1. Introduction
A loss of neurons in our brain could lead to severe defects in our bodily functions [
1]. Several factors, such as protein aggregates and inflammation, could lead to the degeneration of neurons [
2,
3]. Additionally, since our brain is an organ that uses a great deal of energy, deprivation of oxygen and glucose could also cause severe neurodegeneration [
4,
5]. Within a few minutes of oxygen–glucose deprivation (OGD), the electrochemical gradient in neurons collapses, leading to their degeneration [
6].
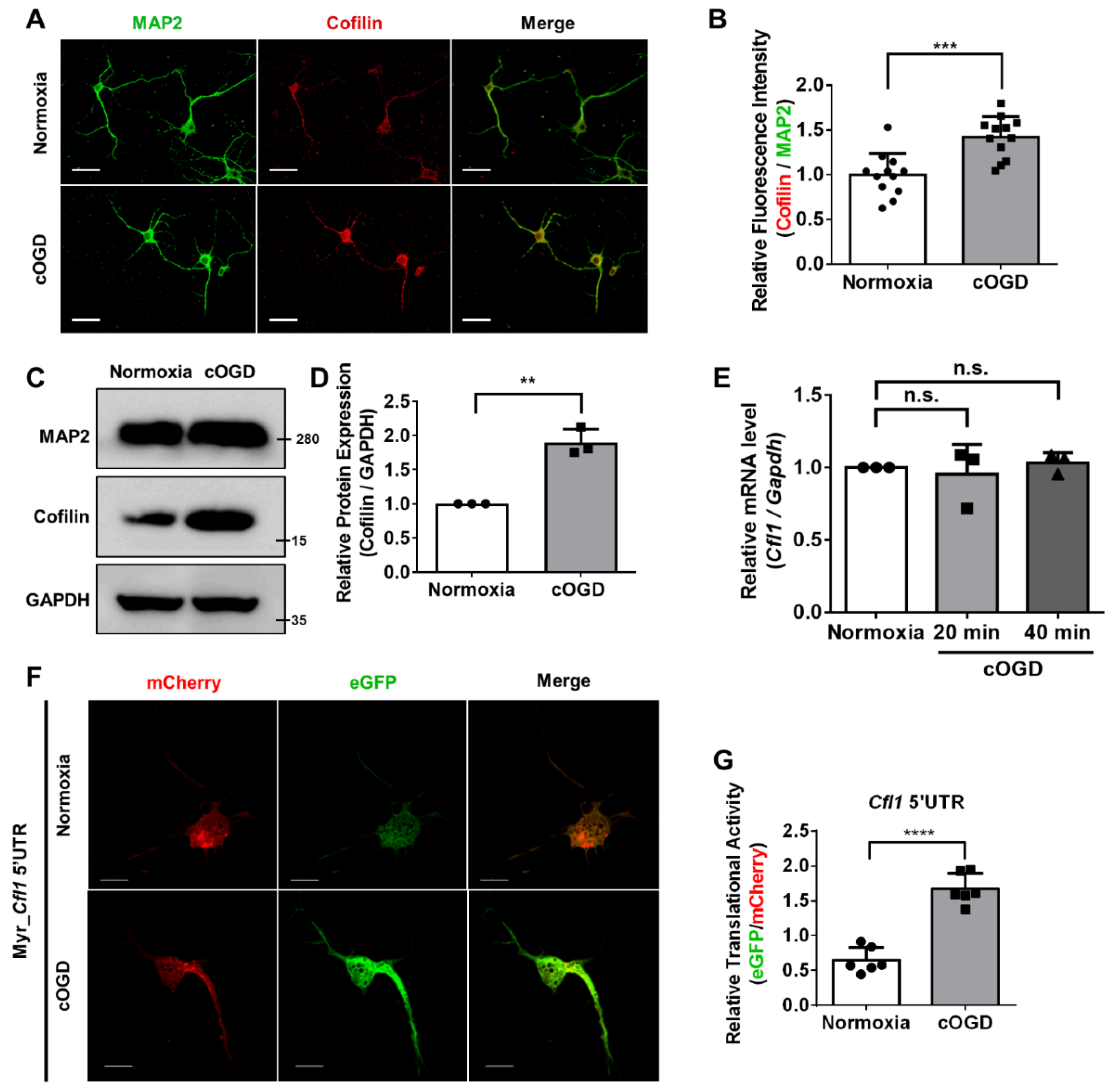
One of the hallmarks found in neurodegeneration caused by oxygen–glucose deprivation is cofilin–actin aggregates [
7]. There are several types of cofilin–actin aggregates found in neurodegenerative disease patients [
8]. These cofilin–actin aggregates are toxic to neurons because they interfere with the intracellular trafficking system, which induces synaptic loss [
9]. Cofilin–actin aggregates are known to be formed when the intracellular level of reactive oxygen species (ROS) rises due to oxidative stress [
10,
11]. Interestingly, a previous study also showed that upregulated expression of
Cofilin (
Cfl1) could induce the formation of the aggregates [
12]. Despite its importance, studies on the regulatory mechanism behind the expression of
Cfl1 under both normal and disease condition are rare. We have shown that the local translation of
Cfl1 mRNA preferentially happens through an internal ribosome entry site (IRES) [
13]. However, the relationship between previously found mechanisms and brain disease remains unclear.
