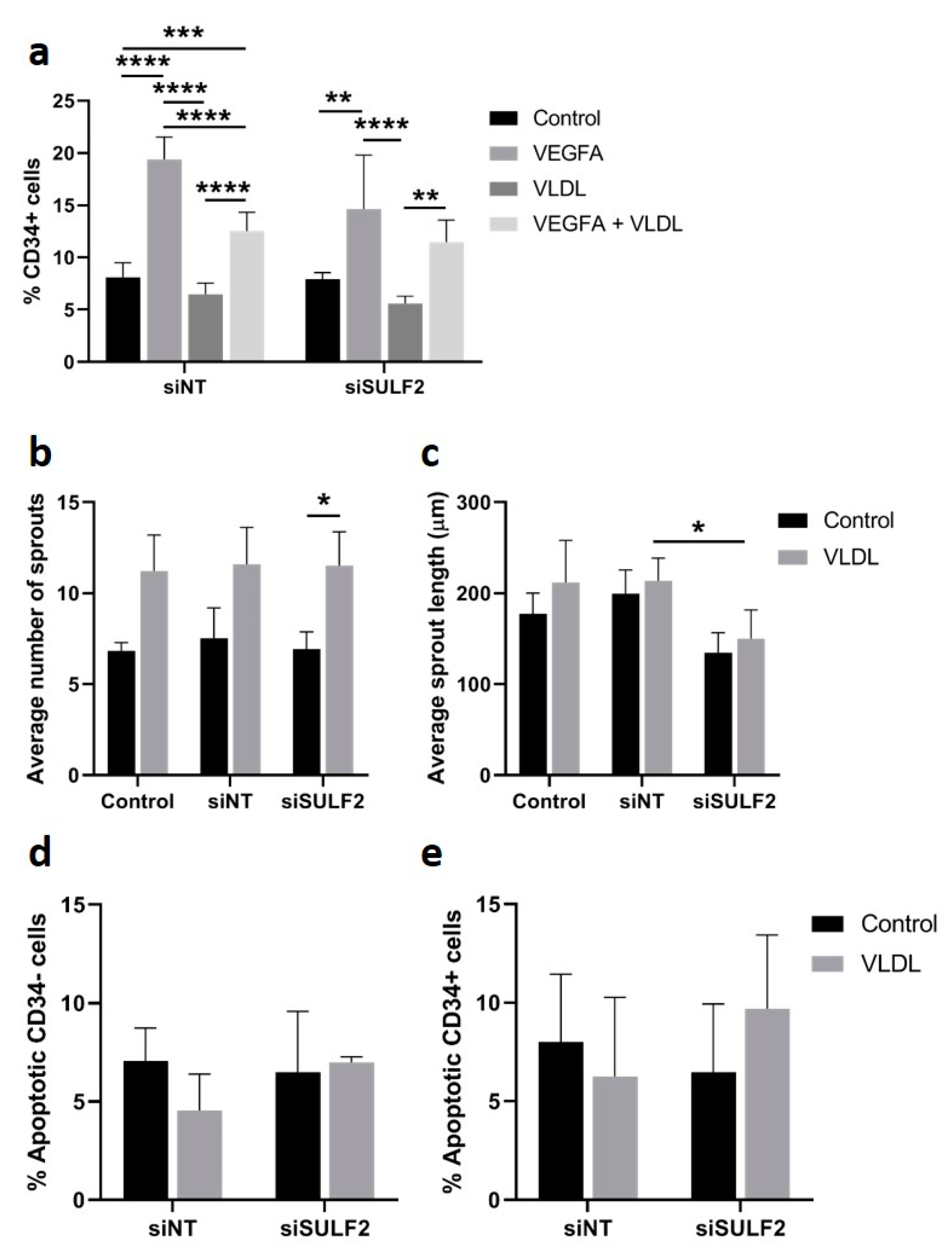
3.4. Role of SULF2 in VLDL Uptake by HUVECs
3.5. VLDL Uptake Does not Affect the Tip Cell Percentage but Enhances the Number of Sprouts
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dallinga, M.G.; Boas, S.E.M.; Klaassen, I.; Merks, R.H.M.; van Noorden, C.J.F.; Schlingemann, R.O. Tip cells in angiogenesis. eLS 2015, 10, 1–10. [Google Scholar] [CrossRef]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Yang, Z.; Mo, X.; Gong, Q.; Pan, Q.; Yang, X.; Cai, W.; Li, C.; Ma, J.X.; He, Y.; Gao, G. Critical effect of VEGF in the process of endothelial cell apoptosis induced by high glucose. Apoptosis 2008, 13, 1331–1343. [Google Scholar] [CrossRef]
- Geretti, E.; Shimizu, A.; Klagsbrun, M. Neuropilin structure governs VEGF and semaphorin binding and regulates angiogenesis. Angiogenesis 2008, 11, 31–39. [Google Scholar] [CrossRef]
- Sarabipour, S.; Mac Gabhann, F. VEGF-A121a binding to Neuropilins—A concept revisited. Cell Adhes. Migr. 2018, 12, 204–214. [Google Scholar] [CrossRef]
- Billings, P.C.; Pacifici, M. Interactions of signaling proteins, growth factors and other proteins with heparan sulfate: Mechanisms and mysteries. Connect. Tissue Res. 2015, 56, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Smorenburg, S.; van Noorden, C.J.F. The complex effects of heparins on cancer progression and metastasis in experimental studies. Pharmacol. Rev. 2001, 53, 93–105. [Google Scholar] [PubMed]
- Ruhrberg, C.; Gerhardt, H.; Golding, M.; Watson, R.; Ioannidou, S.; Fujisawa, H.; Betsholtz, C.; Shima, D.T. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002, 16, 2684–2698. [Google Scholar] [CrossRef] [PubMed]
- Kohn-Luque, A.; de Back, W.; Starruss, J.; Mattiotti, A.; Deutsch, A.; Perez-Pomares, J.M.; Herrero, M.A. Early embryonic vascular patterning by matrix-mediated paracrine signalling: A mathematical model study. PLoS ONE 2011, 6, e24175. [Google Scholar] [CrossRef]
- Pries, A.R.; Secomb, T.W.; Gaehtgens, P. The endothelial surface layer. Pflüg. Arch. 2000, 440, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Gouverneur, M.; Berg, B.; Nieuwdorp, M.; Stroes, E.; Vink, H. Vasculoprotective properties of the endothelial glycocalyx: Effects of fluid shear stress. J. Intern. Med. 2006, 259, 393–400. [Google Scholar] [CrossRef]
- Möckl, L.; Hirn, S.; Torrano, A.A.; Uhl, B.; Bräuchle, C.; Krombach, F. The glycocalyx regulates the uptake of nanoparticles by human endothelial cells In Vitro. Nanomedicine 2017, 12, 207–217. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, M.E.; Saez-Torres, K.L.; Cano, I.; Hu, Z.; Saint-Geniez, M.; Ng, Y.S.; D’Amore, P.A. Glycocalyx regulation of vascular endothelial growth factor receptor 2 activity. FASEB J. 2019, 33, 9362–9373. [Google Scholar] [CrossRef] [PubMed]
