
NETosis in Wound Healing: When Enough Is Enough




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Dark Side of NETosis
3. The Bright Side of NETosis
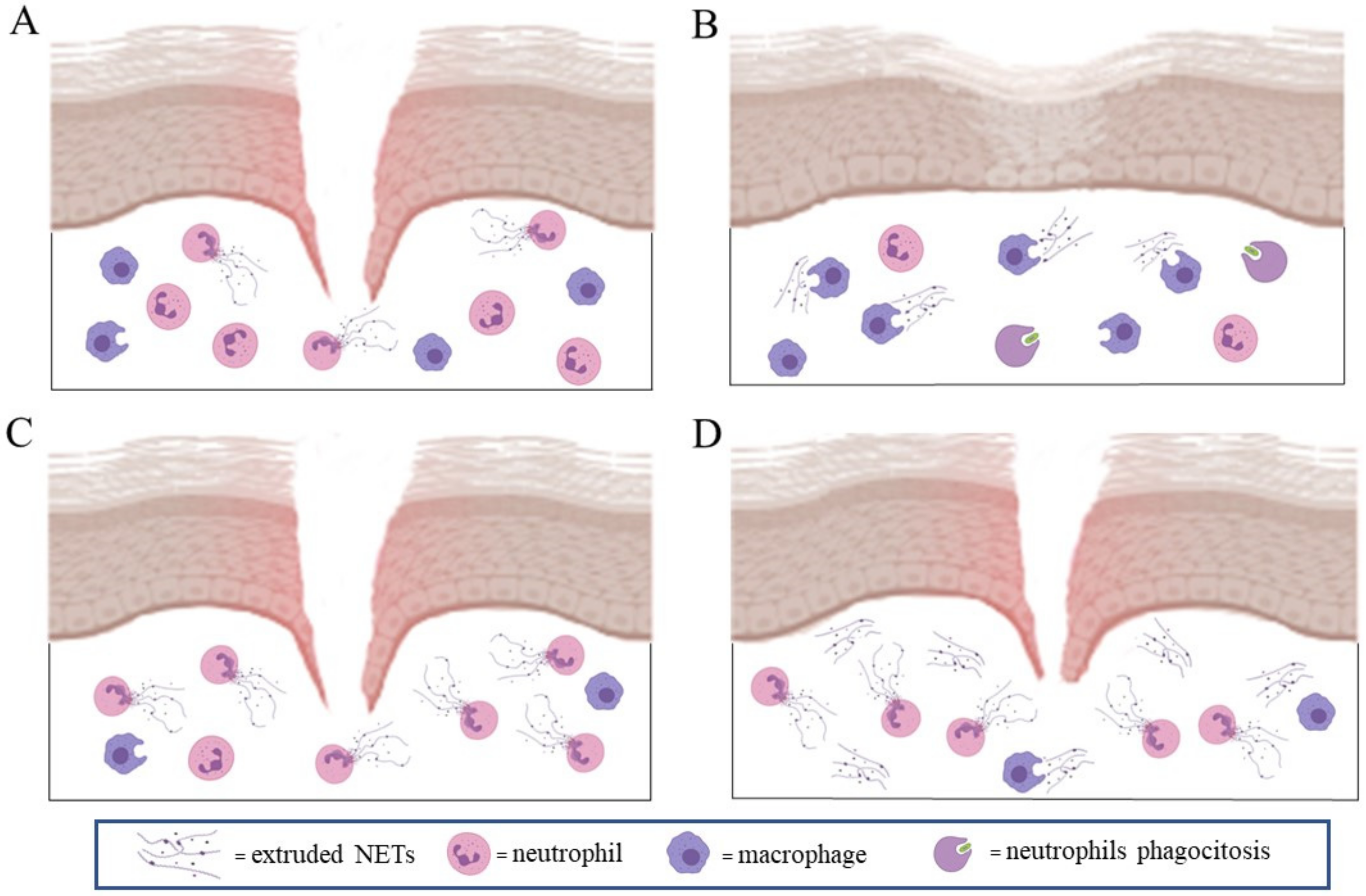
4. NETs Formation in Wound Healing
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hidalgo, A.; Chilvers, E.R.; Summers, C.; Koendermann, L. The Neutrophil Life Cycle. Trends Immunol. 2019, 40, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Scapini, P.; Cassatella, M.A. Social networking of human neutrophils within the immune system. Blood 2014, 124, 710–719. [Google Scholar] [CrossRef]
- Tseng, C.W.; Liu, G.Y. Expanding roles of neutrophils in aging hosts. Curr. Opin. Immunol. 2014, 29, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Marcos, V.Z.; Zhou, A.; Yildirim, Ö.; Bohla, A.; Hector, A.; Vitkov, L.; Wiedenbauer, E.-M.; Krautgartner, W.D.; Stoiber, W.; Belohradsky, B.H.; et al. CXCR2 mediates NADPH oxidase–independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat. Med. 2011, 17, 899. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weirauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [PubMed]
- Hemmers, S.; Teijaro, J.R.; Arandjelovic, S.; Mowen, K.A. PAD4-Mediated Neutrophil Extracellular Trap Formation Is Not Required for Immunity against Influenza Infection. PLoS ONE 2011, 6, e22043. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Zychlinsky, A. NETs: A new strategy for using old weapons. Trends Immunol. 2009, 30, 513–521. [Google Scholar] [CrossRef]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef]
- Ehrlich, H.L. Are gram-positive bacteria capable of electron transfer across their cell wall without an externally available electron shuttle? Geobiology 2008, 6, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Polyanichko, A.M.; Andrushchenko, V.V.; Chikhirzhina, E.V.; Vorob’ev, V.I.; Wieser, H. The effect of manganese(II) on DNA structure: Electronic and vibrational circular dichroism studies. Nucleic Acids Res. 2004, 32, 989–996. [Google Scholar] [CrossRef]
- Juttukonda, L.J.; Skaar, E.P. Manganese homeostasis and utilization in pathogenic bacteria. Mol. Microbiol. 2015, 97, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzman, M.; Wu, K.; Meijndert, H.C.; et al. 1Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Steinberg, B.E.; Grinstein, S. Unconventional roles of the NADPH oxidase: Signaling, ion homeostasis, and cell death. Sci. STKE 2007, 2007, pe11. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef]
- Ponath, V.; Kaina, B. Death of Monocytes through Oxidative Burst of Macrophages and Neutrophils: Killing in Trans. PLoS ONE 2017, 12, e0170347. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Stojkov, D.; Germic, N.; Simon, D.; Wang, X.; Charaf Benarafa, C.; Simon, H.-U. Untangling “NETosis” from NETs. Eur. J. Immunol. 2019, 49, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Gold, J.A.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schimdt, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J.; et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defence. Nat. Med. 2008, 14, 949–995. [Google Scholar] [CrossRef]