To address such a challenge, we used primary hippocampal neurons cultured in chemically oxygen–glucose deprived (cOGD) conditions and a transient cerebral ischemia model mouse induced by transient middle cerebral artery occlusion (tMCAO) as a model for in vivo neurodegeneration and investigated the regulatory mechanism behind the expression of Cfl1.
2. Materials and Methods
2.1. Cell Culture and hnRNP Q1 KO Cells
Neuro2A (n2a) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Hyclone) with 10% fetal bovine serum (FBS) (Hyclone) and 1% penicillin–streptomycin (Welgene). The cells were cultured in a CO
2 incubator at 37 °C. HnRNP Q1 KO cells were previously generated in our laboratory [
14]. Briefly, single-guide RNA (sgRNA) was designed through an online CRISPR design tool at
http://crispr.mit.edu, accessed on 20 July 2019. Then, the double-stranded DNA of our target (hnRNP Q1) was cloned into the pSpCas9(BB)-2A-Puro (PX459) plasmid (Addgene, cat. 62988). PX459 plasmid were then transfected into Neuro2A cells, which were later isolated through puromycin selection (2 μg/mL; Sigma-Aldrich). HnRNP Q1 KO cells were also cultured in DMEM with 10% FBS and 1% penicillin–streptomycin in a CO
2 incubator at 37 °C.
2.2. Plasmid Construction
The plasmids that were used in this study were generated previously by our lab [
13]. Briefly, the PCR product of 5′UTR of mouse
Cfl1 (Primers: forward: ACGCGTCGACGCCGGAAGGCCGCCCCG; reverse: TCCCCCCGGGGTTTCCGGAAACGAAAGGGAGAC) was cloned into the pRF bicistronic vector that contains coding sequences of renilla luciferase (Rluc) and firefly luciferase (Fluc). Plasmids with the deletion of the D1 region of
Cfl1 5′UTR (pRF ΔD1) were also generated similarly. These plasmids were used to measure the translational activity of
Cfl1 5′UTR. The Rluc and Fluc coding sequences of pRF vectors were then replaced with coding sequences of mCherry and eGFP fluorescent protein with a myristoylation signal to measure the translational activity of
Cfl1 5′UTR in the primary hippocampal neuron. 5′UTR of Cfl1 mRNA was also cloned into pSK vectors for in vitro transcription.
2.3. RNA Interference
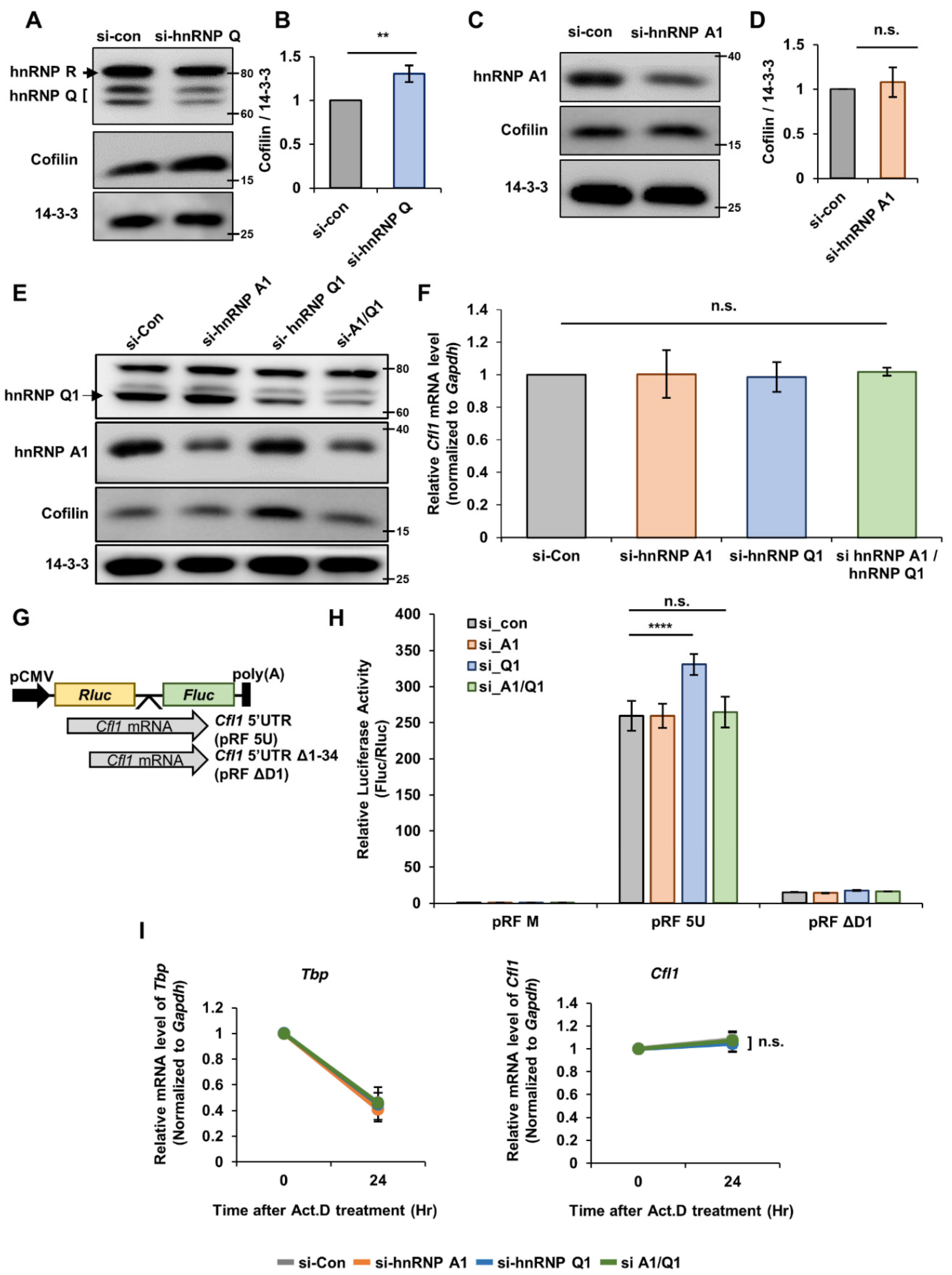
N2a cells were transfected with small interfering RNAs (siRNAs) or short hairpin RNAs (shRNA) by electroporation using NEON™ transfection system (Invitrogen, Waltham, MA, USA) or by Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruction. The siRNAs that were used in this study are as follows: si-control (Bioneer; 5′-CCUACGCCACCAAUUUCGU-3′), si-hnRNP Q (Bioneer; 5′-AGACAGUGAUCUCUCUCAUTT-3′), si-hnRNP Q1 (Bioneer; 5′-GAUCAGAAGAGGAAAGAAATT-3′), si-hnRNP A1 (Bioneer; 5′-GGACUGUAUUUGUGACUAA-3′), and si-nPTB, which was bought from Dharmacon (siGENOME SMARTpool Mouse Ptbp2 siRNA; M-049626). The siRNA sequences of hnRNP Q1 and hnRNP A1 were used to generate shRNA. The oligonucleotides of hnRNP Q1 and hnRNP A1 were annealed and inserted into pLentiLox3.7 (pLL3.7) lentiviral plasmid.
2.4. SDS-Polyacrylamide Gell Electrophoresis (PAGE) and Immunoblotting