- Avizienyte, E.; Cole, C.L.; Rushton, G.; Miller, G.J.; Bugatti, A.; Presta, M.; Gardiner, J.M.; Jayson, G.C. Synthetic Site-Selectively Mono-6-O-Sulfated Heparan Sulfate Dodecasaccharide Shows Anti-Angiogenic Properties In Vitro and Sensitizes Tumors to Cisplatin In Vivo. PLoS ONE 2016, 11, e0159739. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Stringer, S.E.; Rusch, M.A.; Selleck, S.B.; Ekker, S.C. A unique role for 6-O sulfation modification in zebrafish vascular development. Dev. Biol. 2005, 284, 364–376. [Google Scholar] [CrossRef]
- Dai, Y.; Yang, Y.; MacLeod, V.; Yue, X.; Rapraeger, A.C.; Shriver, Z.; Venkataraman, G.; Sasisekharan, R.; Sanderson, R.D. HSulf-1 and HSulf-2 are potent inhibitors of myeloma tumor growth In Vivo. J. Biol. Chem. 2005, 280, 40066–40073. [Google Scholar] [CrossRef]
- Dallinga, M.G.; Dallinga-Thie, G.M. Role of sulfatase 2 in lipoprotein metabolism and angiogenesis. Curr. Opin. Lipidol. 2016, 27, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Hassing, H.C.; Mooij, H.; Guo, S.; Monia, B.P.; Chen, K.; Kulik, W.; Dallinga-Thie, G.M.; Nieuwdorp, M.; Stroes, E.S.; Williams, K.J. Inhibition of hepatic sulfatase-2 In Vivo: A novel strategy to correct diabetic dyslipidemia. Hepatology 2012, 55, 1746–1753. [Google Scholar] [CrossRef] [PubMed]
- Teuwen, L.A.; Draoui, N.; Dubois, C.; Carmeliet, P. Endothelial cell metabolism: An update anno 2017. Curr. Opin. Hematol. 2017, 24, 240–247. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.; Pirillo, A.; Callegari, E.; Hamsten, A.; Catapano, A.; Eriksson, P. Gene expression and intracellular pathways involved in endothelial dysfunction induced by VLDL and oxidised VLDL. Cardiovasc. Res. 2003, 59, 169–180. [Google Scholar] [CrossRef][Green Version]
- Graupera, M.; Potente, M. Regulation of angiogenesis by PI3K signaling networks. Exp. Cell Res. 2013, 319, 1348–1355. [Google Scholar] [CrossRef]
- Kotlinowski, J.; Jozkowicz, A. PPAR Gamma and Angiogenesis: Endothelial Cells Perspective. J. Diabetes Res. 2016, 2016, 8492353. [Google Scholar] [CrossRef] [PubMed]
- Siemerink, M.J.; Klaassen, I.; Vogels, I.M.; Griffioen, A.W.; Van Noorden, C.J.; Schlingemann, R.O. CD34 marks angiogenic tip cells in human vascular endothelial cell cultures. Angiogenesis 2012, 15, 151–163. [Google Scholar] [CrossRef] [PubMed]