- Chow, O.A.; von Köckritz-Blickwede, M.; Bright, A.T.; Hensler, M.E.; Zinkernagel, A.S.; Cogen, A.L.; Gallo, L.R.; Monestier, M.; Yanming, W.; Glass, C.K.; et al. Statins Enhance Formation of Phagocyte Extracellular Traps. Cell Host Microbe 2010, 8, 445–454. [Google Scholar] [CrossRef]
- Lin, A.M.; Rubin, C.J.; Khandpur, R.; Wang, J.Y.; Riblett, M.B.; Yalavarthi, S.; Villanueva, E.C.; Shah, P.; Kaplan, M.J.; Bruce, A.T. Mast Cells and Neutrophils Release IL-17 through Extracellular Trap Formation in Psoriasis. J. Immunol. 2011, 187, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Morshed, M.; Hlushchuk, R.; Simon, D.; Walls, A.F.; Obata-Ninomiya, K.; Karasuyama, H.; Djonov, V.; Eggel, A.; Kaufmann, T.; Simon, H.-U.; et al. NADPH oxidase-independent formation of extracellular DNA traps by basophils. J. Immunol. 2014, 192, 5314–5323. [Google Scholar] [CrossRef] [PubMed]
- Wartha, F.; Henriques-Normark, B. ETosis: A novel cell death pathway. Sci. Signal. 2008, 1, pe25. [Google Scholar] [CrossRef]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, F.; Altamura, S.; Frosali, S.; Conti, P. Key Role of DAMP in Inflammation, Cancer, and Tissue Repair. Clin. Ther. 2016, 38, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Denning, N.L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMP and NETs in sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding Cell Death Signals in Inflammation and Immunity. Cell 2010, 140, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Hasler, P.; Giaglis, S.; Hahn, S. Neutrophil extracellular traps in health and disease. Swiss Med. Wkly. 2016, 146, w14352. [Google Scholar] [CrossRef]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Vorobjeva, N.V.; Pinegin, B.V. Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Health and Disease. Biochemistry 2014, 79, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Phillipson, M.; Kubes, P. The neutrophil in vascular inflammation. Nat. Med. 2011, 17, 1381–1390. [Google Scholar] [CrossRef]
- Gevaert, E.; Zhang, N.; Krysko, O.; Lan, F.; Holtappels, G.; De Ruyck, N.; Nauwynck, H.; Yousefi, S.; Simon, H.-U.; Bachert, C. Extracellular eosinophilic traps in association with Staphylococcus aureus at the site of epithelial barrier defects in patients with severe airway inflammation. J. Allergy Clin. Immunol. 2017, 139, 1849–1860. [Google Scholar] [CrossRef] [PubMed]
- Neubert, E.; Meyer, D.; Kruss, S.; Erpenbeck, L. The power from within—Understanding the driving forces of neutrophil extracellular trap formation. J. Cell Sci. 2020, 133, jcs241075. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaa6676. [Google Scholar] [CrossRef]
- Itakura, A.; McCarty, O.J.T. Pivotal role for the mTOR pathway in the formation of neutrophil extracellular traps via regulation of autophagy. AJP Cell Physiol. 2013, 305, C348–C354. [Google Scholar] [CrossRef]
- Remijsen, Q.; Vanden Berghe, T.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef]
- Liang, X.; Liu, L.; Wang, Y.; Guo, H.; Fan, H.; Zhang, C.; Houa, L.; Liua, Z. Autophagy-driven NETosis is a double-edged sword—Review. Biomed. Pharmacother. 2020, 126, 110065. [Google Scholar] [CrossRef]