A total of 20 or 30 μg of cell lysates (protein in cell lysates) were mixed with 5× sample buffer (0.6% 1M Tris, 50% Glycerol, 10% SDS, 0.5% 2-Mercaptoethanol, and 1% Bromophenol blue) to create loading samples. The samples were loaded onto the Western blot gel and were resolved in electrophoresis chambers (Bio-Rad, Hercules, CA, USA). Then, the proteins in the gel were transferred to nitrocellulose membranes (Pall Corporation, NY, USA) using the same power supply and transfer chamber (Bio-Rad). For immunoblotting, the membranes were incubated with the primary antibodies for 12 h at 4 °C followed by secondary antibody for 2 h at room temperature. The membranes were visualized with the LAS-4000 system (FUJIFILM, Tokyo, Japan) after treating the membrane with enhanced chemiluminescent (ECL) solution.
2.5. RNA Extraction and RT-qPCR
The RNA from harvested cells or brain tissues were extracted using TRI reagent according to the manufacturer’s instructions. Briefly, the cell or brain tissues were homogenized in the TRI reagent before adding chloroform totaling 1/5th of the original volume. Then, the samples were incubated at room temperature for 10 min before centrifugation at 15,000 rpm, at 4 °C, for 10 min. Then, the supernatant of the samples was moved to a fresh e-tube, and an equal volume of isopropanol was added to the sample. After incubating the samples in ice for 10 min, the samples were centrifuged at 15,000 rpm, at 4 °C, for 10 min. After removing isopropanol from the samples, RNA pellets were washed in ethanol and dissolved in DEPC-treated water. Isolated RNAs from the cell or brain tissue were reverse-transcribed with Improm-II reverse transcription system from Promega following the provider’s instructions. The cDNA from RT-PCR was used to measure the RNA level of cells or brain tissue. FastStart Universal SYBR Green Master from Roche was used for the reaction while StepOnePlus Real-Time system was used to measure the level. Different primers of the target genes were used as follows: Cfl1 (mouse), 5′-GCCAACTTCTAACCACAATAG-3′ and 5′-CCTTACTGGTCCTGCTTCC-3′; Gapdh (mouse), 5′-AAATGGTGAAGGTCGGTGTG-3′ and 5′-TGAAGGGGTCGTTGATGG-3′; Rpl32 (mouse), 5′-AACCCAGAGGCATTGACAAC-3′ and 5′-CACCTCCAGCTCCTTGACAT-3′; Tbp (mouse), 5′-CAGCCTTCCACCTTATGCTC-3′ and 5′-TTGCTGCTGCTGTCTTTGTT-3′; Hnrnpa1 (mouse), 5′-CTGTCGAAGCAAGAGATGGC-3′ and 5′-GCCTCCTCCATAACCACCAT-3′; Syncrip (mouse), 5′-ACCACCTCCAGATTCCGTTT-3′ and 5′-GCCTCTTGTGCTGCTTCTTT-3′; Hif1α (mouse), 5′-CTATGGAGGCCAGAAGAGGGTAT-3′, and 5′-CCCACATCAGGTGGCTCATAA-3′.
2.6. Luciferase Assay
Neuro2a cells were co-transfected with pRF vectors and siRNAs and harvested 24 h after transfection. Cells were lysed with the reporter lysis buffer (Promega, Madison, WI, USA), incubated in ice for 10 min and centrifuged at 15,000 rpm, at 4 °C, for 10 min before measuring the activity of luciferase of renilla or firefly. The luciferase activity was measured using Dual-Luciferase Reporter Assay system (Promega) as instructed by the manufacturer.