- van der Schaft, D.W.J.; Toebes, E.A.H.; Haseman, J.R.; Mayo, K.H.; Griffioen, A.W. Bactericidal/permeability-increasing protein (BPI) inhibits angiogenesis via induction of apoptosis in vascular endothelial cells. Blood 2000, 96, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Korff, T.; Augustin, H.G. Integration of endothelial cells in multicellular spheroids prevents apoptosis and induces differentiation. J. Cell Biol. 1998, 143, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Alitalo, K.; Allen, E.; Anisimov, A.; Aplin, A.C.; Auerbach, R.; Augustin, H.G.; Bates, D.O.; van Beijnum, J.R.; Bender, R.H.F.; et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis 2018, 21, 425–532. [Google Scholar] [CrossRef] [PubMed]
- Chieco, P.; Jonker, A.; De Boer, B.A.; Ruijter, J.M.; Van Noorden, C.J. Image cytometry: Protocols for 2D and 3D quantification in microscopic images. Prog. Histochem. Cytochem. 2013, 47, 211–333. [Google Scholar] [CrossRef]
- Rezaee, F.; Casetta, B.; Levels, J.H.; Speijer, D.; Meijers, J.C. Proteomic analysis of high-density lipoprotein. Proteomics 2006, 6, 721–730. [Google Scholar] [CrossRef]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.; Moorman, A.F. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef]
- Ruijter, J.M.; Thygesen, H.H.; Schoneveld, O.J.; Das, A.T.; Berkhout, B.; Lamers, W.H. Factor correction as a tool to eliminate between-session variation in replicate experiments: Application to molecular biology and retrovirology. Retrovirology 2006, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Dallinga, M.G.; Yetkin-Arik, B.; Kayser, R.P.; Vogels, I.M.C.; Nowak-Sliwinska, P.; Griffioen, A.W.; van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. IGF2 and IGF1R identified as novel tip cell genes in primary microvascular endothelial cell monolayers. Angiogenesis 2018, 21, 823–836. [Google Scholar] [CrossRef]
- Berndsen, R.H.; Castrogiovanni, C.; Weiss, A.; Rausch, M.; Dallinga, M.G.; Miljkovic-Licina, M.; Klaassen, I.; Meraldi, P.; van Beijnum, J.R.; Nowak-Sliwinska, P. Anti-angiogenic effects of crenolanib are mediated by mitotic modulation independently of PDGFR expression. Br. J. Cancer 2019, 121, 139–149. [Google Scholar] [CrossRef]
- Fantin, A.; Vieira, J.M.; Plein, A.; Denti, L.; Fruttiger, M.; Pollard, J.W.; Ruhrberg, C. NRP1 acts cell autonomously in endothelium to promote tip cell function during sprouting angiogenesis. Blood 2013, 121, 2352–2362. [Google Scholar] [CrossRef]
- Chen, L.; Jiang, W.; Huang, J.; He, B.C.; Zuo, G.W.; Zhang, W.; Luo, Q.; Shi, Q.; Zhang, B.Q.; Wagner, E.R.; et al. Insulin-like growth factor 2 (IGF-2) potentiates BMP-9-induced osteogenic differentiation and bone formation. J. Bone Miner. Res. 2010, 25, 2447–2459. [Google Scholar] [CrossRef] [PubMed]
- Ho, F.M.; Lin, W.W.; Chen, B.C.; Chao, C.M.; Yang, C.R.; Lin, L.Y.; Lai, C.C.; Liu, S.H.; Liau, C.S. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-kappaB and c-Jun NH2-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cell Signal. 2006, 18, 391–399. [Google Scholar] [CrossRef]