- Neeli, I.; Dwivedi, N.; Khan, S.; Radic, M. Regulation of extracellular release from neutrophils. J. Innate Immun. 2009, 1, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef]
- Németh, T.; Mócsai, A. The role of neutrophils in autoimmune diseases. Immunol. Lett. 2012, 143, 9–19. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, Y.; Zhong, L.; Ye, D.; Zhang, J.; Tu, Y.; Bornstein, S.R.; Zhou, Z.; Lam, K.S.L.; Xu, A. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated withβ-cell autoimmunity in patients with type 1 diabetes. Diabetes 2014, 63, 4239–4248. [Google Scholar] [CrossRef]
- Sørensen, O.E.; Borregaard, N. Neutrophil extracellular traps—The dark side of neutrophils. J. Clin. Investig. 2016, 126, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Li, H.; Li, Y.; Dai, M.; Zhang, L.; Liu, S.; Tan, H.; Deng, P.; Liu, J.; Mao, Z.; et al. NETs promote ALI/ARDS inflammation by regulating alveolar macrophage polarization. Exp. Cell Res. 2019, 382, 111486. [Google Scholar] [CrossRef]
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikrogulis, D.; Kostantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.; et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 2014, 233, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Erpenbeck, L.; Schön, M.P. Neutrophil extracellular traps: Protagonists of cancer progression? Oncogene 2017, 36, 2483–2490. [Google Scholar] [CrossRef] [PubMed]
- Teijeira, Á.; Garasa, S.; Gato, M.; Alfaro, C.; Migueliz, I.; Cirella, A.; de Andrea, C.; Ochoa, M.C.; Otano, I.; Etxeberria, I.; et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 2020, 52, 856–871. [Google Scholar] [CrossRef]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, J.; Olsson, A.-K. Immunity Gone Astray—NETs in Cancer. Trends Cancer. 2016, 2, 633–634. [Google Scholar] [CrossRef] [PubMed]
- Garley, M.; Jabłońska, E.; Dąbrowska, D. NETs in cancer. Tumor Biol. 2016, 37, 14355–14361. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.-K.; Cedervall, J. NETosis in Cancer—Platelet-Neutrophil Crosstalk Promots Tumor-Associated Pathology. Front. Immunol. 2016, 7, article373. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Fadini, G.P.; Menegazzo, L.; Rigato, M.; Scattolini, V.; Poncina, N.; Bruttocao, A.; Ciciliot, S.; Mammano, F.; Ciobutaro, C.D.; Poncina, N.; et al. NETosis Delays Diabetic Wound Healing in Mice and Humans. Diabetes 2016, 65, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Kienhöfer, D.; Hahn, J.; Stoof, J.; Cspregi, J.Z.; Reinwald, C.; Urbonaviciute, V.; Johnsson, C.; Maueröder, C.; Podolska, M.J.; Biermann, M.H.; et al. Experimental lupus is aggravated in mouse strains with impaired induction of neutrophil extracellular traps. JCI Insight 2017, 2, 92920. [Google Scholar] [CrossRef]
- Giaglis, S.; Hahn, S.; Hasler, P. “The NET Outcome”: Are Neutrophil Extracellular Traps of Any Relevance to the Pathophysiology of Autoimmune Disorders in Childhood? Front. Pediatr. 2016, 4, 97. [Google Scholar] [CrossRef] [PubMed]