2.7. mRNA Stability Assay
To measure the stability of an mRNA, n2a cells were treated with actinomycin D (Sigma-Aldrich, St. Louis, MO, USA, A9415) (5 μg/mL), a transcription blocker, for the indicated times. Then, the RNAs were extracted from the cell as explained previously. The mRNA stability was measured through RT-qPCR.
2.8. RNA Immunoprecipitation
The n2a cells were lysed with RNA-IP buffer (10 mM HEPES (pH 7.5), 100 mM of KCl, 5 mM of MgCl2, 0.1% NP-40, protease inhibitor) and the lysates were incubated with mouse IgG, anti-hnRNP Q, or anti-hnRNP A1 at 4 °C for overnight. Then, the mixture was further incubated with Protein G agarose bead (Thermo Fisher Scientific, Waltham, MA, USA) for 4 to 6 h. The beads were washed three times before isolating the protein-bound RNAs using TRI reagent. The level of RNAs that were bound to the protein were quantified by RT-qPCR as explained previously.
2.9. In Vitro RNA Binding Assay
pSK vectors that contain Cfl1 5′UTR were linearized with Xba I restriction enzyme. The linearized vectors were in vitro transcribed using T7 RNA polymerase (Promega) in the presence of biotin-uridine 5′-triphosphate (UTP). The biotinylated RNA transcript of D1 region or D2 region of Cfl1 5′UTR (Cfl1 5′UTR D1 or Cfl1 5′UTR D2) was generated by Bioneer (Daejeon, Korea). The biotinylated RNA transcripts of Cfl1 5′UTR were incubated with normal n2a cell lysates or siRNA-transfected n2a cell lysates and were further subjected to incubation with streptavidin agarose beads (Thermo Fischer Scientific). The proteins that were bound on streptavidin agarose beads were pulled down and analyzed by SDS-PAGE followed by immunoblotting.
2.10. Primary Hippocampal Neuron Culture
Primary hippocampal neuron was prepared from E18 mouse embryos. The hippocampi were dissected and isolated from mouse embryo, which were later dissociated with trypsin (Sigma-Aldrich) and deoxyribonuclease I (Sigma-Aldrich). The dissociated tissues were plated onto poly-L-lysine-coated (Sigma-Aldrich) dishes for biochemical analysis or onto a poly-L-lysine-coated microscope cover glass for image analysis. Primary hippocampal neurons were cultured and maintained with Neurobasal media (Gibco, Waltham, MA, USA) supplemented with B27 (Gibco) and GlutaMAX-I (Gibco) in a humidified CO
2 chamber (5%) at 37 °C. The media was exchanged with fresh media every 3 days. Different DNA vectors or siRNAs were transfected with Lipofectamine
TM 2000 (Invitrogen) at DIV 1 or DIV 2. The chemically oxygen–glucose-deprived (cOGD) neuron was induced between DIV 5~7 as described previously [
7]. Briefly, the Neurobasal media that the neurons were cultured in was exchanged with balanced salt solution (BSS) (1.2 mM of CaCl
2, 0.4 mM of MgSO
4, 5.3 mM of KCl, 0.4 mM of KH
2PO
4, 137 mM of NaCl
2, 0.3 mM of NaHPO
4, 5 mM of glucose, and 10 mM of 1,4-piperazinediethanesulfonate (PIPES) buffer, pH 7.3.) or BSS with 6 mM of 2-deoxyglucose and 10 mM sodium azide (cOGD BSS). After 20 or 40 min of incubation at 37 °C, the control BSS or cOGD BSS were replaced with fresh control BSS and were further incubated at 37 °C for 10 min. Then, the control neuron or cOGD neuron were subjected to different experiments such as Western blot, immunocytochemistry, or fluorescent in situ hybridization. To minimize the effect of glial cells, the cultured plate was treated with 5 µM of cytosine arabinoside (AraC).
2.11. Immunocytochemistry
After siRNA transfection, shRNA transfection, or culturing in cOGD condition, mouse hippocampal neurons were fixed in 4% paraformaldehyde (PFA)–PBS solution for 20 min at room temperature. After several washes with PBS, the hippocampal neurons were permeabilized with 0.5% Triton X-100 dissolved in PBS for 10 min at room temperature, blocked with 5% FBS-PBS solution for 2 h and incubated with primary antibodies for 12 h at 4 °C. Then, the neurons were washed in PBS several times before they were incubated in Alexa Fluor-conjugated secondary antibodies for 1 h at room temperature. Neurons were then mounted with the fluorescent mounting medium (Dako) and were further observed with fluorescent microscope. The “n” represents the number of independent experiments.