- Xu, Y.; Yuan, L.; Mak, J.; Pardanaud, L.; Caunt, M.; Kasman, I.; Larrivee, B.; Del Toro, R.; Suchting, S.; Medvinsky, A.; et al. Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J. Cell Biol. 2010, 188, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Martyn, U.; Schulte-Merker, S. Zebrafish neuropilins are differentially expressed and interact with vascular endothelial growth factor during embryonic vascular development. Dev. Dyn. 2004, 231, 33–42. [Google Scholar] [CrossRef]
- Favier, B.; Alam, A.; Barron, P.; Bonnin, J.; Laboudie, P.; Fons, P.; Mandron, M.; Herault, J.P.; Neufeld, G.; Savi, P.; et al. Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood 2006, 108, 1243–1250. [Google Scholar] [CrossRef]
- Kim, W.H.; Lee, S.H.; Jung, M.H.; Seo, J.H.; Kim, J.; Kim, M.A.; Lee, Y.M. Neuropilin2 expressed in gastric cancer endothelial cells increases the proliferation and migration of endothelial cells in response to VEGF. Exp. Cell Res. 2009, 315, 2154–2164. [Google Scholar] [CrossRef]
- Hassing, H.C.; Surendran, R.P.; Derudas, B.; Verrijken, A.; Francque, S.M.; Mooij, H.L.; Moens, S.J.B.; Hart, L.M.; Nijpels, G.; Dekker, J.M.; et al. SULF2 strongly prediposes to fasting and postprandial triglycerides in patients with obesity and type 2 diabetes mellitus. Obesity 2014, 22, 1309–1316. [Google Scholar] [CrossRef]
- Ausprunk, D.H.; Folkman, J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc. Res. 1977, 14, 53–65. [Google Scholar] [CrossRef]
- Rousseau, S. Vascular Endothelial Growth Factor (VEGF)-driven Actin-based Motility Is Mediated by VEGFR2 and Requires Concerted Activation of Stress-activated Protein Kinase 2 (SAPK2/p38) and Geldanamycin-sensitive Phosphorylation of Focal Adhesion Kinase. J. Biol. Chem. 2000, 275, 10661–10672. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I. Role of Akt Signaling in Vascular Homeostasis and Angiogenesis. Circ. Res. 2002, 90, 1243–1250. [Google Scholar] [CrossRef]
- Kazemi, M.; Carrer, A.; Moimas, S.; Zandona, L.; Bussani, R.; Casagranda, B.; Palmisano, S.; Prelazzi, P.; Giacca, M.; Zentilin, L.; et al. VEGF121 and VEGF165 differentially promote vessel maturation and tumor growth in mice and humans. Cancer Gene Ther. 2016, 23, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Fantin, A.; Maden, C.H.; Ruhrberg, C. Neuropilin ligands in vascular and neuronal patterning. Biochem. Soc. Trans. 2009, 37, 1228–1232. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef]
- Yetkin-Arik, B.; Vogels, I.M.C.; Nowak-Sliwinska, P.; Weiss, A.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci. Rep. 2019, 9, 12608. [Google Scholar] [CrossRef] [PubMed]