- Allam, R.; Kumar, S.V.R.; Darisipudi, M.N.; Anders, H.-J. Extracellular histones in tissue injury and inflammation. J. Mol. Med. 2014, 92, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Kronbichler, A.; Park, D.D.-Y.; Park, Y.M.; Moon, H.; Kim, H.; Choi, J.H.; Choi, Y.S.; Shim, S.; Lyu, I.S.; et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun. Rev. 2017, 161, 1160–1173. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, D.; Shida, H.; Kusunoki, Y.; Miyoshi, A.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. The responses of macrophages in interaction with neutrophils that undergo NETosis. J. Autoimmun. 2016, 67, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.T.; Gao, F.; Gu, K.; Chen, D.K. The role of monocytes and macrophages in autoimmune disease: A comprehensive review. Front. Immunol. 2019, 10, 1140. [Google Scholar] [CrossRef]
- Nakazawa, D.; Desai, J.; Steiger, S.; Müller, S.; Devarapu, S.K.; Mulay, S.R.; Iwakura, T.; Anders, H.-J. Activated platelets induce MLKL-driven neutrophil necroptosis and release of neutrophil extracellular traps in venous thrombosis. Cell Death Discov. 2018, 4, 71–82. [Google Scholar] [CrossRef]
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell. Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Meier, A.; Chien, J.; Hobohm, L.; Patras, K.A.; Nizet, V.; Corriden, R. Inhibition of human neutrophil extracellular trap (NET) production by propofol and lipid emulsion. Front. Pharmacol. 2019, 10, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophils extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef] [PubMed]
- Tomar, B.; Anders, H.J.; Desai, J.; Mulay, S.R. Neutrophils and Neutrophil Extracellular Traps Drive Necroinflammation in COVID-19. Cells 2020, 9, 1383. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar]
- Bonow, R.O.; Fonarow, G.C.; O’Gara, P.T.; Yancy, C.W. Association of Coronavirus Disease 2019 (COVID-19) With Myocardial Injury and Mortality. JAMA Cardiol. 2020, 5, 751–753. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Hahn, J.; Knopf, J.; Maueröder, C.; Kienhöfer, D.; Leppkes, M.; Herrmann, M. Neutrophils and neutrophil extracellular traps orchestrate initiation and resolution of inflammation. Clin. Exp. Rheumatol. 2016, 34, 6–8. [Google Scholar] [PubMed]
- Mitroulis, I.; Kambas, K.; Ritis, K. Neutrophils, IL-1β, and gout: Is there a link? Semin. Immunopathol. 2013, 35, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Reber, L.L.; Gaudenzio, N.; Starkl, P.; Galli, S.J. Neutrophils are not required for resolution of acute gouty arthritis in mice. Nat. Med. 2016, 22, 1382–1384. [Google Scholar] [CrossRef]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weiden, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019, 33, 1401–1414. [Google Scholar] [CrossRef]
- Hoppenbrouwers, T.; Autar, A.A.; Sultan, A.R.; Abraham, T.E.; van Cappellen, W.A.; Houtsmuller, A.B.B.; van Wamel, W.J.B.; van Beusekom, H.M.M.; van Neck, J.W.; de Maat, M.P.M. In vitro induction of NETosis: Comprehensive live imaging comparison and systematic review. PLoS ONE 2017, 12, e0176472. [Google Scholar] [CrossRef]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef]