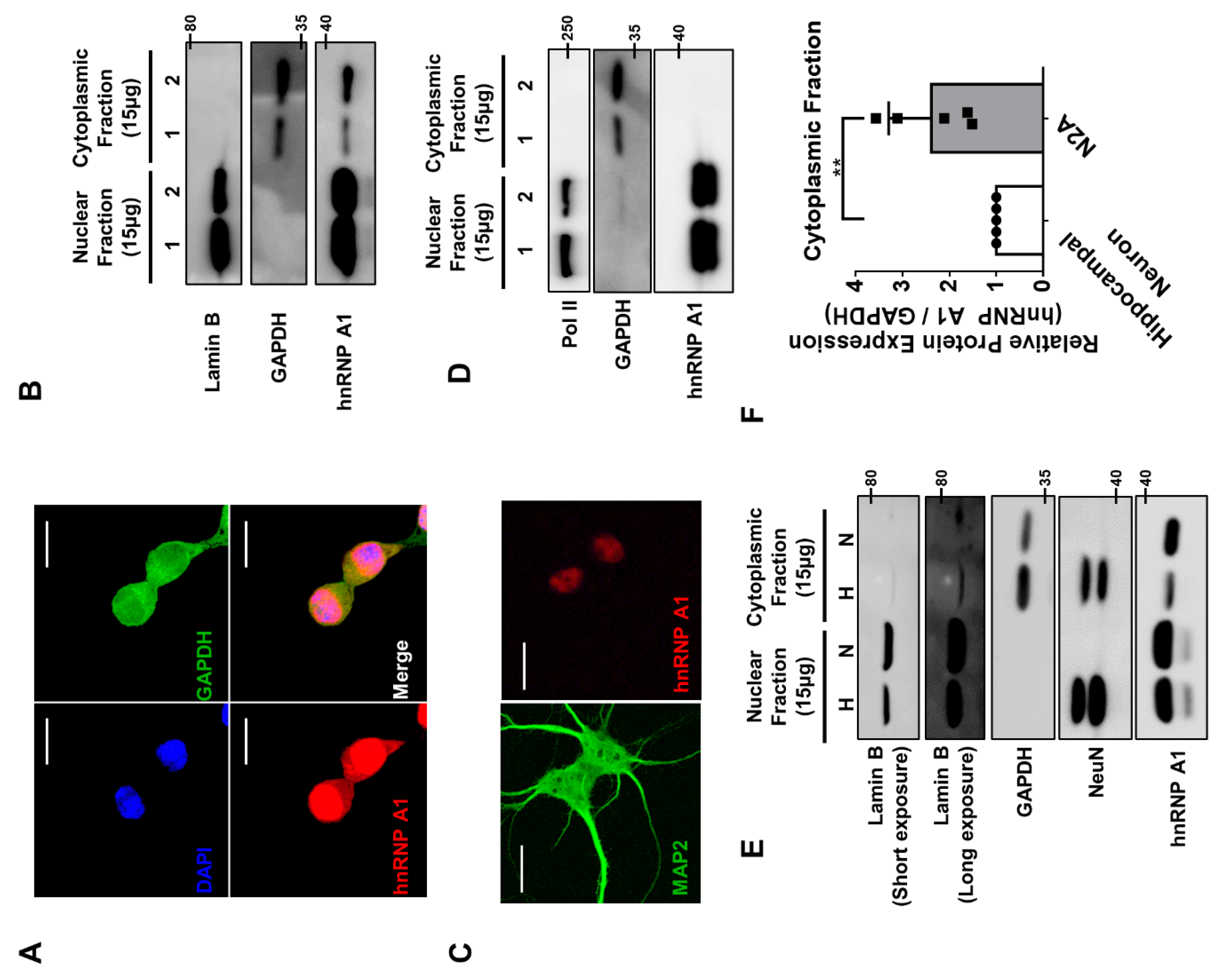
2.12. Fractionation
To divide the nuclear fraction and cytoplasmic fraction of n2a cell or primary hippocampal neuron, the harvested cells or neurons were re-suspended in fractionation buffer (10 mM of HEPES, 10 mM of KCl, and 0.05% Np-40; pH 7.4). After 20 min of incubation in ice, re-suspended cells or neurons were centrifuged at 15,000 rpm at 4 °C for 10 min. After centrifugation, the cells or neurons are divided into supernatants and pellets. The supernatant was moved to a fresh e-tube and was used to analyze the cytoplasmic fraction of the cells and neurons. The pellet was re-suspended in radioimmunoprecipitation assay (RIPA) buffer and was used to analyze the nuclear fraction of the cells and neurons. Both the cytoplasmic fraction and nuclear fraction of cell lysates were further resolved by SDS-PAGE, immunoblotted with different antibodies, and visualized with the LAS-4000 system. To isolate the axonal fraction of neuron from the soma fraction, we cultured hippocampal neuron on a hanging insert (SPL) with filters (pore size 3.0 µm). Then, the top side of the filters were cleaned with cotton swab to remove the soma of the neuron. The filters were then isolated by cutting around the circumference of the hanging insert, and they were lysed with lysis buffer (axonal fraction). The samples were further resolved by SDS-PAGE, immunoblotted with different antibodies, and visualized with the LAS-4000 system.
2.13. RNA Fluorescence In Situ Hybridization (FISH)
RNA FISH was performed with Stellaris® RNA FISH products from Biosearch Technologies (Hoddesdon, UK) according to the manufacturer’s instructions. Briefly, after the incubation with secondary antibodies from immunocytochemistry, the primary hippocampal neurons were washed several times with PBS. Then, the neurons were fixed in 4% PFA once again for 20 min at room temperature. Then, the neurons are washed with Wash Buffer A (Biosearch Technologies) for 5 min at room temperature, after several washes with PBS. While washing, custom probes that target Cfl1 mRNA (Biosearch Technologies) and Hybridization Buffer (HB; Biosearch Technologies) were mixed in the ratio noted by the instructions. After the wash, the neurons are incubated with the probe–HB solution for 4 h at 37 °C. Then, the neurons were washed with Wash Buffer A for 30 min and with Wash Buffer B (Biosearch Technologies) for an additional 5 min at room temperature, before being mounted on the slide glass. “n” represents the number of independent experiments.