- Yetkin-Arik, B.; Vogels, I.M.C.; Neyazi, N.; van Duinen, V.; Houtkooper, R.H.; van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. Endothelial tip cells In Vitro are less glycolytic and have a more flexible response to metabolic stress than non-tip cells. Sci. Rep. 2019, 9, 10414. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.W.; Lo, Y.H.; Chen, C.H.; Lin, C.Y.; Tsai, C.H.; Chen, P.J.; Yang, Y.F.; Wang, C.H.; Tan, C.H.; Hou, M.F.; et al. VLDL and LDL, but not HDL, promote breast cancer cell proliferation, metastasis and angiogenesis. Cancer Lett. 2017, 388, 130–138. [Google Scholar] [CrossRef]
- Christianson, H.C.; Belting, M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol. 2014, 35, 51–55. [Google Scholar] [CrossRef]
- Dumas, S.J.; García-Caballero, M.; Carmeliet, P. Metabolic Signatures of Distinct Endothelial Phenotypes. Trends Endocrinol. Metab. 2020. [Google Scholar] [CrossRef]
- Barker, A.L.; Konopatskaya, O.; Neal, C.R.; Macpherson, J.V.; Whatmore, J.L.; Winlove, C.P.; Unwin, P.R.; Shore, A.C. Observation and characterisation of the glycocalyx of viable human endothelial cells using confocal laser scanning microscopy. Phys. Chem. Chem. Phys. 2004, 6, 1006–1011. [Google Scholar] [CrossRef]
- Goveia, J.; Rohlenova, K.; Taverna, F.; Treps, L.; Conradi, L.C.; Pircher, A.; Geldhof, V.; de Rooij, L.; Kalucka, J.; Sokol, L.; et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell 2020, 37, 21–36.e13. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dallinga, M.G.; Habani, Y.I.; Schimmel, A.W.M.; Dallinga-Thie, G.M.; van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. The Role of Heparan Sulfate and Neuropilin 2 in VEGFA Signaling in Human Endothelial Tip Cells and Non-Tip Cells during Angiogenesis In Vitro. Cells 2021, 10, 926. https://doi.org/10.3390/cells10040926
Dallinga MG, Habani YI, Schimmel AWM, Dallinga-Thie GM, van Noorden CJF, Klaassen I, Schlingemann RO. The Role of Heparan Sulfate and Neuropilin 2 in VEGFA Signaling in Human Endothelial Tip Cells and Non-Tip Cells during Angiogenesis In Vitro. Cells. 2021; 10(4):926. https://doi.org/10.3390/cells10040926
Chicago/Turabian StyleDallinga, Marchien G., Yasmin I. Habani, Alinda W. M. Schimmel, Geesje M. Dallinga-Thie, Cornelis J. F. van Noorden, Ingeborg Klaassen, and Reinier O. Schlingemann. 2021. "The Role of Heparan Sulfate and Neuropilin 2 in VEGFA Signaling in Human Endothelial Tip Cells and Non-Tip Cells during Angiogenesis In Vitro" Cells 10, no. 4: 926. https://doi.org/10.3390/cells10040926
APA StyleDallinga, M. G., Habani, Y. I., Schimmel, A. W. M., Dallinga-Thie, G. M., van Noorden, C. J. F., Klaassen, I., & Schlingemann, R. O. (2021). The Role of Heparan Sulfate and Neuropilin 2 in VEGFA Signaling in Human Endothelial Tip Cells and Non-Tip Cells during Angiogenesis In Vitro. Cells, 10(4), 926. https://doi.org/10.3390/cells10040926