- Hoste, E.; Maueröder, C.; van Hove, L.; Catrysse, L.; Vikkula, H.-K.; Sze, M.; Maes, B.; Karjosukarso, D.; Martens, L.; Gonçalves, A.; et al. Epithelial HMGB1 Delays Skin Wound Healing and Drives Tumor Initiation by Priming Neutrophils for NET Formation. Cell Rep. 2019, 29, 2689–2701. [Google Scholar] [CrossRef] [PubMed]
- Roth, F.R.J.; Czech, M.P. NETs and traps delay wound healing in diabetes. Trends Endocrinol. Metab. 2015, 26, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; Wagner, D.D. Peptidylarginine deiminase 4: A nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 2018, 32, 6258–6370. [Google Scholar] [CrossRef] [PubMed]
- Segoviano-Ramirez, J.C.; Lopez-Altamirano, D.F.; Garcia-Juarez, J.; Aguirre-Garza, J.E.S.; Cárdenas-Estrada, E.; Ancer-Rodriguez, J. The Diethylcarbamazine Delays and Decreases the NETosis of Polymorphonuclear Cells of Humans with DM Type 2. J. Diabetes Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kaur, T.; Dumoga, S.; Koul, V.; Singh, N. Modulating neutrophil extracellular traps for wound healing. Biomater. Sci. 2020, 8, 3212–3223. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef]
- Menegazzo, L.; Scattolini, V.; Cappellari, R.; Bonora, B.M.; Albiero, M.; Bortolozzi, M.; Romanato, F.; Ceolotto, G.; de Kreutzeberg, S.V.; Avogaro, A.; et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. 2018, 55, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, P.; Gao, M.; Yu, T.; Shi, Y.; Zhang, M.; Yao, M.; Liu, Y.; Zhang, X. NLRP3 activation induced by neutrophil extracellular traps sustains inflammatory response in the diabetic wound. Clin. Sci. 2019, 133, 565–582. [Google Scholar] [CrossRef]
- Huang, W.; Jiao, J.; Liu, J.; Huang, M.; Hu, Y.; Ran, W.; Yan, L.; Xiong, Y.; Li, M.; Quan, Z.; et al. MFG-E8 accelerates wound healing in diabetes by regulating “NLRP3 inflammasome-neutrophil extracellular traps” axis. Cell Death Discov. 2020, 6, 84–99. [Google Scholar] [CrossRef]
- Yang, S.; Gu, Z.; Lu, C.; Zhang, T.; Guo, X.; Xue, G.; Zhang, L. Neutrophils extracellular traps are markers of wound healing impairment in patients with diabetic foot ulcers treated in a multidisciplinary setting. Adv. Wound Care 2020, 9, 16–27. [Google Scholar] [CrossRef]
- Tesmer, L.A.; Lundy, S.K.; Sarkar, S.; Fox, D.A. Th17 cells in human disease. Immunol. Rev. 2008, 223, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.J.; Boniface, K.; Chan, J.R.; McKenzie, B.S.; Blumenschein, W.M.; Mattson, J.D.; Basham, B.; Smith, K.; Chen, T.; Morel, F.; et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 2007, 8, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Anderson, D.E.; Baecher-Allan, C.; Hastings, W.D.; Bettelli, E.; Oukka, M.; Kuchroo, V.K.; Hafler, D.A. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature 2008, 454, 350–352. [Google Scholar] [CrossRef]