2.14. Mice and Transient Middle Cerebral Artery Occlusion (tMCAO)
Mice that were used in this study was used with protocols approved by the Institutional Animal Care and Use Committees of Sookmyung Women’s University. Transient middle cerebral artery occlusion (tMCAO) was performed to induce the transient cerebral ischemia model mouse. Before the MCA occlusion, a mouse was first narcotized with 2.0% isoflurane (VS Pharm, Hanam, Korea) in 30% oxygen and 70% nitrous oxide using a facemask. Every mouse was placed on a heating pad during the entire surgical procedure to maintain the body temperature. Then, the midline of neck of the mouse was incised to expose the left common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA). After clipping the CCA, two knots were made on either side of the ECA. After cutting in between the two knots of the ECA, nylon monofilament suture (Filament size 6–0, diameter 0.09–0.11 mm, length 20 mm; Doccol Corporation, Sharon, MA, USA) was inserted into the lumen of ECA, which was further advanced to the lumen of ICA, to block the blood flow. After 45 min of blocking the blood flow by tMCAO, the mouse brain was re-perfused as the nylon suture was removed. Two knots of the ECA were closed, and the clip was removed from CCA. The midline of the neck of the mouse was closed by stitching. The sham mice were surgically operated on in a similar way except for the cutting of the ECA and insertion of nylon suture. The mice were sacrificed 24 h after the tMCAO surgery.
2.15. Immunohistochemistry
The brains of tMCAO mice were isolated and fixed with 4% PFA-PBS solution. Then, the brains were dehydrated through a sequential wash with different percentages of ethanol before they were frozen with O.C.T. compound Tissue-Tek (Sakura, Torrance, CA, USA, 4583). The 30-micrometer-thick coronal sections of frozen tMCAO brains were collected using a cryostat (Leica CM1850). The brain sections were washed three times with PBS and blocked with blocking buffer (5% Fetal Bovine serum, 3% bovine serum albumin, 0.3% Triton X-100 in PBS) for 1 h at room temperature. Then, the brain sections were incubated with primary antibodies that were dissolved in blocking buffer for 12 h at 4 °C. After several washes with PBS, the brain sections were incubated with Alexa Fluor™ (Invitrogen)-conjugated secondary antibodies for 1 h at room temperature. Then, the brain sections were stained with Hoechst (5 μg/mL) for 10 min after several washes with PBS. The brain sections were then mounted with the fluorescence mounting medium (Dako, Thermo Fischer) before imaging with Zeiss LSM 800 Epifluorescence microscope. “n” represents the number of mice.
2.16. Imaging
For immunocytochemistry analysis and RNA FISH, FV1000 Confocal microscope (Olympus) or FV3000 Confocal Laser Scanning microscope (Olympus, Tokyo, Japan) was used. The fluorescent images were obtained using Coherent® High Performance OBIS™ laser with wavelengths of 405 nm, 488 nm, and 561 nm. The z-stack images were also taken using the same imaging system (11~12 stacks, 0.480 μm/slices, Olympus). The images were exported using the FV31S-SW program. For immunohistochemistry analysis, the fluorescent signals were detected with Zeiss LSM 800 Epifluorescence microscope with excitation and emission wavelengths of 358/461 nm, 495/519 nm, and 590/617 nm. The intensity or co-localization of fluorescent signals of immunocytochemistry images or immunohistochemistry images were analyzed using ImageJ (NIH, Bethesda, MD, USA).
2.17. Antibody
For primary antibodies in Western blot analysis, Anti-hnRNP Q (1:1000; Sigma-Aldrich, R5653), Anti-hnRNP A1 (1:1000; Santa Cruz, sc-32301), Anti-Cofilin (1:250; Abcam, ab54532, ab42824), Anti-14-3-3ζ (1:1000; Santa Cruz, sc-1657, sc-1019), Anti-GAPDH (1:1000; Bethyl, A300-641A), normal Anti-Mouse IgG (1:1000; Santa Cruz, sc-2025), Anti-nPTB (1:1000; Abcam, ab154787), Anti-Lamin B (1:1000; Santa Cruz, sc-6216), Anti-RNA PolII (1:1000; Abcam, ab5408), and Anti-NeuN (1:1000; Sigma-Aldrich, MAB377) were used. For secondary antibodies in Western blot analysis, horseradish peroxidase (HRP)-conjugated Anti-Mouse IgG (1:10,000; Invitrogen, 31430), HRP-conjugated Anti-Rat IgG (1:10,000; Bethyl, A110-105P), HRP-conjugated Anti-Goat IgG (1:10,000; Bethyl, A50-101P), and HRP-conjugated Anti-Rabbit IgG (1:10,000; Promega, W4018) were used. For primary antibodies in immunocytochemistry and immunohistochemistry analysis, Anti-Cofilin (1:100; Abcam, ab54532, ab42824), Anti-hnRNP Q (1:200; Sigma-Aldrich, R5653), Anti-hnRNP A1 (1:200; Santa Cruz, sc-32301), Anti-MAP2 (1:500; Abcam, ab5392), Anti-Tau (1:300; Abcam, ab64193), and Anti-NeuN (1:100; Sigma-Aldrich, MAB377) were used. For secondary antibodies in immunocytochemistry and immunohistochemistry analysis, Anti-Mouse IgG conjugated with Alexa Fluor™ 488 (1:1000; Invitrogen, A-11,001), Alexa Fluor™ 405 (1:1000; Invitrogen, A-31553), or Alexa Fluor™ 594 (1:1000; Invitrogen, A-11005) was used. Additionally, Anti-Rabbit IgG conjugated with Alexa Fluor™ 488 (1:1000; Invitrogen, A-11008), Alexa Fluor™ 405 (1:1000; Invitrogen, A-31556), or Alexa Fluor™ 594 (1:1000; Invitrogen, A-11012) and Anti-Chicken IgY conjugated with Alexa Fluor™ 488 (1:1000; Abcam, ab150169). To detect cofilin–actin rods in primary hippocampal neurons, Alexa Fluor™ 488-conjugated phalloidin (Invitrogen, A12379) was used to stain filamentous actin (F-Actin) according to the manufacturer’s instructions.