- Manel, N.; Unutmaz, D.; Littman, D.R. The differentiation of human T (H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat. Immunol. 2008, 9, 641–649. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Frangou, E.; Chrysanthopoulou, A.; Mitsios, A.; Kambas, K.; Arelaki, S.; Angelidou, I.; Arampatzioglou, A.; Gakiopoulou, H.; Bertsias, G.K.; Verginis, P.; et al. REDD1/autophagy pathway promotes thromboinflammation and fibrosis in human systemic lupus erythematosus (SLE) through NETs decorated with tissue factor (TF) and interleukin-17A (IL-17A). Ann. Rheum Dis 2019, 78, 238–248. [Google Scholar] [CrossRef]
- Ahmed, S.; Misra, D.P.; Agarwal, V. Interleukin-17 pathways in systemic sclerosis-associated fibrosis. Rheumatol. Int. 2019, 39, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.P.; Hunziker, P. Atherosclerosis: Insights into Vascular Pathobiology and Outlook to Novel Treatments. J. Cardiovasc. Transl. Res. 2020, 13, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Safi, R.; Kallas, R.; Bardawil, T.; Mehanna, C.J.; Abbas, O.; Hamam, R.; Imad Uthman, I.; Kibbic, A.-G.; Nassar, D. Neutrophils contribute to vasculitis by increased release of neutrophil extracellular traps in Behçet’s disease. J. Dermatol. Sci. 2018, 92, 143–150. [Google Scholar] [CrossRef]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.; Satchell, S.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.H.; Berden, J.H.M.; et al. Neutrophil Extracellular Traps Drive Endothelial to-Mesenchymal Transition. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef]
- Aldabbous, L.; Abdul-Salam, V.; McKinnon, T.; Duluc, L.; Pepke-Zaba, J.; Southwood, M.; Ainscough, A.J.; Hadinnapola, C.; Wilkins, M.R.; Toshner, M.; et al. Neutrophil Extracellular Traps Promote Angiogenesis Evidence from vascular pathology in pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; DiPietro, L.A. Toll-Like Receptor Function in Acute Wounds. Adv. Wound Care 2017, 6, 344–355. [Google Scholar] [CrossRef]
- Lebre, M.C.; van der Aar, A.M.G.; van Baarsen, L.; van Capel, T.M.M.; Schuitemaker, J.H.N.; Kapsenberg, M.L.; de Jong, E.C. Human Keratinocytes Express Functional Toll-Like Receptor 3, 4, 5, and 9. J. Investig. Dermatol. 2007, 127, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Guo, S.; Ranzer, M.J.; DiPietro, L.A. Toll-Like receptor 4 Has an essential role in early skin wound healing. J. Investig. Dermatol. 2013, 133, 258–267. [Google Scholar] [CrossRef]
- Singh, K.; Agrawal, N.K.; Gupta, S.K.; Mohan, G.; Chaturvedi, S.; Singh, K. Increased expression of endosomal members of toll-like receptor family abrogates wound healing in patients with type 2 diabetes mellitus. Int. Wound. J. 2016, 13, 927–935. [Google Scholar] [CrossRef]
- Ewald, S.E.; Lee, B.L.; Lau, L.; Wickliffe, K.E.; Shi, G.P.; Chapman, H.A.; Barton, G.M. The ectodomain of toll-like receptor 9 is cleaved to generate a functional receptor. Nature 2008, 456, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Schoenemeyer, A.; Visintin, A.; Fitzgerald, K.A.; Monks, B.G.; Knetter, C.F.; Lien, E.; Nilsen, N.J.; Espevik, T.; Golenbock, D.T. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat. Immunol. 2004, 5, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Tripodo, C.; Gong, M.; Sangaletti, S.; Colombo, M.P.; Coffman, R.L.; Barrat, F.J. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J. Exp. Med. 2010, 207, 2931–2942. [Google Scholar] [CrossRef]
- Takeshita, F.; Leifer, C.A.; Gursel, I.; Ishii, K.J.; Takeshita, S.; Gursel, M.; Klinman, D.M. Cutting edge: Role of Toll-like receptor 9 in CpG DNA-induced activation of human cells. J. Immunol. 2001, 167, 3555–3558. [Google Scholar] [CrossRef]
- Morizane, S.; Yamasaki, K.; Mühleisen, B.; Kotol, P.F.; Murakami, M.; Aoyama, Y.; Iwatsuki, K.; Hata, T.; Gallo, R.L. Cathelicidin antimicrobial peptide LL-37 in psoriasis enables keratinocyte reactivity against TLR9 ligands. J. Investig. Dermatol. 2012, 132, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Tonello, S.; Rizzi, M.; Migliario, M.; Rocchetti, V.; Renò, F. Low concentrations of neutrophil extracellular traps induce proliferation in human keratinocytes via NF-kB activation. J. Dermatol. Sci. 2017, 88, 110–116. [Google Scholar] [CrossRef]
- Tseng, C.W.; Kyme, P.A.; Arruda, A.; Ramanujan, V.K.; Tawackoli, W.; Liu, G.Y. Innate immune dysfunctions in aged mice facilitate the systemic dissemination of methicillin-resistant S. aureus. PLoS ONE 2012, 7, e41454. [Google Scholar] [CrossRef]
- Hazeldine, J.; Harris, P.; Chapple, I.L.; Grant, M.; Greenwood, H.; Livesey, A.; Sapey, E.; Lord, J.M. Impaired neutrophil extracellular trap formation: A novel defect in the innate immune system of aged individuals. Aging Cell 2014, 13, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Bhongir, R.K.V.; Kasetty, G.; Papareddy, P.; Mörgelin, M.; Herwald, H.; Egesten, A. DNA-fragmentation is a source of bactericidal activity against Pseudomonas aeruginosa. Biochem. J. 2017, 474, 411–425. [Google Scholar] [CrossRef]
- Vollmer, J.; Weeratna, R.; Payette, P.; Jurk, M.; Schetter, C.; Laucht, M.; Sapey, E.; Lord, J.M. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur. J. Immunol. 2004, 34, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.B.; Lad, A.; Bharath Prasad, A.S.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013, 587, 2241. [Google Scholar] [CrossRef]
- Arampatzioglou, A.; Papazoglou, D.; Konstantinidis, T.; Chrysanthopoulou, A.; Mitsios, A.; Angelidou, I.; Maroulakou, I.; Ritis, K.; Skendros, P. Clarithromycin Enhances the Antibacterial Activity and Wound Healing Capacity in Type 2 Diabetes Mellitus by Increasing LL-37 Load on Neutrophil Extracellular Traps. Front. Immunol. 2018, 9, 2064. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabbatini, M.; Magnelli, V.; Renò, F. NETosis in Wound Healing: When Enough Is Enough. Cells 2021, 10, 494. https://doi.org/10.3390/cells10030494
Sabbatini M, Magnelli V, Renò F. NETosis in Wound Healing: When Enough Is Enough. Cells. 2021; 10(3):494. https://doi.org/10.3390/cells10030494
Chicago/Turabian StyleSabbatini, Maurizio, Valeria Magnelli, and Filippo Renò. 2021. "NETosis in Wound Healing: When Enough Is Enough" Cells 10, no. 3: 494. https://doi.org/10.3390/cells10030494
APA StyleSabbatini, M., Magnelli, V., & Renò, F. (2021). NETosis in Wound Healing: When Enough Is Enough. Cells, 10(3), 494. https://doi.org/10.3390/cells10030494