2.18. Experimental Design and Statistical Analysis
All cell-based data are the results of at least three independent experiments performed with cells from different passage. All microscopy experiments with primary hippocampal neurons were repeated more than 3 times with neurons from different biological samples (mice). The comparison between the two groups were statistically analyzed by unpaired Student’s t tests. Comparisons between three or more groups with one independent variable were analyzed by ordinary one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. When there were two independent variables in the experiment, ordinary two-way ANOVA with Tukey’s multiple comparison test was used for analysis. All quantitative data are presented as means ± SD. p values greater than 0.05 were not considered significant. The significance of the statistical analysis was indicated as such: n.s., not significant, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, and **** p ≤ 0.0001.
4. Discussion
Although a relationship between the expression of
Cfl1 and the cofilin–actin aggregates formation was previously suggested [
12], the regulatory mechanism behind the expression of
Cfl1 under OGD conditions was unclear. Here, we revealed an important regulatory mechanism of
Cfl1 expression during OGD that could promote the formation of the cofilin aggregates. We initially found that the translational activity of
Cfl1 mRNA is increased in cOGD neurons and that two RNA-binding proteins, hnRNP Q and hnRNP A1, interact with the 5′UTR of
Cfl1 mRNA at similar locations. However, the mechanism behind how these proteins bind at similar locations of
Cfl1 mRNA is yet to be found. Previous studies revealed that RNA-binding proteins usually interact with specific motif sequences of RNA [
27]. In this case, hnRNP Q and hnRNP A1 may share similar binding motifs, which exist on the D1 region of
Cfl1 mRNA. Additionally, this may be why less hnRNP A1 interact with
Cfl1 mRNA in the presence of hnRNP Q. The binding motif for hnRNP A1 may not be available when
Cfl1 mRNA is bound by hnRNP Q, a relatively bigger-sized protein. Two proteins may also form a complex, like other RNA-binding protein complexes that regulate the translation [
16,
17]. In the presence of hnRNP Q, hnRNPA1 may be recruited to hnRNP Q, instead of
Cfl1 5′UTR. All in all, further investigations are needed to reveal how they interact at
Cfl1 mRNA.
We further illustrated that hnRNP Q inhibits the interaction between hnRNP A1 and
Cfl1 mRNA and further suppresses the translational activity of
Cfl1 mRNA. However, there was a clear discrepancy in this mechanism between n2a cells and primary hippocampal neurons. Unlike the results found with n2a cells, hnRNP Q or hnRNP A1 did not alter the level of cofilin nor the translation of
Cfl1 5′UTR in primary hippocampal neurons. This divergence may come from differences in the characteristics of the cell line and primary cells. Many times, the scientists use the immortalized cells because they are easier to maintain and use during experiments. However, the problem with the cells is that they are sometimes misidentified over multiple passage of culturing [
22,
23]. Additionally, the environment in which the cells are cultured is often different from the actual tissue which they were isolated from. This difference may affect the whole protein profiling of the cells and reduce its biological relevance [
24,
25]. This may be the case with hnRNP A1 in n2a cell and primary hippocampal neurons. In n2a cells, hnRNP A1 localizes in both the nucleus and cytoplasm, while it mainly localizes in the nucleus of neurons. Due to the difference, the interaction between hnRNP A1 and
Cfl1 mRNA could not have been increased by the knockdown of hnRNP Q in neurons since there is almost no hnRNP A1 to interact with in the cytoplasm.
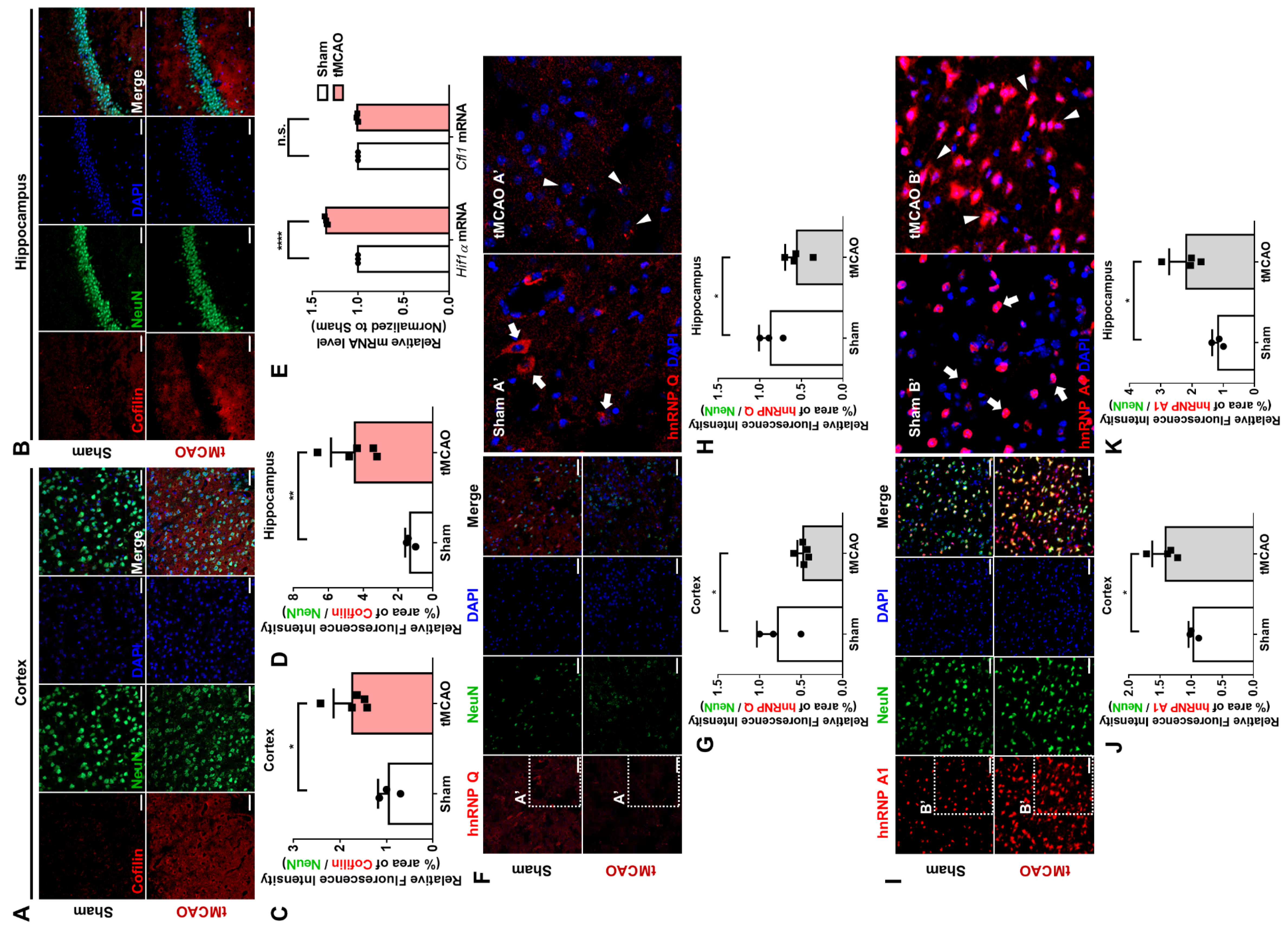
We then demonstrated that hnRNP Q and hnRNP A1 re-localizes under stress caused by OGD and alters the co-localization between the proteins and
Cfl1 mRNA. The interaction between hnRNP A1 and
Cfl1 mRNA especially increases significantly under cOGD conditions. We found that hnRNP Q re-localizes to the granule-like spots in the soma, while hnRNP A1 re-localizes from the nucleus to the soma and axon. Previous studies show that proteins may change their localization under high oxidative stress conditions [
28,
29]. A study revealed that arsenite-induced oxidative stress could induce the cytoplasmic re-localization of hnRNP A1, which then participates in mRNA translation [
28]. Others revealed that oxidative stress re-localizes hnRNP Q from the axonal transport granule to the cytoplasmic stress granule located in the soma [
29]. Since cOGD neurons and the tMCAO mouse mimic the stressful environment of neurodegeneration, hnRNP Q and hnRNP A1 could surely change their location. Additionally, we cannot exclude the effects of glial cells on the response of hippocampal neurons to a stressful environment. Although we added cytosine arabinoside (AraC) to minimize the effect of glial cells, we could not completely eliminate them. Thus, further experiments examining the mechanism behind the re-localization of proteins, which may be responses of neurons to a stressful environment, should be conducted.
Finally, we showed that altering the expression of hnRNP Q or hnRNP A1 to inhibit the translation of
Cfl1 mRNA significantly alleviates the formation of cofilin–actin aggregates. Cofilin–actin aggregates are toxic to neurons as they can induce neurodegeneration by interrupting the intracellular vesicular transport system and synaptic function [
12,
30]. These toxic aggregates are transiently formed under OGD conditions, such as an ischemic injury [
31]. Our study revealed a key mechanism behind the formation of the toxic aggregations, which suggests a possible therapeutic approach. We found that increasing the level of hnRNP Q or decreasing the level of hnRNP A1 inhibits the formation of cofilin–actin aggregates. Like other RNAi systems used in different disorders [
32,
33], RNAi therapy targeting hnRNP A1 may alleviate the aggregate formation in the neurons and prevent the neurodegeneration. Altogether, our study revealed a novel regulatory mechanism behind the translation of
Cfl1 mRNA and the formation of cofilin–actin aggregates during oxygen–glucose deprivation.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}